

Abstract
Lysosomal diseases are inherited metabolic disorders caused by defects in a wide spectrum of lysosomal and a few non-lysosomal proteins. In most cases a single type of primary storage material is identified, which has been used to name and classify the disorders: hence the terms sphingolipidoses, gangliosidoses, mucopolysaccharidoses, glycoproteinoses, and so forth. In addition to this primary storage, however, a host of secondary storage products can also be identified, more often than not having no direct link to the primary protein defect. Lipids - glycosphingolipids and phospholipids, as well as cholesterol - are the most ubiquitous and best studied of these secondary storage materials. While in the past typically considered nonspecific and nonconsequential features of these diseases, newer studies suggest direct links between secondary storage and disease pathogenesis and support the view that understanding all aspects of this sequestration process will provide important insights into the cell biology and treatment of lysosomal disease.
Keywords: sphingolipid, glycosphingolipid, ganglioside, lactosylceramide, glucosylceramide, sphingomyelin, phospholipid, bis(monoacylglycero)phosphate, cholesterol
1. Introduction
Lysosomal diseases represent a complex family of nearly 60 inherited metabolic disorders linked by defects in specific proteins critical for lysosomal function [1]. A majority of these disorders are severe, and many are characterized by a progressive neurodegenerative and mental deterioration. Collectively, their estimated incidence ranges between 1/5000 to 1/9000 live births. Most are due to the deficiency of a specific hydrolytic lysosomal enzyme (or its ancillary “activator” protein) directly responsible for degradation of complex molecules, or of a lysosomal membrane transport protein involved in egress from lysosomes of one of the final products. They can also be the consequence of a multiple deficiency of lysosomal enzymes due to a defect in ER- and Golgi/TGN-associated enzymes involved in a common processing/trafficking step. Finally they can be due to defects in soluble or transmembrane lysosomal proteins of still partially unclear function believed essential for substrate degradation, vesicle fusion, pH regulation, substrate salvage, trafficking/ sorting of lipids and proteins and so forth. Indeed, defects in no less than 50 different proteins have been implicated to date as causing lysosomal dysfunction, and new proteins linked to lysosomal disease continue to be identified. Defects in lysosomal function typically lead to lysosomal storage and in the case of single hydrolytic enzyme deficiencies, primary storage materials can usually be readily identified. Yet in reality the storage process affecting cells is far more complex than suggested by this simple one enzyme-one substrate relationship, with multiple substrate storage being typically characteristic. While in some cases this heterogeneity can be explained by a metabolic link, with multiple compounds sensitive to the same enzyme, most examples of secondary storage appear unrelated to this and more likely arise through other, often still poorly defined mechanisms. Importantly, considerable evidence suggests that secondary storage compounds can themselves be actively involved in disease pathogenesis. In order to gain a full understanding of the cell biology and pathogenesis of lysosomal disease, it is essential to understand the basis for these complex metabolic events leading to multiple substrate storage. Various types of compounds may accumulate. Some of them are particularly important in certain diseases, such as the c-subunit of mitochondrial ATP synthase or saposins in ceroid lipofuscinoses, whereas others are more ubiquitous, among which lipids constitute the most common and best studied category and are therefore the focus of this review.
2. Patterns of secondary lipid accumulation
Secondarily accumulating lipids in lysosomal disease can belong to any of the major classes, cholesterol, phospholipids and glycosphingolipids (GSLs), with different patterns in brain and in visceral organs often occurring in the same disease. Most of the changes have been initially described using biochemical methods, first in tissues of patients and then in various animal models. More recently, studies using antibodies (especially against GSLs) or cytochemical staining for cholesterol have provided new insights into the cellular site of accumulation. Ancillary studies in living cells have also been useful to pinpoint some secondary metabolic blocks.
2.1 Glycosphingolipids and free sphingoid bases
Glycosphingolipids in brain
A secondary accumulation of GM2 and GM3 gangliosides is a common feature associated with neuropathology in a number of lysosomal storage diseases, principally Niemann-Pick diseases and mucopolysaccharidoses, but also prosaposin deficiency, as well as some glycoproteinoses and ceroid lipofuscinoses. In human brain, GM2 and GM3 are normally very minor components and constitute no more than 1-2% of the total gangliosides. Their proportion is even smaller in normal mouse brain. The largest increase, affecting both gangliosides but more particularly GM2, has been found in autopsy brain of patients with Niemann-Pick disease type A (but not type B) [2-7], and in patients with Niemann-Pick C disease (NPC) [7-13]. Very similar changes have been found in sphingomyelinase-deficient and in NPC1- or NPC2-deficient models in mice or cats [14,15]. For either Niemann-Pick A or C diseases, abnormalities were absent in human brain at the foetal stage [16-18]. However, for both diseases, ganglioside storage occurs early, as shown by studies in mutant mice. In the npc1nih mouse brain, abnormal levels of GM2 were already present in 10-day old animals, with a later increase of GM3 [15; L. Verot, P. Lobel and MT Vanier, unpublished]. Abnormalities were observed for both GM2 and GM3 in the brain of a 3-month old child with NPC [13]. Pronounced abnormalities have also been reported in prosaposin deficiency (a combined defect in the sphingolipid activator proteins saposins A, B, C and D), both in humans and the mouse model [19,20], and very recently in the cathepsin D deficient mouse (model of CLN10) [21]. In Niemann-Pick A and C as well as in prosaposin deficiency, the ganglioside increase is accompanied by a prominent increase in glucosylceramide and lactosylceramide [9,13,18-20]. Clearly abnormal levels of GM3 and GM2, although significantly lower, are further seen in mucopolysaccharidosis (MPS) type III brain, in human, mice and emu bird [22-26], and similar but even milder changes occur in MPS I (Hurler type) brain [22,23] and α-iduronidase deficient cats, dogs and mice [28-31] (Fig. 1). In MPS I and MPS IIIB mice, ganglioside abnormalities appear around one month of age, and quantitatively, GM3 appears more affected than GM2. Only few data exist regarding glycoproteinoses but definite abnormalities have been reported for α-mannosidosis in the cat [29]. No increase was found in I-cell disease [32]. In many other lysosomal and some non-lysosomal diseases, a small increase in the proportion of GM2 and GM3 is often seen; however, this is usually more conspicuous by immunocytochemical methods than by biochemical measurement. This can be explained by the fact that biochemistry will take into account the global amount of gangliosides, irrespective of its subcellular localization, while immunocytochemistry can pinpoint even tiny amounts, when present in vesicular structures in cells.
Figure 1.

Gangliosides GM3 and GM2 in brain from mouse models of several lysosomal storage diseases. All mice were studied in the laboratory of one of us (MTV) using the same procedure. Results are expressed in nmol ganglioside/g wet weight of tissue. WT: wild type; PSAP: prosaposin deficiency; ctsd: cathepsin D deficiency (CLN10); Hex-0: Hexosaminidase A + B deficiency (GM2-gangliosidosis, Sandhoff variant); NPC1: NPC1 deficiency (Niemann-Pick C); MPSI: α-iduronidase deficiency; MPSIIIb: α-N-acetylglucosaminidase (NAGLU) deficiency; NPA: acid sphingomyelinase deficiency (Niemann-Pick A); Hex-A: hexosaminidase A deficiency (GM2-gangliosidosis, Tay-Sachs variant). Age at study varied from 1 month to 10 months, as indicated; Note that the GM2 level in the Sandhoff mouse at 1 month of age is already higher than in the Tay-Sachs model at 10 months of age.
The availability of well characterized antibodies to GM2 and GM3 gangliosides, coupled with biochemical data described above indicating only a minimal presence of these two gangliosides in normal brain, has meant that their localization in storage diseases could be identified in individual cell types in brain, and indeed, to individual vesicles within neurons. It has also been found that in some cases probing tissues with antibodies fails to readily reveal individual gangliosides in specific sites (e.g., the plasmalemma). This inaccessibility most likely is due to the blocking action of associated proteins, or simply because the ganglioside molecules do not occur in sufficient abundance to be labeled and identified. This cryptic feature of gangliosides in terms of immunocytochemistry again indicates the importance of combined biochemical and morphological techniques. Immunostaining studies applied to lysosomal diseases undergoing secondary lysosomal accumulation of GM2 and GM3 have consistently shown that these gangliosides are sequestered in vesicles, appearing as punctate, granular structures within the cytoplasm of cells. This labeling has been documented in a wide variety of lysosomal diseases, including Niemann-Pick type A [Walkley, unpublished] and type C (both NPC1 and NPC2 deficiencies) [15,33], MPS diseases including type I, II, IIIA, VI, and VII [34,35], mucolipidosis type IV [36], several of the Batten disorders, including CLN2 [Walkley, unpublished], CLN6 and CLN10 diseases [21], and α-mannosidosis [37,38]. In most such diseases, GM2 and GM3 have been found to accumulate in a variety of neurons and glia, with both gangliosides typically occurring within the same cells. Many other patterns of staining, however, are also evident. For example, in mice with Niemann-Pick C disease Purkinje cells accumulate GM2 ganglioside but not GM3 [15]. In mice lacking cathepsin D (CLN10 disease) it appeared that neurons accumulated GM2 whereas glia primarily harbored GM3 storage [21]. In the glycoproteinosis, α-mannosidosis, all neurons exhibited storage of water soluble oligosaccharides as expected, whereas only scattered numbers of pyramidal and GABAergic neurons in the cerebral cortex also exhibited conspicuous accumulation of GM2 and GM3 gangliosides [37]. As discussed later, this accumulation of gangliosides in α-mannosidosis revealed a close correlation between the abnormal presence of GM2 ganglioside in pyramidal neurons and ectopic dendritogenesis, a phenomenon originally discovered in GM2 gangliosidosis [39] and now known to be unique to lysosomal diseases with GSL storage.
The availability of appropriately fixed tissues from large and small animal models of lysosomal disease, coupled with immunofluorescence and confocal microscopy, has permitted detailed analysis of the subcellular localization of gangliosides. Unexpectedly, GM2 and GM3 gangliosides, while often being sequestered within the same neurons, have been consistently found to reside in separate vesicle populations in those cells [34,40] (Fig. 2). As described below, these labeled vesicles also often occur independently of detectable amounts of unesterified cholesterol visualized by filipin labeling. That GM2 and GM3 are not co-sequestered has been interpreted to mean either that these gangliosides are sequentially processed in separate compartments within the endosomal/lysosomal system, or that these two gangliosides are being generated by separate and independent processes, e.g., in synthetic vs. degradative compartments in cells [40,41].
Figure 2.
