

Abstract
The Androgen Receptor (AR) is a critical oncogene in prostate cancer (PCa) development and progression. Here we demonstrate cell cycle dependent regulation of AR activity, localization, and phosphorylation. We show that on three AR target genes, androgen-stimulated AR transactivation is highest in G1, decreased in S-phase, and abrogated in G2/M. This change in AR transactivation parallels changes in AR localization and phosphorylation. A combination of imaging techniques and quantitative analysis shows nuclear AR localization during interphase and the exclusion of the majority, but not all, AR from chromatin in mitosis. Flow cytometry analyses using a phospho-S308 AR specific antibody in asynchronous and chemically enriched G2/M PCa cells revealed ligand-independent induction of S308 phosphorylation in mitosis when CDK1 is activated. Consistent with our flow cytometry data, IP-Western blotting showed an increase in S308 phosphorylation in G2/M and an in vitro kinase assay demonstrated that CDK1 was able to phosphorylate the AR on S308. Pharmacological inhibition of CDK1 activity resulted in decreased S308 phosphorylation in PCa cells. Importantly, using a combination of anti-total AR and phospho-S308 specific antibodies in immunofluorescence experiments, we show that the AR is excluded from condensed chromatin in mitotic cells when phosphorylated on S308. In summary, we show that the phosphorylation of the AR on S308 by CDK1 in mitosis regulates AR localization and correlates with changes in AR transcriptional activity. These findings have important implications for understanding AR function as an oncogene.
Keywords: Androgen Receptor, Cell Cycle, Phosphorylation, Localization, Mitosis
Introduction
Inhibition of AR function is standard therapy for the initial presentation of disseminated prostate cancer (George & Moul 2011). However, treatment almost invariably fails leading to the emergence of castration-resistant prostate cancer (CRPC). The clinical importance of targeting AR function in CRPC is best illustrated by the FDA approval of abiraterone, a CYP17 inhibitor, and the anti-androgen, Enzalutamide. Unfortunately, the clinical benefit of these new therapies is not durable (Joseph et al. 2013). These observations further emphasize the importance of AR signaling in PCa development and progression. Only a thorough understanding of AR biology will provide novel insights into how to therapeutically target this critical driver of PCa.
The AR functions as a driver of G1 progression through cross-communication with the cell cycle machinery and regulation of transcription of genes that control the G1-S transition (Balk & Knudsen 2008). Upon androgen withdrawal, prostate cancer cells arrest in early G1 with hypo-phosphorylated RB suppressing E2F activity (Knudsen et al. 1998; Xu et al. 2006). Stimulation with androgen leads to the accumulation of cyclin D1 and activation of CDK4, which promotes phosphorylation of RB (Xu et al. 2006). Furthermore, AR-induced expression of p21 and degradation of p27 enhance CycD/CDK4 and CycE/CDK2-dependent phosphorylation and inactivation of RB allowing expression of E2F target genes (Knudsen et al. 1998; Lu et al. 1999). Thus, androgen-induced alterations in CDK activity enable expression of genes critical for S-phase entry (Knudsen 2006).
Cross-talk between AR signaling and the cell cycle machinery is not limited to androgen effects on the G1-S transition as several components of the cell cycle machinery have been shown to modulate AR function. It was first noted in fibroblasts that AR activity is regulated as a function of the cell cycle; this study suggested that AR transcriptional activity is lowest at the G1/S transition, when Cyclin D1 levels and CDK4 activity are at their peak (Martinez & Danielsen 2002). Cyclin D1 represses AR transcriptional activity independently of CDK4 by directly binding the coactivator-binding/AR dimerization motif in the AR AF-1 (Knudsen et al. 1999; Reutens et al. 2001; Martinez & Danielsen 2002; Petre et al. 2002). This interaction competes with AR coactivators such as p300/CAF and interferes with N/C-terminal AR interactions (Knudsen et al. 1999; Reutens et al. 2001; Burd et al. 2006). Thus, cyclin D1 can act in a negative feedback loop attenuating AR activity. This cyclin D1 repression is disrupted at multiple levels in human tumors facilitating increased AR activity (Burd et al. 2006; Knudsen 2006; Comstock & Knudsen 2007). Cyclin E has also been shown to associate with the AR AF-1 to enhance AR transcription independently of CDK2 (Yamamoto et al. 2000). In addition, CDK6 negates the ability of cyclin D1 to suppress AR function, and can serve to heighten AR activity independent of its kinase function (Lim et al. 2005). However, surprisingly little has been reported on the role of the AR in G2 or mitosis.