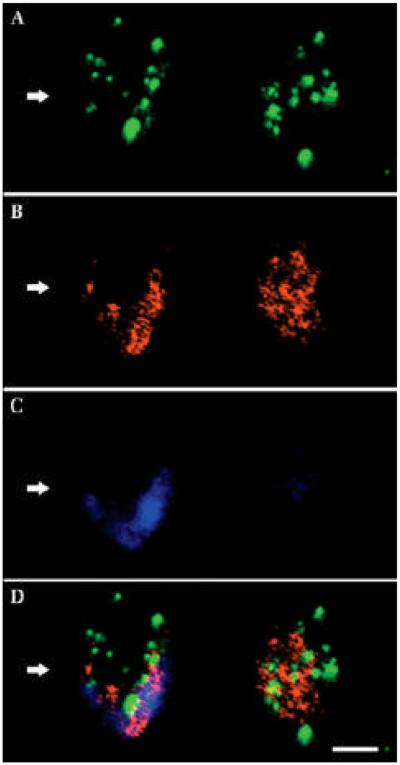
Gangliosides GM3 and GM2 and cholesterol in two adjacent cerebrocortical neurons in murine MPS IIIA disease. Confocal analysis reveals the subcellular localization of GM2, GM3, and cholesterol. A: GM2 immunofluorescence. B: GM3 immunofluorescence. C: Filipin histochemistry (or unesterified cholesterol). D: Merged image. As is evident, most labeled vesicles in A and B contained either GM2 or GM3 gangliosides, but very few showed significant co-labeling (D). While most cholesterol also appeared to occur independently of ganglioside-containing vesicles, GM3 ganglioside and cholesterol exhibited similar subcellular distribution, as shown by the neuron on left (arrow). Note that the cell on the right in C (and D) does not exhibit significant cholesterol accumulation (it is largely filipin-negative). Scale bar in D equals 10 microns and applies to A–D. (From ref. 34, used with permission).
Other secondary changes in brain ganglioside composition have been reported in lysosomal storage diseases such as globoid cell leukodystrophy (Krabbe disease) [50] or the infantile type of CLN1 [51], where they seem to reflect invasion of the brain by cells of mesenchymal origin, prominent astrogliosis or prominent loss of gray matter (for CLN1 disease), rather than lysosomal dysfunction.
Glycosphingolipids in liver and spleen
With regard to secondary GSL storage in non-neural organs, only few diseases with prominent hepatosplenomegaly have been studied in detail. Interestingly, both the sphingomyelinase-deficient types (A and B) of Niemann-Pick diseases and Niemann-Pick type C again share common features, with an increase of the major ganglioside in these organs (GM3 in humans or cats, GM2 in mice) as well as several neutral glycolipids, glucosylceramide, lactosylceramide, globotriaosylceramide and globoside [44-46]. GM3 is also typically increased in liver and spleen from Gaucher disease [53,54]. Lactosylceramide alterations in lysosomal storage diseases have recently been reviewed [49]. Most prominent increases have been described in Niemann-Pick diseases and prosaposin deficiency.
Free sphingoid bases
The pathophysiological role of free sphingoid bases, sphingolipid metabolism intermediates normally present in minute amounts but showing many-fold elevation in certain lysosomal diseases, may have been underestimated and is currently being revisited (see below). Sphingosine and sphinganine share the common feature of a free amino group with lysosphingolipids. The latter have been shown to be highly cytotoxic, and a specific increase of the lysocompound corresponding to the main substrate for a defective sphingohydrolase – and thus closely linked to the primary defect - has been well documented in Krabbe [50-52] and Gaucher disease [47,53,54], but was also reported in GM2 gangliosidosis [55,56], metachromatic leukodystrophy [57], sphingomyelinase deficiencies [6] and Fabry disease [58]. In Krabbe disease brain, galactosylsphingosine is currently still considered as the primary offending metabolite [59] (see below). With regard to free sphingoid bases, a large increase was shown to occur in liver of the npcnih mouse and of NPC patients [60-62]. This finding was recently extended to the NPC1 cat model as well as to NPC2-deficient patients and mouse [Vanier and coworkers, unpublished data]. Subcellular fractionation disclosed a lysosomal localization of the accumulated compounds in NPC liver, and the sphingosine/sphinganine ratios indicated that the compounds originated from sphingolipid degradation and not biosynthesis [60,61]. Cultured skin fibroblasts from classical NPC patients also showed elevated amounts of these metabolites, and data on phorbol ester binding suggested a deleterious effect on protein kinase C (PKC) [63]. While in Niemann-Pick C disease the sphingosine increase appeared disproportionately high in relation with the concomitant sphingolipid increase, it did not appear specific to this disorder, although few data are yet available [6,63]. However, only a very modest increase was observed in Niemann-Pick C brain [at most 3 to 5-fold normal values in humans, mouse and cat] compared to the 20-50 fold elevation occurring in liver or spleen [6; Vanier and coworkers, unpublished data]. Biochemical data in brain of other diseases are essentially lacking today. Further, the lack of suitable antibodies for intermediate sphingolipid metabolites has meant that more refined studies using morphological (confocal) techniques have not been possible. More recent studies regarding the potential deleterious role of sphingoid bases in Niemann-Pick C disease is discussed below [see section 3.2]
2.2 Cholesterol
From our current knowledge on the function of the NPC1 or NPC2 proteins, the prominent storage of unesterified cholesterol occurring in liver and spleen tissue in Niemann-Pick C disease should be considered as a primary consequence of the gene defect. In the liver, interestingly, the mouse model accumulates much larger cholesterol amounts (up to 8-10 fold the normal level) [64] than found in human patients in biopsies or at autopsy (generally 2-3 fold the normal level)[46]. An enhanced secretion of lipoproteins and increased lipid synthesis has been demonstrated in NPC1-deficient mouse hepatocytes [65] and studies in the mouse model suggest that the late endosomal/lysosomal content of unesterified cholesterol correlates with cell damage [64] .On the other hand, the marked progressive increase in cholesterol concentrations observed in liver and spleen of patients with Niemann-Pick A or B is clearly secondary to the deficiency in acid sphingomyelinase. An important issue with regard to several lysosomal storage diseases is actually the coordination of cholesterol metabolism with that of sphingomyelin and of glycosphingolipids. Sphingomyelin levels correlate closely with the amounts of cholesterol in different membranes and it has long been hypothesized that the two lipids might form a complex. The presence of sphingomyelin is also known to influence cholesterol movement and its distribution among membranes [66]. Conversely, lysosomal accumulation of unesterified cholesterol secondarily affects sphingomyelin metabolism (see below). More recently, it has further been shown that cholesterol plays a major part in regulating traffic of sphingolipids along the endocytic pathway. Indeed, lysosomal cholesterol overload correlates with abnormalities observed in Niemann-Pick C cells, but quite unexpectedly, these studies also showed that cholesterol homeostasis was perturbed in a number of sphingolipidoses, secondary to sphingolipid accumulation [67,68]. Finally, free sphingoid bases, progesterone and many cationic amphiphiles known to elicit drug-induced lipidoses have all been shown to affect intracellular cholesterol trafficking [69-73]. Indeed, a significant increase in cholesterol concentrations is typically part of the storage pattern observed in drug-induced lipidoses in the peripheral organs, but not in brain [74]. Similarly, and despite the putative functions on cholesterol transport of the NPC1 and NPC2 proteins, biochemical studies in brain tissue from NPC patients or mouse models have failed to demonstrate a significant mass increase of unesterified cholesterol. It has been argued that this might be explained by the concomitant myelin loss. This is certainly a valid argument for studies conducted on whole brain tissue, like those in mice. However, by biochemical measurements, no significant cholesterol increase was found in dissected human gray matter from patients, a result that should alleviate such a bias [13,75]. To a large extent, this probably reflects the exclusive local source of brain cholesterol [76-78], but also a possible imbalance between neuronal bodies and distal axons [79] (see below).
While total cholesterol levels in brain are apparently not increased in lysosomal diseases, in situ labeling using filipin histochemistry reveals the presence of conspicuous sequestration of unesterified cholesterol in individual brain cells (Fig. 3). This accumulation occurs as storage-like granules in cell bodies of neurons and glia and has been reported in a wide spectrum of lysosomal diseases, including not only Niemann-Pick C disease [15,33] but also GM1 and GM2 gangliosidosis, , α-mannosidosis [80], as well as MPS I, II, IIIA, and VI diseases [34,35]. In addition to cholesterol, each of these disorders also exhibits either primary or secondary accumulation of GSLs, suggesting some type of linkage between sequestration of these two classes of compounds. Importantly, however, the accumulating cholesterol as visualized with filipin labeling does not systematically co-localize with each of the sequestered gangliosides. For example, in MPS disease, while vesicular GM3 storage appears closely allied with areas of filipin labeling in cells, GM2 does not reveal a similar association (Fig. 2). For Niemann-Pick type C disease in which defects in the cholesterol-binding proteins, NPC1 or NPC2 are causative [15], cholesterol is believed to be sequestered as a result of an induced block in its retroendocytic movement from late endosomes and lysosomes to other sites in the cell [81]. The presence of accumulating unesterified cholesterol in the cell bodies of neurons and glia in brain, in view of the absence of increases in total cholesterol (as described above), have suggested possible shifts in the distribution of the cholesterol rather than absolute increases. Evidence for this can be found in studies examining cholesterol localization in neuronal cell bodies vs. axons in culture which appear to show elevated perikaryal cholesterol but decreased axonal cholesterol [76]. Such findings, as well as others described below suggestive of functional cholesterol deficits in NPC disease, indicate that the abnormal sequestration of materials in lysosomal disease may have significant consequences for neuron function.
Figure 3.

Filipin histochemical staining for unesterified cholesterol in cerebrocortical neurons in multiple lysosomal diseases. A: Wt. (12 weeks old) B: MPS IIIA disease (12 weeks old). C: Niemann-Pick disease type C (8 weeks old). D: GM1 gangliosidosis (12 weeks old). Note that neurons in Wt brain exhibit no significant filipin labeling of somata, whereas in each of the lysosomal diseases there is substantial filipin labeling of individual neurons. In MPS IIIA disease, some neurons are clearly more affected than others, whereas in Niemann-Pick C and GM1 gangliosidosis all neurons are positive. Filipin staining in GM1 was substantial and appeared to exceed that of Niemann-Pick C in late stage disease. Calibration bar in C equals 12 µm and applies to all.
As described above, cholesterol sequestration in Niemann-Pick C disease is also accompanied by accumulation of GSLs, including GM2 and GM3 gangliosides. Abnormal cholesterol sequestration was reported to be already present in the brain of 9 days-old npc1 mutant mice [80]. In the latter study, no ganglioside increase could be demonstrated at this age. However, significantly increased levels of GM2 ganglioside were repeatedly documented in the brain of 10-days-old mice in the authors’ laboratories (see above, section 2.1). In a manner remarkably similar to that reported for MPS disease [34], immunocytochemical studies of neurons in Niemann-Pick C have revealed the presence of GM2 and GM3 gangliosides in independent populations of vesicles which in turn (particularly for GM2) have very little overlap with cholesterol-sequestering vesicles [Walkley and co-workers, unpublished]. Interestingly, while this GM2 and GM3 accumulation has generally been considered to occur secondary to cholesterol storage in NPC disease, two independent studies have nonetheless shown that limiting expression of complex gangliosides in this condition dramatically reduces this intracellular cholesterol sequestration in neurons lacking NPC1 [83,84]. In both cases this work was carried out using double mouse mutants lacking the NPC1 protein and GalNAc transferase, the enzyme responsible for synthesis of all complex gangliosides beyond GM3 and GD3, accompanied by filipin staining to reveal intraneuronal sequestration of cholesterol. Thus while this and related GSL synthetic mutants, as well as this double mutant, lacks demonstrable impact on cholesterol homeostasis overall [85], the types of gangliosides expressed by neurons lacking NPC1 appear to ultimately dictate the degree of intraneuronal cholesterol sequestration [83,84, Walkley and co-workers, unpublished].
2.3 Phospholipids
The two phospholipids of particular interest in the context of this review are sphingomyelin and bis(monoacylglycero)phosphate (BMP), also named (quite incorrectly) lysobisphosphatidic acid (LBPA) by some authors, an anionic phospholipid enriched in internal membranes of multivesicular endosomes and lysosomes [86]. A striking accumulation of BMP was first described in liver and spleen of patients with Niemann-Pick disease type A [87] a finding extended to Niemann-Pick B and C [10,46,75, 88,89]. BMP is also a major component of the storage in non–neural organs of drug-induced lipidosis [90], but no major increase has been found to occur in Gaucher disease. Data regarding BMP in brain are lacking for most lysosomal diseases. A small increase had been reported in the brain of patients with infantile NCL (CLN1) [91,92], and a significant elevation was recently found in the brain of the cathepsin D deficient mouse (model of CLN10) [21]. Interestingly, in Niemann-Pick diseases A or C (as well as in drug-induced lipidosis), no noticeable elevation of the concentration of BMP has been found in brain [15,87,93].
Historically, all sphingomyelin-storage diseases were included under the eponym “Niemann-Pick disease”, until the early eighties and the seminal discovery by the group of P. Pentchev demonstrating abnormal intracellular trafficking of cholesterol in Niemann-Pick C disease. Early work by Crocker and Farber [94] had already pointed out that liver and spleen from patients with types C and D showed much less sphingomyelin storage than those with types A and B. It nevertheless took a long time to clearly demonstrate that in Niemann-Pick C (now including type D) disease sphingomyelin accumulation is secondary to lysosomal cholesterol storage. The first piece of evidence came from studies in cultured fibroblasts from patients showing that the typical partial deficiency of sphingomyelinase activity could be modulated by the presence or absence of LDL in the cultivating medium [95,96], and this is also supported by comparative studies of cholesterol and sphingomyelin in liver from type A and type C foetuses (Fig. 4). Note than in human Niemann-Pick type C disease, sphingomyelin accumulation remains very moderate in liver and is much higher in spleen [46] , while liver is a major site of accumulation in the mouse models [15]. Very importantly, no secondary increase of sphingomyelin as so far been described in brain for any lysosomal storage disease (including Niemann-Pick C), and this does not occur either in drug-induced lipidoses.
Figure 4.

Lipid storage in liver from 20 week-old foetuses with Niemann-Pick type A (NPA) (3 cases) and type C (NPC) (3 cases) diseases. This was compared to concentrations in foetuses with lysosomal disorders not affecting the liver (controls, 5 cases). All results are expressed as mean values for each group. In NPA, the primary stored lipid, sphingomyelin, is already very elevated, and is accompanied by a prominent secondary increase of cholesterol. In NPC, massive cholesterol storage occurs, with a modest sphingomyelin increase.
3. Mechanisms and Consequences of Secondary Storage
3.1. Complex mechanisms underlie heterogeneous substrate accumulation in lysosomal disease
As outlined above, many studies have revealed that the types of sequestered compounds and their distribution in lysosomal disease is marked by heterogeneity and in most cases no clear connection has yet been established between the primary lysosomal defect and the sequestration of secondary compounds that become involved in the storage process. Of particular focus here is the accumulation of GSLs, phospholipids, and unesterified cholesterol across a wide spectrum of lysosomal disease. Given that a principal role of the endosomal/lysosomal system is to degrade and recycle substrates derived from the cell surface by endocytosis and from inside the cell by way of autophagy, it would only be logical that single lysosomal protein defects might lead to overall dysfunction of this system and therefore to storage of multiple substrates. It might also be argued that these multiple substrates would be comingled within the same individual storage bodies accumulating within cells, a view supported by early work on Tay-Sachs disease. Cell fractionation studies of brain tissue from Tay-Sachs disease examining components in the fraction enriched in storage bodies (membranous cytoplasmic bodies) showed that one third of the dry weight to be ganglioside, mostly GM2 (but also detectable amounts of asialo-GM2, lactosylceramide, and glucocerebrosides. Parallel findings were also reported for GM1 gangliosidosis [97]. Later studies similarly revealed evidence that cytosomes in Tay-Sachs disease were characterized by accumulation of not only GM2 ganglioside but also phospholipids, cerebrosides, sphingomyelin, cholesterol and cholesterol esters, and so forth [Reviewed in 98]. While these biochemical studies could be interpreted to suggest that all storage components in lysosomal disease were co-sequestered within the same individual storage vesicles, the use of electron microscopy consistently demonstrated a significant degree of heterogeneity in the ultrastructure of cytoplasmic storage bodies. The latter finding is consistent with a differential or uneven distribution of individual storage materials. This heterogeneity of storage at the subcellular level has been further elucidated using modern confocal and immunofluorescence methods. For example, as described earlier, in MPS disease neurons accumulate not only the primary glycosaminoglycan (GAG) storage compounds secondary to individual lysosomal enzyme defects but also GM2 and GM3 gangliosides and unesterified cholesterol [34]. Importantly, these multiple substrates, as shown by fluorescent markers and confocal microscopy, are not all systematically co-localized within single types of storage vacuoles (Fig. 2). For example, the GM2 and GM3 gangliosides that accumulate in a host of lysosomal diseases largely reside in separate compartments within the cell, with GM2 showing minimal, if any, co-sequestration with cholesterol. This is similarly true in neurons affected by Niemann-Pick C disease in which cholesterol accumulation is believed primary and ganglioside sequestration secondary [Walkley and colleagues, unpublished]. Even the primary storage compound in MPS IIIa disease, heparan sulfate, does not appear consistently co-sequestered with each secondary storage compound [34].
It might be argued that such secondary sequestration of materials in neurons is of little real significance, since it occurs downstream from the primary disease event. However, the tight correlation between some specific features in the pathogenic cascade (e.g., ectopic dendritogenesis) and involvement of particular classes of secondary storage materials (in this case GSLs, particularly GM2 ganglioside) [29], provide clear evidence that understanding why secondary storage events occur likely holds important insight for these diseases as a whole. In thinking about why the defective function of a single lysosomal enzyme or other protein could lead to storage of multiple substrates, several possible mechanisms can be suggested. These could include inhibition of lysosomal enzymes by an accumulating substrate (e.g., GAGs) or some other critical disruption of the internal environment of late endosomes and lysosomes (pH change, lack of saposin-substrate interaction, etc.) leading to reduced degradation of additional substrates. It is also conceivable that lysosomal storage may in some manner compromise the egress of substrates from late endosomes/lysosomes leading to prolonged persistence at this site. Additionally, sequestration of compounds may disrupt normal trafficking or fusion of vesicles in the extended endosomal pathway, including early endosomes, recycling endosomes as well as late endosomes. This could result in compromise in movement of a variety of materials to sites for further degradation. Finally, induction of autophagy, or its blockage, may further complicate vesicle fusion and trafficking events. All of these mechanisms have been suggested to contribute to sequestration of materials in different lysosomal diseases. Understanding precisely which of these events occurs in a given lysosomal disease is important since one mechanism versus another may provide insight into additional downstream consequences, from induction of dendritogenesis to formation of axonal spheroids, two key features of the cellular pathology of many lysosomal diseases. Such understanding may also reveal close linkage between different types of lysosomal diseases, including issues of importance for therapy.
One of the earliest mechanisms explored as a cause of secondary storage of gangliosides (e.g., GM2, GM3, and GD3) was in MPS disease and involved the possibility that accumulating GAGs were binding to and inhibiting the activity of several additional lysosomal hydrolases. One early study [99] demonstrated partial deficiencies of β-galactosidase, α-galactosidase, and arylsufatase A in the livers of patients with MPS disease, possibly as a result of secondary GAG binding and isoelectric points being made more acid. Experimentally mixing chondroitin sulfate and β-galactosidase also diminished its activity providing further support to this view. Similar results were reported in another study showing that 21 different lysosomal enzyme activities in leukocytes were strongly inhibited by mixing them in vitro with various GAGs, with the mechanism again believed to be possibly secondary to GAG-enzyme binding [100]. A third study showed that neuraminidase activities toward GD1a, GD3 and GM3 were diminished in fibroblasts of MPS diseases, possibly due to binding of sulfated GAGs with the enzyme [101]. While a plausible mechanism for disrupting ganglioside degradation, it is not readily apparent why different MPS disorders with different types of GAG storage each result in storage of essentially the same pattern of gangliosides. As described earlier, this accumulation consists primarily of GM2 and GM3 gangliosides, yet blockage of GM2 degradation would in itself suggest the presence of less GM3. While inhibition of neuraminidase could explain the GM3 elevation, β-hexosaminidase (for degrading GM2 ganglioside) has not been reported to be inhibited in the MPS diseases. Similarly, GM1 has not been reported to accumulate in spite of the reported sensitivity of β-galactosidase to the proposed GAG-induced inhibition. Finally, the pattern of GM2 and GM3 sequestration in separate vesicles [34] is not readily explained by a generalized enzyme inhibition mechanism, but oddly, is also found in other lysosomal diseases (without known GAG storage) [40]. These studies raise significant questions about whether GAG-induced inhibition of lysosomal enzymes is occurring in vivo as the mechanism underlying ganglioside storage in MPS disease.