The effect of the cell cycle on AR protein expression during the cell cycle has been examined in one study where it was suggested that AR protein expression is lost in mitosis and that the AR functions as a mitotic licensing factor (Litvinov et al. 2006). However, others have reported that the AR is bound to condensed chromatin during mitosis (Kumar et al. 2008). Thus, little is known about the AR in G2/M and what is postulated about the AR in mitosis is conflicting.
In this study, we examined endogenous AR transcriptional activity, protein levels, localization, and phosphorylation during the cell cycle. We found that for a subset of AR-dependent genes, transcription is highest in the G1 phase of the cell cycle, reduced in S phase, and essentially abrogated in G2/M. This change in transcription was not due to a reduction in AR levels during cell cycle progression. AR localization changes in mitotic cells compared to interphase cells. This change in AR localization and reduction in transactivation correlated with AR phosphorylation on S308 in mitosis. AR S308 phosphorylation occurs in mitosis coincident with peak CDK1 activity and pharmacologic inhibition of CDK1 abrogated S308 phosphorylation. Moreover, CDK1 phosphorylated the AR on S308 in an in vitro kinase assay. Using a combination of phospho-S308-specific and anti-total AR antibodies, we show that: 1) only mitotic PCa cells express AR phosphorylated on S308; and 2) AR phosphorylated on S308 is excluded from chromatin. The data reported herein suggests that AR phosphorylation on S308 by CDK1 regulates AR localization in mitosis. We further propose that this CDK1-mediated change in the AR localization may regulate AR transcriptional activity. These observations have important implications for understanding AR biology and elucidating possible mechanisms of PCa progression.
Materials and Methods
Cell culture and treatment conditions
Prostate cancer cell lines LNCaP and C4-2 were cultured in T-Media supplemented with 5% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA). For experiments cells were switched for phenol-red free RPMI supplemented with 5% Charcoal-stripped serum (CSS) (Invitrogen). PC3 and COS7 cell lines were maintained in DMEM with 5%FBS. LHS cells were cultured in PrEBM media supplemented with growth factors (Lonza). RWPE-1 cells were maintained in KLM media supplemented with pituitary factors. The identity of our cell lines was verified by DNA fingerprinting using commercial kits containing multiple STR markers (DDC Medical). Cells were synchronized in G2/M by aphidicolin-block–release and nocodazole block: cells were treated with 2µg/ml aphidicolin for 24hrs, washed, and treated with 50ng/ml nocodazole for 16hrs. For ImageStream experiments, cells were treated with aphidicolin and released for 12hs.
Cell sorting
For protein analysis, growing cells were stained with 4ug/ml Hoechsit3342, and collected for sorting. For RNA, cells were treated with 1nm DHT for 2hrs, washed, collected, and fixed in cold methanol for 20min. Then cells were washed with PBS and stained with 4ug/ml Hoechst3342 at 4C O/N. RNaseA was added into samples at concentration 100µg/ml. Cells were sorted in cell cycle compartments based on DNA content using Vantage flow cytometer. Sorted cells were centrifuged and cell pellets were subjected to lysis for protein or RNA isolation as previously described (Gordon et al. 2010; Whitworth et al. 2012).
qRT-PCR
qRT-PCR experiments were done as previously described (Gordon et al. 2010; Whitworth et al. 2012). The primers are as follows: SNAI2 forward 5′-CTCCATCTGACACCTCCT-3′, SNAI2 reverse 5′-ACTGTAGTCTTTCCTCTTCATC-3′, SGK1 forward 5′-GGATGGGTCTGAACGACTTT-3′, SGK1reverse 5′-GAAGGACTTGGTGGAGGAGA-3′, UBE2C forward 5′-TGGTCTGCCCTGTATGATGT-3′, UBE2C reverse 5′-AAAAGCTGTGGGGTTTTTCC-3′ PSMB6 forward 5′-CAAACTGCACGGCCATGATA-3′, PSMB6 reverse 5′-GAGGCATTCACTCCAGACTGG-3′, GUS forward 5′– CTCATTTGGAATTTTGCCGATT-3′, GUS reverse 5′–CCGAGTGAAGATCCCCTTTTTA-3′. Expression levels of analyzed genes were normalized to housekeeping genes PSMB6 (Proteasome subunit beta type-6) and GUS (beta-glucuronidase).