As mentioned earlier, lysosomal cholesterol homeostatic events have been found to be a significant aspect not only in Niemann-Pick C cells, but also in a number of sphingolipidoses, secondary to sphingolipid accumulation [67,68]. Given that cholesterol and glycosphingolipids, as well as sphingomyelin, are constituents of membrane specializations referred to as “rafts” has led to the provocative view that lysosomal diseases may be essentially collections of non-degraded rafts constituents within lysosomes [102]. In diseases like Niemann-Pick C, cholesterol accumulation is believed to appear first, as a result of defective function of the NPC1 or NPC2 proteins, and to be subsequently accompanied by GSL sequestration. In GM1 or GM2 gangliosidosis, the ganglioside storage would presumably be the initial event, followed by cholesterol sequestration. The extent to which this primary vs. secondary accumulation drives subsequent steps in the disease cascade is not clearly understood. Importantly, recent studies of NPC cells, and cells treated with U188666A, which is believed to mimic NPC disease [69], failed to show an alteration in the raft to non-raft membrane ratio, a finding believed to argue against the raft accumulation hypothesis [103]. This finding is also consistent with the confocal microscopy studies described earlier which indicate a significant degree of separation of cholesterol/GSL storage components within vesicular domains of lysosomal disease-affected neurons [34]. What was found in the in vitro studies instead of evidence for raft accumulation, was that cholesterol (and presumably any accompanying GSLs), leads to an inhibition of intra-endosomal membrane dynamics. Importantly, these intraluminal vesicles are believed critical for degradative processing, during which time in normal cells, the content of unesterified cholesterol has been shown to decrease while the negatively charged phospholipid, BMP, increases. Efficient degradation of GSLs at these sites requires, in addition to an acidic pH, water soluble hydrolases, sphingolipid activator proteins and anionic phospholipids like BMP [104]. The focus on abnormalities at the intra-endosomal membrane is further supported by the ultrastructure of storage vacuoles in NPC disease, known as polymembranous cytoplasmic bodies, which in essence resemble abnormal multivesicular bodies (i.e., late endosomes) [15,105]. Along this same line is the finding that in NPC fibroblasts, high cholesterol levels markedly impaired the normal co-localization of glucosylceramidase, saposin C and BMP and altered both the stability and activity of the enzyme [106]. Interestingly, recent studies have also revealed that exogenous application of BMP (LBPA) to cells with the NPC cellular phenotype has the ability to reverse the cholesterol accumulation [107].
3.2 Consequences of lysosomal storage
To a certain extent the question posed here – What are the consequences of secondary lipid storage in lysosomal disease? – is applicable to not just secondary events, but also to primary storage. For example, correlations have been drawn above between secondary storage of GM2 ganglioside in diseases like α-mannosidosis and MPS I disease and ectopic dendritogenesis [29,108]. Yet, this remarkable phenomenon was discovered in Tay-Sachs and related GM2 gangliosidoses [39] in which the storage of GM2 is the primary event. Therefore, the correlation between GM2 sequestration and the induced growth of ectopic dendrites suggests that the reason for the GM2 elevation may not matter. Rather it is the involvement of GM2 and related GSLs, coupled with lysosomal dysfunction, that appear in some way to stimulate new dendrite growth, not whether the cause of the GM2 elevation is primary or secondary. But how might such an alteration in GLS catabolism, synthesis, or localization lead to downstream consequences of disease such as ectopic dendritogenesis? A parallel question can be raised for cholesterol, that is, is there evidence that its sequestration in lysosomal disease directly relates to pathogenesis? The possible deficit of cholesterol in axons secondary to somatic sequestration [79], mentioned earlier, surely would have consequences on myelination and action potential propagation. In the Niemann-Pick C disease mouse model the sequestration of cholesterol also appears to lead to deficient production of the cholesterol-derived neurosteroid, allopregnanolone [109]. Presumably, this occurs as a result of the blockage of cholesterol movement out of late endosomes to mitochondria where enzyme systems are present for neurosteroid biosynthesis. This finding suggests that one aspect of the pathogenic cascade in lysosomal disease may relate to deficiency conditions, rather than simple overabundance of stored compounds [41]. An unanswered question related to this finding is whether the mechanism causing cholesterol sequestration matters in terms of this outcome. That is, in other conditions, like GM1 or GM2 gangliosidosis in which significant cholesterol sequestration also occurs (here not primary but clearly secondary to ganglioside storage) is allopregnanolone synthesis similarly compromised?
These types of complex events involving primary and secondary lipid accumulation in lysosomal disease clearly suggest that it is not simply dysfunction of the lysosome that is central to pathogenesis, but rather that the entire endosomal/lysosomal system, and its salvage pathways, is in some manner compromised. Endocytosis, along with involvement in regulating the expression of components of neuronal membranes through, for example, the internalization of receptor-ligand complexes, also plays a critical role in attenuating and integrating a wide variety of signaling events affecting many cellular functions [110]. Endocytic events in neurons govern a variety of signaling mechanisms, from those linked to growth factors to neurotransmitters. Growth factor receptors are internalized by endocytosis in neurons, both in the axonal and soma-dendritic domains and signaling events are believed dependent on the specifics of the endocytic route and vesicle type [111]. The finding that growth factors like brain-derived neurotrophic factor (BDNF), a TrkB ligand, when applied to young cortical neurons cause exuberant dendrite growth [112] raises the question of whether growth factor signaling might be abnormal in lysosomal disease as a result of ganglioside sequestration and thus account for the phenomenon of ectopic dendritogenesis. There is indeed one published work suggesting that Trk B signaling in Niemann-Pick C disease may be abnormal [113 should be a new 113].
Endocytic mechanisms are also known to control the availability of neurotransmitter receptors at excitatory synapses. AMPA (α-amino-3-hydroxy-5-methylisoazole-4-proprionic acid) receptors, which regulate the majority of fast excitatory neurotransmission and are therefore critical for synaptic plasticity, are known to undergo rapid constitutive internalization as a consequence of synaptic activity. Once internalized, AMPA receptors are sorted from early endosomes to either specialized recycling endosomes for re-insertion in the plasmalemma or, are trafficked to late endosomes for fusion with lysosomes and resulting elimination [114,115]. Subunits forming AMPARs (GluR1-4) are well documented to assemble as homo- or hetero-dimers, with subunit composition being critical for their functional properties. The GluR2 subunit, unlike GluR1, 3 and 4, is Ca2+-impermeable, making it the key determinant of AMPAR function [116]. Importantly, over-expression of the GluR2 subunit in cultured neurons induces ectopic dendritic spine formation, consistent with AMPA receptor expression and localization playing a major role in dendrite and synapse plasticity [117]. These findings suggest that the endosomal/lysosomal system, through control of AMPA receptor availability, is positioned to have significant influence over dendritic plasticity and raise the question of whether GSL accumulation in early or recycling endosomes is affecting this mechanism and causing ectopic dendritogenesis in lysosomal disease [41].
In addition to possible signal transduction derangements secondary to ganglioside accumulation in the endosomal/lysosomal system, other major consequences of secondary lipid sequestration can also be raised. For example, does “leakage” or altered trafficking of gangliosides out of the endosomal/lysosomal system to other sites in the cell normally devoid of significant levels of gangliosides (e.g., the endoplasmic reticulum) cause consequences for neuron function. It has been proposed, for example, that in GM1 gangliosidosis that the GM1 ganglioside mislocalizes to membrane domains normally containing little or no ganglioside, like the ER, resulting in depletion of Ca++ stores, activation of the ER stress response and eventually apoptosis and neuron death [118]. Similarly, in GM2 gangliosidosis it has been shown that GM2 ganglioside increases in microsomal membranes and inhibits the activity of SERCA, which in turn has the capacity to cause ER stress and apoptosis [119]. Parallel events in dysregulation of intracellular stores of Ca++ secondary to inappropriate ganglioside buildup in internal cell membranes may similarly be occurring in Gaucher, Niemann-Pick A and other lysosomal diseases.
A second issue in terms of the consequences of storage is whether the accumulating compounds are themselves toxic to the cells resulting in acute cell death. The best example here is the psychosine (galactosylsphingosine) hypothesis in Krabbe disease [see for review, 59]. Minute amounts of this compound have been shown to be aberrantly synthesized by oligodendrocytes. Normally psychosine will be degraded by galactosylceramidase. In Krabbe disease, in which this enzyme is deficient, galactosylsphingosine levels progressively increase, and assumedly the local concentration soon reaches a toxic level, resulting in dysfunction and finally death of the oligodendrocyte. This explains why galactosylceramide accumulates only in globoid cells and to a globally very minor amount. The cytotoxicity of this compound has been well established, but despite a number of studies following the hypothesis of Hannun and Bell on PKC inhibition [120], the exact mechanism remains unclear and is probably complex. While all experimental data seem to confirm the role of galactosylsphingosine in pathogenesis of Krabbe disease, that of other lysosphingolipids appear much weaker, with the notable exception of glucosylsphingosine in Gaucher neurons [see 59 for review]. The potential pathogenic role of free sphingoid bases in Niemann-Pick type C has recently been revisited. In NPC1 fibroblasts, one group showed hypophosphorylation of vimentin as a result of PKC inhibition, leading to entrapment of rab9 and alterations in lipid trafficking [121]. In a very recent paper [122], NPC1-mutant cells were found to have a large reduction in the acidic compartment calcium store compared to wild-type cells. The authors conclude that sphingosine storage is an initiating factor in NPC1 disease pathogenesis that causes altered calcium homeostasis, leading to the secondary storage of sphingolipids and cholesterol. [122]. No data, however, are so far available from neuronal cells, which are the primary target in the disease and store much lesser amounts of free sphingosine.
In terms of neuron death, mentioned above, it is recognized that lysosomal diseases are typically referred to in the literature as neurodegenerative. The actual role of neuron death, however, in clinical disease onset and progression is not well established. In most cases individuals with lysosomal disease appear normal in early life and only subsequently exhibit loss of developmental milestones or, if later onset, changes in cognition, behavior, motor/sensory function, and so forth. The extent to which death of neurons per se underlies this clinical change is not known and deserves greater study. Importantly, there is ample evidence indicating that neurons can survive for years, indeed decades, with some level of intraneuronal storage in lysosomal disease, so the storage process itself need not be acutely cytotoxic. It may well be the host of pathogenic cascade events, from signal transduction defects to deranged features of dendrites and axons that largely underlie chronic neuronal dysfunction. This question is made all the more relevant by therapeutic attempts designed to alleviated microglial activation and thereby, presumably, reduce the elimination of damaged neurons. This may or may not prove neurologically beneficial. On the other hand, selective neuronal vulnerability also is evident in lysosomal diseases, with Purkinje cell loss in the cerebellum in Niemann-Pick C disease perhaps being one of the best examples. It has been argued elsewhere that Purkinje cell death may be secondary to axonal spheroid formation, a predominate feature of the cellular pathology of this neuron not only in Niemann-Pick C but also other lysosomal diseases [123]. But what renders Purkinje cells so vulnerable to this event? While in Niemann-Pick C Purkinje cells are known to accumulate cholesterol and GM2 ganglioside, these same neurons in other storage diseases (e.g., α-mannosidosis) with this exact same pattern of cell loss, accumulate other materials, arguing for a more generic defect in endosomal/lysosomal function as the cause. In this case, knowledge of the secondary lipid storage components in the affected neurons has not provided a clue as to why the neurons die early.
Yet a third possible mechanism leading to compromise in cell function is whether the sequestration of materials within neurons, e.g., simple gangliosides like GM2 and GM3, are leading to states of induced “starvation” secondary to the sequestration of precursors for GSL synthesis. This possibility is somewhat parallel to the studies mentioned earlier which report allopregnanolone deficiency in Niemann-Pick C mice secondary to cholesterol sequestration [109]. GSLs may represent a second class of compounds impacted by sequestration of metabolic precursors, a view suggested many years ago by Sandhoff and colleagues [124]. This provocative idea draws support from the finding that 70-90% of the GSL pool in neurons is salvaged prior to complete degradation in the endosomal/lysosomal system [125]. Thus the sequestration of gangliosides like GM1, GM2, or GM3 in lysosomal disease may result in a deficit of precursor material in the Golgi/TGN for renewal of complex gangliosides at the plasmalemma. A possible consequence of such an event is an up-regulation GSL synthesis, an event that in turn could alter the expression of specific gangliosides or related components of membrane rafts in the plasmalemma. Such enhanced GSL synthesis likely would not solve the neuron’s deficit in precursor material, however, but rather would simply add further to the storage process. Yet another possible solution to metabolic precursor deficits might be for the neuron to “mine” resources within the cell through a mechanism known as macroautophagy. Recent studies have shown that maintaining a basal level of autophagy is critical for normal neuronal function [126,127] so in this respect they are not really different from other cells in which starvation-induced stress elicits autophagy [128]. The involvement of autophagy in lysosomal disease has recently come under increased focus [129-132], with current reports supporting both induction and blockage. Presumably, blockage in autophagy would be detrimental to neurons, given their reliance on this mechanism, but even if autophagy is increased, this again may not provide benefit since ultimately the same primary catabolic defect, at least in the case of lysosomal enzyme deficiencies, would limit degradation of the stored gangliosides in the autophagolysosomes.
4. Secondary storage compounds as biomarkers to evaluate therapy
The secondary increase in brain gangliosides GM2 and GM3 has been used as a biomarker to evaluate efficacy of gene therapy by intracerebral injection of an adeno-associated virus (AAV) coding for the defective enzyme in the MPS1 models of mouse and dog [133,134] and MPSIIIB and MPS VII mice [135,136], and of bone marrow transplantation in the feline MPSI model [31]. An excellent correlation was found between the reduction or normalization of these markers and the amelioration of primary biochemical markers or clinical features. Similarly, the beneficial effect of injection at 7 postnatal days of allopregnanolone in ß-cyclodextrin in the npcnih mouse was reflected by a significant decrease in the proportion of these two gangliosides [109]. The secondary increase of minor brain gangliosides thus appears as a good biomarker for monitoring of therapeutic efficacy in animal models of a number of lysosomal storage diseases. A major limitation is that brain tissue analysis is not applicable to patients. No study has been published regarding the levels of these gangliosides in cerebrospinal fluid of human patients with mucopolysaccharidoses or NPC. Using tandem mass spectrometry, minute amounts of compounds can now be reliably measured. However, whether the detected changes would reflect what happens in brain tissue remains unknown. The relevance to neuropsychological manifestations of lipid markers in plasma, such as increases of glucosylceramide in NPC patients [45], of GM3 in Gaucher patients [48], or modifications in the pattern of BMP species recently reported for several lysosomal diseases [136] may be even more limited. Although a considerable effort is currently pursued, as of today, the lack of suitable biomarkers constitutes a major problem for clinical trials in lysosomal diseases affecting the central nervous system.
5. Conclusions
Lysosomal diseases are complex conditions caused by a wide range of gene and protein defects and characterized by intracellular sequestration of multiple substrates, including GSLs, phospholipids and cholesterol. Some of these storage materials bear no direct relation to the primary metabolic defect and are therefore considered as secondary, yet ample evidence suggests they nonetheless play key roles in disease pathogenesis. GSLs, for example, are characteristically increased in a number of lysosomal diseases without primary defects in GSL catabolism and the intraneuronal accumulation of GM2 ganglioside correlates with ectopic dendritogenesis, a phenomenon unique to lysosomal disease. Cholesterol accumulation also occurs along with GSLs in many lysosomal diseases and evidence suggests that it may interfere with normal endosomal/lysosomal processing as well as lead to cholesterol deficits elsewhere in neurons, with consequences for neuronal function. Fully understanding the types, the cellular and subcellular locations, and the reasons for sequestration of the many secondary storage compounds characterizing lysosomal disease is critical for the full elucidation of the cell biology of these conditions, and may also provide new clues for biomarker identification and therapeutic advances.
Acknowledgments
The authors would like to thank their many colleagues who have contributed to research on storage-related events in lysosomal disease, many of whom were not mentioned in this review due to its limited space. Mice with GM1 gangliosidosis used for the study shown in Figure 3 were generously provided by Dr. Allesandra d’Azzo. We also thank the National Institutes of Health (NS053677 [SUW] and HD045561 [SUW]), the Institut National de la Santé et de la Recherche Médicale, the Ara Parseghian Medical Research Foundation, Vaincre les Maladies Lysosomales, and Dana’s Angels Research Trust for support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Platt FM, Walkley SU. Lysosomal Defects and Storage. In: Platt FM, Walkley SU, editors. Lysosomal Disorders of the Brain. Oxford University Press; Oxford: 2004. pp. 32–49. [Google Scholar]
- [2].Cumings JN. Abnormalities in lipid metabolism in two members of a amily with Niemann-Pick disease. In: Aronson SM, Volk BW, editors. Cerebral lipidoses. Academic Press; New York: 1962. pp. 171–178. [Google Scholar]
- [3].Kamoshita S, Aron AM, Suzuki K, Suzuki K. Infantile Niemann-Pick disease. A chemical study with isolation and characterization of membranous cytoplasmic bodies and myelin. Am. J. Dis. Child. 1969;117:379–394. [PubMed] [Google Scholar]
- [4].Martin JJ, Philippart M, Van Hauwaert J, Callahan JW, Deberdt R. Niemann-Pick disease (Crocker's group A). Late onset and pigmentary degeneration resembling Hallervorden-Spatz syndrome. Arch. Neurol. 1972;27:45–51. doi: 10.1001/archneur.1972.00490130047007. [DOI] [PubMed] [Google Scholar]
- [5].Brunngraber EG, Berra B, Zambotti V. Altered levels of tissue glycoproteins, gangliosides, glycosaminoglycans and lipids in Niemann-Pick's disease. Clin. Chim. Acta. 1973;48:173–181. doi: 10.1016/0009-8981(73)90363-x. [DOI] [PubMed] [Google Scholar]
- [6].Rodriguez-Lafrasse C, Vanier MT. Sphingosylphosphorylcholine in Niemann-Pick disease brain: accumulation in type A but not in type B. Neurochem. Res. 1999;24:199–205. doi: 10.1023/a:1022501702403. [DOI] [PubMed] [Google Scholar]
- [7].Crocker AC. The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. J. Neurochem. 1961;7:69–80. doi: 10.1111/j.1471-4159.1961.tb13499.x. [DOI] [PubMed] [Google Scholar]
- [8].Philippart M, Martin L, Martin JJ, Menkes JH. Niemann-Pick disease. Morphologic and biochemical studies in the visceral form with late central nervous system involvement (Crocker's group C) Arch. Neurol. 1969;20:227–238. doi: 10.1001/archneur.1969.00480090015001. [DOI] [PubMed] [Google Scholar]
- [9].Tjiong HB, Seng PN, Debuch H, Wiedemann HR. Brain lipids of a case of juvenile Niemann-Pick disease. J. Neurochem. 1973;21:1475–1485. doi: 10.1111/j.1471-4159.1973.tb06031.x. [DOI] [PubMed] [Google Scholar]
- [10].Harzer K, Schlote W, Peiffer J, Benz HU, Anzil AP. Neurovisceral lipidosis compatible with Niemann-Pick disease type C: morphological and biochemical studies of a late infantile case and enzyme and lipid assays in a prenatal case of the same family. Acta Neuropathol. (Berl) 1978;43:97–104. doi: 10.1007/BF00685003. [DOI] [PubMed] [Google Scholar]
- [11].Elleder M, Jirasek A, Smid F, Ledvinova J, Besley GT. Niemann-Pick disease type C. Study on the nature of the cerebral storage process. Acta Neuropathol. (Berl) 1985;66:325–336. doi: 10.1007/BF00690966. [DOI] [PubMed] [Google Scholar]
- [12].Elleder M. Niemann-Pick disease. Pathol. Res. Pract. 1989;185:293–328. doi: 10.1016/S0344-0338(89)80006-8. [DOI] [PubMed] [Google Scholar]
- [13].Vanier MT. Lipid changes in Niemann-Pick disease type C brain: personal experience and review of the literature. Neurochem. Res. 1999;24:481–489. doi: 10.1023/a:1022575511354. [DOI] [PubMed] [Google Scholar]
- [14].Pentchev PG, Gal AE, Booth AD, Omodeo-Sale F, Fouks J, Neumeyer BA, Quirk JM, Dawson G, Brady RO. A lysosomal storage disorder in mice characterized by a dual deficiency of sphingomyelinase and glucocerebrosidase. Biochim. Biophys. Acta. 1980 Sep 8;619:669–679. doi: 10.1016/0005-2760(80)90116-2. [DOI] [PubMed] [Google Scholar]