Flow cytometry
For flow cytometry experiments using anti-phospho Histone-H3 (Cell Signaling) and anti-AR antibodies, cells were fixed in freshly prepared 4%(w/v)PFA (paraformaldehyde), pH7.2, for 15min and then permeabilized with cold methanol for 20min. Cells were blocked in blocking buffer containing 5% goat serum and then incubated with mix of mouse anti-phospho Histone-H3 (Cell Signaling) and rabbit anti-AR (antigen–first 20 N-terminal amino acids; (Gordon et al. 2010)) primary antibodies followed by separate incubations with mix of secondary goat Fluorescein (FITC)-conjugated anti-mouse antibody (Jackson ImmunoResearch) and goat R-phycoerythrin (PE)-conjugated anti-rabbit antibody (Invitrogen, Molecular Probes). Alternative anti-AR antibody used in flow cytometry studies was mouse antibody from BD Pharmingen. DNA was stained with TO-PRO3 iodide (Invitrogen). Primary rabbit anti-phospho-S308 antibody was from Santa Cruz. Data were collected on BD FACSCalibur flow cytometer (BD Biosciences). FITC and PE fluorescence was detected in the FL1 and FL2 channels, respectively, and TO-PRO-3 fluorescence was detected in FL4 channel, with CellQUEST Pro Software (Becton-Dickinson). Data were analyzed using FlowJo 8.6.6 software. For the ImageStream experiments, samples were stained similarly as for flow cytometry and imaged on an Imagestream imaging cytometer (Amnis, EMD Millipore) with data analysis using IDEAS 4.0 software.
Fluorescence microscopy
For indirect immunofluorescence microscopy, cells were grown on glass coverslips in 6-well dishes, treated with 50ng/ml nocodazole for 16hrs and fixed in freshly prepared 4%PFA for 20min. Samples were rinsed with PBS, blocked and permeabilized in blocking/antibody dilution buffer containing 5%-normal goat serum and 0.3%-Triton X in PBS for 1hr. Samples were incubated with anti-AR primary antibodies followed by secondary goat Alexa Flour 594-conjugated anti-rabbit (Invitrogen, Molecular Probes) or secondary goat FITC-conjugated anti-mouse antibody (Jackson ImmunoResearch). Anti-AR antibodies used were rabbit antibody developed against N-terminal domain (Gordon et al. 2010), mouse antibodies from Santa Cruz (AR441) and BD Pharmingen. Optimal dilutions of the antibodies were determined in titration experiments. The samples were mounted in Vectashield mounting medium containing DAPI (4′,6-diamidino-2-phenylindole) for nuclear staining (Vector Laboratories Inc.) and visualized by confocal microscopy using the LSM510. A Zeiss Plan-Apo 40×1.40 NA oil immersion objective was used for image acquisition. Co-localization of AR and DNA was quantified by calculating the percentage of signal overlap as described in (Koryakina et al. 2012).
In vitro kinase assay and Western blots
The in vitro kinase assays and Western blots were done as described in (Gordon et al. 2010; Whitworth et al. 2012) and analyzed using an Odyssey (Licor) imaging system.
Statistical methods
Differences between groups were analyzed using one-way ANOVA. Where multiple groups were compared using ANOVA, a post hoc Tukey test was used to enable multiple comparisons between groups.
Results
AR transcriptional activity changes during the cell cycle
AR transactivation of endogenous genes was assessed by performing qRT-PCR analyses of the AR target genes TMPRSS2, SGK1, and SNAI2 in LNCaP cells sorted into the G1, S-early, S-late, and G2/M cell cycle compartments. LNCaP cells were sorted based on DNA content (Figure 1A). We found that androgen-dependent transcription decreases at these AR target genes during the cell cycle (Figure 1). Transcription of SGK, SNAI2, and TMPRSS2 increased in response to DHT with the greatest induction in G1, decreased during S-phase, and for SGK1 and SNAI2, essentially abrogated in G2/M. This decrease in transcription was not due to a global effect on transcription; UBE2C, which encodes an ubiquitin-conjugating ligase necessary for degradation of mitotic cyclins demonstrated the inverse pattern of expression where UBE2C expression was minimal in G1, increased in S-phase, and showed the highest levels of expression in G2/M consistent with its function in mitosis (Figure 1D). Additionally, the housekeeping gene PSMB6 encoding the proteasome subunit, beta type 6 (and the housekeeping gene beta-glucoronidase, data not shown) did not change during the cell cycle (Figure 1E).