- [15].Sleat DE, Wiseman JA, El Banna M, Price SM, Verot L, Shen MM, Tint GS, Vanier MT, Walkley SU, Lobel P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5886–5891. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wenger DA, Kudoh T, Sattler M, Palmieri M, Yudkoff M. Niemann-Pick disease type B: prenatal diagnosis and enzymatic and chemical studies on fetal brain and liver. Am. J. Hum. Genet. 1981;33:337–344. [PMC free article] [PubMed] [Google Scholar]
- [17].Vanier MT, Boue J, Dumez Y. Niemann-Pick disease type B: first-trimester prenatal diagnosis on chorionic villi and biochemical study of a foetus at 12 weeks of development. Clin. Genet. 1985;28:348–354. doi: 10.1111/j.1399-0004.1985.tb00409.x. [DOI] [PubMed] [Google Scholar]
- [18].Vanier MT, Pentchev PG, Rousson R. Pathophysiological approach of Niemann-Pick disease type C; definition of a biochemical heterogeneity and reevaluation of the lipid storage process. In: Salvayre R, Douste-Blazy L, Gatt S, editors. Vol. 150. Plenum Press; New York: 1988b. pp. 175–185. Lipid Storage Disorders: Biological and Medical Aspects. Series A: Life Sciences 150 (1988) 175-185. [Google Scholar]
- [19].Fujita N, Suzuki K, Vanier MT, Popko B, Maeda N, Klein A, Henseler M, Sandhoff K, Nakayasu H, Suzuki K. Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum. Mol Genet. 1996;5:711–725. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- [20].Hulkova H, Cervenkova M, Ledvinova J, Tochackova M, Hrebicek M, Poupetova H, Befekadu A, Berna L, Paton BC, Harzer K, Boor A, Smid F, Elleder M. A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum. Mol. Genet. 2001 Apr 15;10:927–940. doi: 10.1093/hmg/10.9.927. [DOI] [PubMed] [Google Scholar]
- [21].Jabs S, Quitsch A, Kakela R, Koch B, Tyynela J, Brade H, Glatzel M, Walkley S, Saftig P, Vanier MT, Braulke T. Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J. Neurochem. 2008;106:1415–1425. doi: 10.1111/j.1471-4159.2008.05497.x. [DOI] [PubMed] [Google Scholar]
- [22].Constantopoulos G, Dekaban AS. Neurochemistry of the mucopolysaccharidoses: brain lipids and lysosomal enzymes in patients with four types of mucopolysaccharidosis and in normal controls. J. Neurochem. 1978;30:965–973. doi: 10.1111/j.1471-4159.1978.tb12388.x. [DOI] [PubMed] [Google Scholar]
- [23].Constantopoulos G, Iqbal K, Dekaban AS. Mucopolysaccharidosis types IH, IS, II, and IIIA: glycosaminoglycans and lipids of isolated brain cells and other fractions from autopsied tissues. J. Neurochem. 1980;34:1399–1411. doi: 10.1111/j.1471-4159.1980.tb11220.x. [DOI] [PubMed] [Google Scholar]
- [24].Van Dessel G, Lagrou A, Martin JJ, Ceuterick C, Dierick W. Two cases of mucopolysaccharidosis type III (Sanfilippo). A biochemical study. J. Neurol. Sci. 1979;40:77–86. doi: 10.1016/0022-510x(79)90193-x. [DOI] [PubMed] [Google Scholar]
- [25].Hara A, Kitazawa N, Taketomi T. Abnormalities of glycosphingolipids in mucopolysaccharidosis type III B. J. Lipid Res. 1984;25:175–184. [PubMed] [Google Scholar]
- [26].Li HH, Yu WH, Rozengurt N, Zhao HZ, Lyons KM, Anagnostaras S, Fanselow MS, Suzuki K, Vanier MT, Neufeld EF. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc. Natl. Acad. Sci. U. S. A. 1999;96:14505–14510. doi: 10.1073/pnas.96.25.14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bermudez AJ, Johnson GC, Vanier MT, Schroder M, Suzuki K, Stogsdill PL, Johnson GS, O'Brien D, Moore CP, Fry WW. Gangliosidosis in emus (Dromaius novaehollandiae) Avian Dis. 1995;39:292–303. [PubMed] [Google Scholar]
- [28].Constantopoulos G, Shull RM, Hastings N, Neufeld EF. Neurochemical characterization of canine alpha-L-iduronidase deficiency disease (model of human mucopolysaccharidosis I) J. Neurochem. 1985;45:1213–1217. doi: 10.1111/j.1471-4159.1985.tb05544.x. [DOI] [PubMed] [Google Scholar]
- [29].Siegel DA, Walkley SU. Growth of ectopic dendrites on cortical pyramidal neurons in neuronal storage diseases correlates with abnormal accumulation of GM2 ganglioside. J. Neurochem. 1994;62:1852–1862. doi: 10.1046/j.1471-4159.1994.62051852.x. [DOI] [PubMed] [Google Scholar]
- [30].Russell C, Hendson G, Jevon G, Matlock T, Yu J, Aklujkar M, Ng KY, Clarke LA. Murine MPS I: insights into the pathogenesis of Hurler syndrome. Clin. Genet. 1998;53:349–361. doi: 10.1111/j.1399-0004.1998.tb02745.x. [DOI] [PubMed] [Google Scholar]
- [31].Ellinwood NM, Colle MA, Weil MA, Casal ML, Vite CH, Wiemelt S, Hasson CW, O'Malley TM, He X, Prociuk U, Verot L, Melniczek JR, Lannon A, Aguirre GD, Knox VW, Evans SM, Vanier MT, Schuchman EH, Walkley SU, Haskins ME. Bone marrow transplantation for feline mucopolysaccharidosis I. Mol. Genet. Metab. 2007;91:239–250. doi: 10.1016/j.ymgme.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dacremont G, Kint JA, Cocquyt G. Brain sphingolipids in I cell disease (mucolipidosis II) J. Neurochem. 1974;22:599–602. doi: 10.1111/j.1471-4159.1974.tb06900.x. [DOI] [PubMed] [Google Scholar]
- [33].Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropathol. Exp. Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- [34].McGlynn R, Dobrenis K, Walkley SU. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp Neurol. 2004 Dec 20;480:415–426. doi: 10.1002/cne.20355. [DOI] [PubMed] [Google Scholar]
- [35].Walkley SU, Thrall MA, Haskins ME, Mitchell TW, Wenger DA, Brown DE, Dial S, Seim H. Abnormal neuronal metabolism and storage in mucopolysaccharidosis type VI (Maroteaux-Lamy) disease. Neuropathol. Appl. Neurobiol. 2005;31:536–544. doi: 10.1111/j.1365-2990.2005.00675.x. [DOI] [PubMed] [Google Scholar]
- [36].Micsenyi MC, Dobrenis K, Stephney G, Pickel J, Vanier MT, Slaugenhaupt SA, Walkley SU. Mucolipidosis type IV: Neuropathology of the Mcoln1−/− Knockout Mouse Model. submitted. [DOI] [PMC free article] [PubMed]
- [37].Goodman LA, Livingston PO, Walkley SU. Ectopic dendrites occur only on cortical pyramidal cells containing elevated GM2 ganglioside in α-mannosidosis. Proc. Natl. Acad. Sci. USA. 1991;88:11330–11334. doi: 10.1073/pnas.88.24.11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Walkley SU. Pyramidal neurons with ectopic dendrites in storage diseases exhibit increased GM2 ganglioside immunoreactivity. Neuroscience. 1995;68:1027–1035. doi: 10.1016/0306-4522(95)00208-z. [DOI] [PubMed] [Google Scholar]
- [39].Purpura DP, Suzuki K. Distortion of neuronal geometry and formation of aberrant synapses in neuronal storage disease. Brain Res. 1976;116:1–21. doi: 10.1016/0006-8993(76)90245-6. [DOI] [PubMed] [Google Scholar]
- [40].Walkley SU. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 2004;15:433–444. doi: 10.1016/j.semcdb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- [41].Walkley SU. Pathogenic mechanisms in lysosomal disease: a reappraisal of the role of the lysosome. Acta Paediatr. Suppl. 2007;96:26–32. doi: 10.1111/j.1651-2227.2007.00202.x. [DOI] [PubMed] [Google Scholar]
- [42].Vanier MT, Svennerholm L. Chemical pathlogy of Krabbe’s disease. III. Ceramide-hexosides and gangliosides of brain. Ata Paediatr. Scand. 1975;64:641–648. doi: 10.1111/j.1651-2227.1975.tb03896.x. [DOI] [PubMed] [Google Scholar]
- [43].Svennerholm L, Fredman P, Jungbjer B, Månsson JE, Rynmark BM, Boström K, Hagberg B, Noren L, Santavuori P. Large alterations in ganglioside and neutral glycosphingolipid patterns in brains from cases with infantile neuronal ceroid lipofuscinosis/polyunsaturated fatty acid lipidosis. J. Neurochem. 1987;49:1772–1783. doi: 10.1111/j.1471-4159.1987.tb02435.x. [DOI] [PubMed] [Google Scholar]
- [44].Philippart M. Glycolipid, mucopolysaccharide and carbohydrate distribution in tissues, plasma and urine from glycolipidoses and other disorders. In: Zambotti V, Tettamanti G, Arrigoni M, editors. Adv. Exp. Med. Biol. Vol. 25. Plenum Press; New York: 1972. pp. 231–254. [Google Scholar]
- [45].Dacremont G, Kint JA, Carton D, Cocquyt G. Glucosylceramide in plasma of patients with Niemann-Pick disease. Clin. Chim. Acta. 1974;52:365–367. doi: 10.1016/0009-8981(74)90124-7. [DOI] [PubMed] [Google Scholar]
- [46].Vanier MT. Biochemical studies in Niemann-Pick disease. I. Major sphingolipids of liver and spleen. Biochim. Biophys. Acta. 1983;750:178–184. doi: 10.1016/0005-2760(83)90218-7. [DOI] [PubMed] [Google Scholar]
- [47].Nilsson O, Månsson JE, Håkansson G, Svennerholm L. The occurrence of psychosine and other glycolipids in spleen and liver from the three major types of Gaucher's disease. Biochim. Biophys. Acta. 1982;712:453–463. doi: 10.1016/0005-2760(82)90272-7. [DOI] [PubMed] [Google Scholar]
- [48].Ghauharali-van der Vlugt K, Langeveld M, Poppema A, Kuiper S, Hollak CE, Aerts JM, Groener JE. Prominent increase in plasma ganglioside GM3 is associated with clinical manifestations of type I Gaucher disease. Clin. Chim. Acta. 2008;389:109–113. doi: 10.1016/j.cca.2007.12.001. [DOI] [PubMed] [Google Scholar]
- [49].Hulkova H, Ledvinova J, Asfaw B, Koubek K, Kopriva K, Elleder M. Lactosylceramide in lysosomal storage disorders: a comparative immunohistochemical and biochemical study. Virchows Arch. 2005;447:31–44. doi: 10.1007/s00428-005-1246-y. [DOI] [PubMed] [Google Scholar]
- [50].Vanier M, Svennerholm L. Chemical pathology of Krabbe disease: the occurrence of psychosine and other neutral sphingoglycolipids. Adv. Exp. Med. Biol. 1976;68:115–126. doi: 10.1007/978-1-4684-7735-1_8. [DOI] [PubMed] [Google Scholar]
- [51].Svennerholm L, Vanier MT, Månsson JE. Krabbe disease: a galactosylsphingosine (psychosine) lipidosis. J. Lipid Res. 1980;21:53–64. [PubMed] [Google Scholar]
- [52].Igisu H, Suzuki K. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science. 1984;224:753–755. doi: 10.1126/science.6719111. [DOI] [PubMed] [Google Scholar]
- [53].Nilsson O, Svennerholm L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982;39:709–718. doi: 10.1111/j.1471-4159.1982.tb07950.x. [DOI] [PubMed] [Google Scholar]
- [54].Orvisky E, Park JK, LaMarca ME, Ginns EI, Martin BM, Tayebi N, Sidransky E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol. Genet. Metab. 2002;76:262–270. doi: 10.1016/s1096-7192(02)00117-8. [DOI] [PubMed] [Google Scholar]
- [55].Neuenhofer S, Conzelmann E, Schwarzmann G, Egge H, Sandhoff K. Occurrence of lysoganglioside lyso-GM2 (II3-Neu5Ac-gangliotriaosylsphingosine) in GM2 gangliosidosis brain. Biol. Chem. Hoppe Seyler. 1986;367:241–244. doi: 10.1515/bchm3.1986.367.1.241. [DOI] [PubMed] [Google Scholar]
- [56].Rosengren B, Månsson JE, Svennerholm L. Composition of gangliosides and neutral glycosphingolipids of brain in classical Tay-Sachs and Sandhoff disease: more lyso-GM2 in Sandhoff disease? J. Neurochem. 1987;49:834–840. doi: 10.1111/j.1471-4159.1987.tb00969.x. [DOI] [PubMed] [Google Scholar]
- [57].Toda K, Kobayashi T, Goto I, Ohno K, Eto Y, Inui K, Okada S. Lysosulfatide (sulfogalactosylsphingosine) accumulation in tissues from patients with metachromatic leukodystrophy. J. Neurochem. 1990;55:1585–1591. doi: 10.1111/j.1471-4159.1990.tb04942.x. [DOI] [PubMed] [Google Scholar]
- [58].Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, van Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox-Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2812–2817. doi: 10.1073/pnas.0712309105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Suzuki K. Twenty five years of the "psychosine hypothesis": a personal perspective of its history and present status. Neurochem. Res. 1998;23:251–259. doi: 10.1023/a:1022436928925. [DOI] [PubMed] [Google Scholar]
- [60].Goldin E, Roff CF, Miller SP, Rodriguez-Lafrasse C, Vanier MT, Brady RO, Pentchev PG. Type C Niemann-Pick disease: a murine model of the lysosomal cholesterol lipidosis accumulates sphingosine and sphinganine in liver. Biochim Biophys Acta. 1992;1127:303–311. doi: 10.1016/0005-2760(92)90236-o. [DOI] [PubMed] [Google Scholar]
- [61].Rodriguez-Lafrasse C, Rousson R, Pentchev PG, Louisot P, Vanier MT. Free sphingoid bases in tissues from patients with type C Niemann-Pick disease and other lysosomal storage disorders. Biochim Biophys Acta. 1994;1226:138–144. doi: 10.1016/0925-4439(94)90021-3. [DOI] [PubMed] [Google Scholar]
- [62].te Vruchte D., Lloyd-Evans E, Veldman RJ, Neville DC, Dwek RA, Platt FM, van Blitterswijk WJ, Sillence DJ. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J. Biol. Chem. 2004;279:26167–26175. doi: 10.1074/jbc.M311591200. [DOI] [PubMed] [Google Scholar]
- [63].Rodriguez-Lafrasse C, Rousson R, Valla S, Antignac P, Louisot P, Vanier MT. Modulation of protein kinase C by endogenous sphingosine: inhibition of phorbol dibutyrate binding in Niemann-Pick C fibroblasts. Biochem. J. 1997;325:787–791. doi: 10.1042/bj3250787. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Beltroy EP, Liu B, Dietschy JM, Turley SD. Lysosomal unesterified cholesterol content correlates with liver cell death in murine Niemann-Pick type C disease. J. Lipid Res. 2007;48:869–881. doi: 10.1194/jlr.M600488-JLR200. [DOI] [PubMed] [Google Scholar]
- [65].Kulinski A, Vance JE. Lipid homeostasis and lipoprotein secretion in Niemann-Pick C1-deficient hepatocytes. J. Biol. Chem. 2007;282:1627–1637. doi: 10.1074/jbc.M610001200. [DOI] [PubMed] [Google Scholar]
- [66].Slotte JP, Pörn I, Härmälä AS. Flow and distribution of cholesterol-effects of phospholipids. In: Hoekstra D, editor. Cell Lipids. Current Topics in membranes. Vol. 40. Acad. press; New York: 1994. pp. 483–502. [Google Scholar]
- [67].Puri V, Watanabe R, Dominguez M, Sun X, Wheatley CL, Marks DL, Pagano RE. Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat. Cell Biol. 1999;1:386–388. doi: 10.1038/14084. [DOI] [PubMed] [Google Scholar]
- [68].Pagano RE. Endocytic trafficking of glycosphingolipids in sphingolipid storage diseases. Philos. Trans. R. Soc. Lond B Biol. Sci. 2003 May 29;358:885–891. doi: 10.1098/rstb.2003.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Liscum L, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J. Biol. Chem. 1989;264:11796–11806. [PubMed] [Google Scholar]
- [70].Rodriguez-Lafrasse C, Rousson R, Bonnet J, Pentchev PG, Louisot P, Vanier MT. Abnormal cholesterol metabolism in imipramine-treated fibroblast cultures. Similarities with Niemann-Pick type C disease. Biochim Biophys Acta. 1990;1043:123–128. doi: 10.1016/0005-2760(90)90284-5. [DOI] [PubMed] [Google Scholar]
- [71].Roff CF, Goldin E, Comly ME, Cooney A, Brown A, Vanier MT, Miller SP, Brady RO, Pentchev PG. Type C Niemann-Pick disease: use of hydrophobic amines to study defective cholesterol transport. Dev. Neurosci. 1991;13:315–319. doi: 10.1159/000112179. [DOI] [PubMed] [Google Scholar]
- [72].Butler JD, Blanchette-Mackie J, Goldin E, O'Neill RR, Carstea G, Roff CF, Patterson MC, Patel S, Comly ME, Cooney A. Progesterone blocks cholesterol translocation from lysosomes. J. Biol. Chem. 1992;267:23797–23805. [PubMed] [Google Scholar]
- [73].Lange Y, Ye J, Rigney M, Steck T. Cholesterol movement in Niemann-Pick type C cells and in cells treated with amphiphiles. J. Biol. Chem. 2000;275:17468–17475. doi: 10.1074/jbc.M000875200. [DOI] [PubMed] [Google Scholar]
- [74].Fredman P, Klinghardt GW, Nilsson O, Svennerholm L. Lipid accumulation in liver, spleen, lungs and kidneys of miniature-pigs after chloroquine treatment. Biochem. J. 1982;201:581–588. doi: 10.1042/bj2010581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Besley GT, Elleder M. Enzyme activities and phospholipid storage patterns in brain and spleen samples from Niemann-Pick disease variants: a comparison of neuropathic and non-neuropathic forms. J. Inherit. Metab Dis. 1986;9:59–71. doi: 10.1007/BF01813904. [DOI] [PubMed] [Google Scholar]
- [76].Morell P, Jurevics H. Origin of cholesterol in myelin. Neurochem. Res. 1996;21:463–470. doi: 10.1007/BF02527711. [DOI] [PubMed] [Google Scholar]
- [77].Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- [78].Vance JE, Karten B, Hayashi H. Lipid dynamics in neurons. Biochem. Soc. Trans. 2006;34:399–403. doi: 10.1042/BST0340399. [DOI] [PubMed] [Google Scholar]
- [79].Karten B, Vance DE, Campenot RB, Vance JE. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J. Neurochem. 2002;83:1154–1163. doi: 10.1046/j.1471-4159.2002.01220.x. [DOI] [PubMed] [Google Scholar]
- [80].Reid PC, Sakashita N, Sugii S, Ohno-Iwashita Y, Shimada Y, Hickey WF, Chang TY. A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J. Lipid Res. 2004;45:582–591. doi: 10.1194/jlr.D300032-JLR200. [DOI] [PubMed] [Google Scholar]
- [81].Crawley AC, Walkley SU. Developmental analysis of CNS pathology in the lysosomal storage disease alpha-mannosidosis. J. Neuropathol. Exp. Neurol. 2007;66:687–697. doi: 10.1097/nen.0b013e31812503b6. [DOI] [PubMed] [Google Scholar]
- [82].Pentchev PG, Blanchette-Mackie EJ, Dawidowicz EA. The NP-C gene: a key to pathways of intracellular cholesterol transport. Trends Cell Biol. 1994;4:365–369. doi: 10.1016/0962-8924(94)90086-8. [DOI] [PubMed] [Google Scholar]
- [83].Gondre-Lewis MC, McGlynn R, Walkley SU. Cholesterol accumulation in NPC1-deficient neurons is ganglioside dependent. Curr. Biol. 2003;13:1324–1329. doi: 10.1016/s0960-9822(03)00531-1. [DOI] [PubMed] [Google Scholar]
- [84].Liu Y, Wu YP, Wada R, Neufeld EB, Mullin KA, Howard AC, Pentchev PG, Vanier MT, Suzuki K, Proia RL. Alleviation of neuronal ganglioside storage does not improve the clinical course of the Niemann-Pick C disease mouse. Hum. Mol. Genet. 2000;9:1087–1092. doi: 10.1093/hmg/9.7.1087. [DOI] [PubMed] [Google Scholar]
- [85].Li H, Turley SD, Liu B, Repa JJ, Dietschy JM. GM2/GD2 and GM3 gangliosides have no effect on cellular cholesterol pools or turnover in normal or NPC1 mice. J Lipid Res. 2008;49:1816–1828. doi: 10.1194/jlr.M800180-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kobayashi T, Beuchat MH, Lindsay M, Frias S, Palmiter RD, Sakuraba H, Parton RG, Gruenberg J. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1999;1:113–118. doi: 10.1038/10084. [DOI] [PubMed] [Google Scholar]
- [87].Rouser G, Kritchevsky G, Knudson AG, Jr., Simon G. Accumulation of a glycerolphospholipid in classical niemann-pick disease. Lipids. 1968;3:287–290. doi: 10.1007/BF02531203. [DOI] [PubMed] [Google Scholar]
- [88].Wiedemann HR, Debuch H, Lennert K, Caesar R, Blumcke S, Harms D, Tolksdorf M, Seng PN, Korenke HD, Gerken H, Freitag F, Dorner K. [An infantile-juvenile, subchronically progressive lipoidosis of the sphingomyelinoses (Niemann-Pick) form--a new type? Clinical, pathohistological, electron microscopic and biochemical studies] Z. Kinderheilkd. 1972;112:187–225. [PubMed] [Google Scholar]
- [89].Wenger DA, Barth G, Githens JH. Nine cases of sphingomyelin lipidosis, a new variant in Spanish-American Children. Juvenile variant of Niemann-Pick Disease with foamy and sea-blue histiocytes. Am. J. Dis. Child. 1977;131:955–961. doi: 10.1001/archpedi.1977.02120220021002. [DOI] [PubMed] [Google Scholar]
- [90].Lüllmann-Rauch R. Drug-induced lysosomal storage disorders. In: Dingle JT, Jacques PJ, Shaw H, editors. Lysosomes in applied biology and therapeutics. Vol. 6. North Holland; Amsterdam: 1979. pp. 49–130. [PubMed] [Google Scholar]
- [91].Kahma K, Brotherus J, Haltia M, Renkonen O. Low and moderate concentrations of lysobisphosphatidic acid in brain and liver of patients affected by some storage diseases. Lipids. 1976;11:539–544. doi: 10.1007/BF02532899. [DOI] [PubMed] [Google Scholar]
- [92].Käkelä R, Somerharju P, Tyynela J. Analysis of phospholipid molecular species in brains from patients with infantile and juvenile neuronal-ceroid lipofuscinosis using liquid chromatography-electrospray ionization mass spectrometry. J. Neurochem. 2003;84:1051–1065. doi: 10.1046/j.1471-4159.2003.01602.x. [DOI] [PubMed] [Google Scholar]
- [93].Klinghardt GW, Fredman P, Svennerholm L. Chloroquine intoxication induces ganglioside storage in nervous tissue: a chemical and histopathological study of brain, spinal cord, dorsal root ganglia, and retinal in the miniature pig. J. Neurochem. 1981;37:897–908. doi: 10.1111/j.1471-4159.1981.tb04477.x. [DOI] [PubMed] [Google Scholar]
- [94].Crocker AC, Farber S. Niemann-Pick disease: a review of eighteen patients. Medicine (Baltimore) 1958;37:1–95. doi: 10.1097/00005792-195802000-00001. [DOI] [PubMed] [Google Scholar]
- [95].Thomas GH, Tuck-Muller CM, Miller CS, Reynolds LW. Correction of sphingomyelinase deficiency in Niemann-Pick type C fibroblasts by removal of lipoprotein fraction from culture media. J. Inherit. Metab Dis. 1989;12:139–151. doi: 10.1007/BF01800716. [DOI] [PubMed] [Google Scholar]
- [96].Vanier MT, Rodriguez-Lafrasse C, Rousson R, Gazzah N, Juge MC, Pentchev PG, Revol A, Louisot P. Type C Niemann-Pick disease: spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim. Biophys. Acta. 1991;1096:328–337. doi: 10.1016/0925-4439(91)90069-l. [DOI] [PubMed] [Google Scholar]
- [97].Suzuki K, Suzuki K, Kamoshita S. Chemical pathology of GM1 gangliosidosis (Generalized gangliosidosis) J. Neuropath. Exp. Neurol. 1969;28:27–73. doi: 10.1097/00005072-196901000-00003. [DOI] [PubMed] [Google Scholar]
- [98].Adachi M, Volk BW. The Gangliosidoses. Plenum; New York: 1975. pp. 125–158. 1975, pp. 125-158. [DOI] [PubMed] [Google Scholar]
- [99].Kint JA, Dacremont G, Carton D, Orye E, Hooft C. Mucopolysaccharidosis: secondarily induced abnormal distribution of lysosomal isoenzymes. Science. 1973;181:352–354. doi: 10.1126/science.181.4097.352. [DOI] [PubMed] [Google Scholar]
- [100].Avila JL, Convit J. Inhibition of leucocytic lysosomal enzymes by glycosaminoglycans in vitro. Biochem. J. 1975;152:57–64. doi: 10.1042/bj1520057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Baumkotter J, Cantz M. Decreased ganglioside neuraminidase activity in fibroblasts from mucopolysaccharidosis patients. Inhibition of the activity in vitro by sulfated glycosaminoglycans and other compounds. Biochim. Biophys. Acta. 1983;761:163–170. doi: 10.1016/0304-4165(83)90225-8. [DOI] [PubMed] [Google Scholar]
- [102].Simons K, Gruenberg J. Jamming the endosomal system: lipid rafts and lysosomal storage diseases. Trends Cell Biol. 2000;10:459–462. doi: 10.1016/s0962-8924(00)01847-x. [DOI] [PubMed] [Google Scholar]
- [103].Sobo K, Blanc Le I, Luyet PP, Fivaz M, Ferguson C, Parton RG, Gruenberg J, van der Goot FG. Late endosomal cholesterol accumulation leads to impaired intra-endosomal trafficking. PLoS. ONE. 2007;2:e851. doi: 10.1371/journal.pone.0000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kolter T, Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- [105].Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta. 2004;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- [106].Salvioli R, Scarpa S, Ciaffoni F, Tatti M, Ramoni C, Vanier MT, Vaccaro AM. Glucosylceramidase mass and subcellular localization are modulated by cholesterol in Niemann-Pick disease type C. J Biol. Chem. 2004;279:17674–17680. doi: 10.1074/jbc.M313517200. [DOI] [PubMed] [Google Scholar]
- [107].Chevallier J, Chamoun Z, Jiang G, Prestwich G, Sakai N, Matile S, Parton RG, Gruenberg J. Lysobisphosphatidic acid controls endosomal cholesterol levels. J. Biol. Chem. 2008 doi: 10.1074/jbc.M801463200. [DOI] [PubMed] [Google Scholar]
- [108].Walkley SU. Neurobiology and cellular pathogenesis of glycolipid storage diseases. Philos. Trans. R. Soc. Lond B Biol. Sci. 2003;358:893–904. doi: 10.1098/rstb.2003.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med. 2004;10:704–711. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- [110].Polo S, Di Fiore PP. Endocytosis conducts the cell signaling orchestra. Cell. 2006;124:897–900. doi: 10.1016/j.cell.2006.02.025. [DOI] [PubMed] [Google Scholar]
- [111].Bronfman FC, Escudero CA, Weis J, Kruttgen A. Endosomal transport of neurotrophins: roles in signaling and neurodegenerative diseases. Dev. Neurobiol. 2007;67:1183–1203. doi: 10.1002/dneu.20513. [DOI] [PubMed] [Google Scholar]
- [112].McAllister AK. Cellular and molecular mechanisms of dendrite growth. Cereb. Cortex. 2000;10:963–973. doi: 10.1093/cercor/10.10.963. [DOI] [PubMed] [Google Scholar]
- [113].Henderson LP, Lin L, Prasad A, Paul CA, Chang TY, Maue RA. Embryonic striatal neurons from Niemann-Pick type C mice exhibit defects in cholesterol metabolism and neurotrophin responsiveness. J. Biol. Chem. 2000;275:20179–20187. doi: 10.1074/jbc.M001793200. [DOI] [PubMed] [Google Scholar]
- [114].Barry MF, Ziff EB. Receptor trafficking and the plasticity of excitatory synapses. Curr. Opin. Neurobiol. 2002;12:279–286. doi: 10.1016/s0959-4388(02)00329-x. [DOI] [PubMed] [Google Scholar]
- [115].Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003 Oct 9;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- [116].Isaac JT, Ashby M, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- [117].Passafaro M, Nakagawa T, Sala C, Sheng M. Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature. 2003;424:677–681. doi: 10.1038/nature01781. [DOI] [PubMed] [Google Scholar]
- [118].d'Azzo A, Tessitore A, Sano R. Gangliosides as apoptotic signals in ER stress response. Cell Death. Differ. 2006;13:404–414. doi: 10.1038/sj.cdd.4401834. [DOI] [PubMed] [Google Scholar]
- [119].Ginzburg, Kacher Y, Futerman AH. The pathogenesis of glycosphingolipid storage disorders. Semin. Cell Dev. Biol. 2004;15:417–431. doi: 10.1016/j.semcdb.2004.03.003. [DOI] [PubMed] [Google Scholar]
- [120].Hannun YA, Bell RM. Lysosphingolipids inhibit protein kinase C: implications for the sphingolipidoses. Science. 1987;235:670–674. doi: 10.1126/science.3101176. [DOI] [PubMed] [Google Scholar]
- [121].Walter M, Chen FW, Tamari F, Wang R, Ioannou YA. Endosomal lipid accumulation in NPC1 leads to inhibition of PKC, hypophosphorylation of vimentin and Rab9 entrapment. Biol. Cell. 2008 Aug 6; doi: 10.1042/BC20070171. [DOI] [PubMed] [Google Scholar]
- [122].Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- [123].Walkley SU. Pathogenic cascades and brain dysfunction. In: Platt FM, Walkley SU, editors. Lysosomal Disorders of the Brain. Oxford University Press; Oxford: 2004. pp. 290–324. [Google Scholar]
- [124].Schwarzmann, Sandhoff K. Metabolism and intracellular transport of glycosphingolipids. Biochemistry. 1990;29:10865–10871. doi: 10.1021/bi00501a001. [DOI] [PubMed] [Google Scholar]
- [125].Tettamanti G, Bassi R, Viani P, Riboni L. Salvage pathways in glycosphingolipid metabolism. Biochimie. 2003;85:423–437. doi: 10.1016/s0300-9084(03)00047-6. [DOI] [PubMed] [Google Scholar]
- [126].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- [127].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- [128].Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- [129].Pacheco CD, Lieberman AP. Lipid trafficking defects increase Beclin-1 and activate autophagy in Niemann-Pick type C disease. Autophagy. 2007;3:487–489. doi: 10.4161/auto.4586. [DOI] [PubMed] [Google Scholar]
- [130].Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007;16:1495–1503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- [131].Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, Ballabio A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- [132].Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, Cotman SL. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J. Biol. Chem. 2006;281:20483–20493. doi: 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- [133].Desmaris N, Verot L, Puech JP, Caillaud C, Vanier MT, Heard JM. Prevention of neuropathology in the mouse model of Hurler syndrome. Ann Neurol. 2004;56:68–76. doi: 10.1002/ana.20150. [DOI] [PubMed] [Google Scholar]
- [134].Ciron C, Desmaris N, Colle MA, Raoul S, Joussemet B, Verot L, Ausseil J, Froissart R, Roux F, Cherel Y, Ferry N, Lajat Y, Schwartz B, Vanier MT, Maire I, Tardieu M, Moullier P, Heard JM. Gene therapy of the brain in the dog model of Hurler's syndrome. Ann. Neurol. 2006;60:204–213. doi: 10.1002/ana.20870. [DOI] [PubMed] [Google Scholar]
- [135].Cressant A, Desmaris N, Verot L, Brejot T, Froissart R, Vanier MT, Maire I, Heard JM. Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. J. Neurosci. 2004;24:10229–10239. doi: 10.1523/JNEUROSCI.3558-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Frisella WA, O'Connor LH, Vogler CA, Roberts M, Walkley S, Levy B, Daly TM, Sands MS. Intracranial injection of recombinant adeno-associated virus improves cognitive function in a murine model of mucopolysaccharidosis type VII. Mol. Ther. 2001;3:351–358. doi: 10.1006/mthe.2001.0274. [DOI] [PubMed] [Google Scholar]
- [137].Meikle PJ, Duplock S, Blacklock D, Whitfield PD, Macintosh G, Hopwood JJ, Fuller M. Effect of lysosomal storage on bis(monoacylglycero)phosphate. Biochem. J. 2008;411:71–78. doi: 10.1042/BJ20071043. [DOI] [PubMed] [Google Scholar]