Fig. 1. Regulation of AR target genes during the cell cycle.

LNCaP cells were grown in T-Media supplemented with 5% FBS. Media was switched to phenol red free RPMI with 5% CSS. Cells were treated with either with 1 nm DHT or vehicle, fixed, stained with Hoechst 3342, and sorted into cell cycle compartments by flow cytometry based on DNA content. Expression of AR target genes in different cell cycle compartments was analyzed by qRT-PCR after cell sorting. (A) Histogram shows gating on cell cycle compartments during flow sort based on DNA content. AR-dependent genes SGK1 (B), SNAI2 (C), and TMPRSS2 (D) show cell cycle specific regulation. (E) Transcription of UBE2C is increased in G2/M and is not changed in response to DHT. (F) Transcription of the housekeeping gene PSMB6 is uniform during the cell cycle.
AR levels do not change during the cell cycle
We initially hypothesized that the change in AR transcriptional activity could be due to changes in AR levels during the cell cycle since a previous study suggested that the AR is degraded during mitosis (Litvinov et al. 2006). We performed a series of flow cytometry experiments using two different anti-AR antibodies targeting different epitopes. To distinguish cell cycle compartments, DNA was stained with TO-PRO3. Histone H3 phosphorylation on Ser10 is necessary for chromosome condensation in mitosis (Van Hooser et al. 1998) and was used as a mitotic marker in our studies. Dual-parametric analysis of AR fluorescence and DNA fluorescence did not reveal differences in the AR levels during the cell cycle in LNCaP and C4-2 cells (Figure 2A, 2B). Gating on mitotic phospho-histone H3 positive and interphase phospho-histone H3 negative populations to analyze AR levels in mitotic and interphase cells (Figure 2C), we found that approximately 0.7-1 % of asynchronous LNCaP cells expressed phospho-histone H3. AR levels in interphase and mitotic cells were very similar as shown in Figures 2D and 2E in LNCaP and C4-2 cells, respectively.
Fig. 2. Analyses of AR protein levels during the cell cycle by flow cytometry.
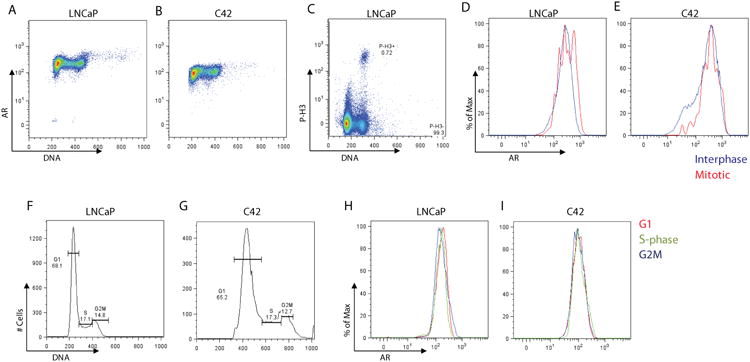
Cells were fixed and stained with two different anti-AR antibodies. Phospho-histone H3 staining was used to identify the mitotic population. DNA was stained with TO-PRO3. Dual-parametric analyses of AR and DNA fluorescence in LNCaP (A) and C4-2 (B) cells show similar levels of AR during the cell cycle. (C) Dual parametric fluorescence analysis of phospho-histone-H3 signal vs DNA shows mitotic phospho-histone H3-positive, and interphase phospho-histone H3-negative cells in LNCaP cells. Histograms in (D) and (E) show AR levels in mitotic (red) and interphase (blue) cells gated on phospho-histone H3 staining in LNCaP and C4-2 cells, respectively. Histograms in (F) and (G) show gating on G1, S-phase, and G2M cell cycle compartments based on DNA staining in LNCaP (E) and C4-2 (F) cells. Histograms illustrate AR protein levels in G1 (red), S-phase (green), and G2/M (blue) in LNCaP (H) and C4-2 (I) cells using anti-AR antibody directed against the first 21 amino acids of the AR (AR21).
AR levels during the cell cycle were analyzed in further detail by gating on cell populations in G1, S, and G2/M phases of the cell cycle using DNA staining (Figures 2F, 2G). As shown in Figures 2H and I, AR levels in G1, S, and G2M overlap in both prostate cancer cell lines. Using a second anti-AR antibody, AR levels were similar in G1, S, and G2/M in both prostate cancer cell lines (Supplemental Figure S1). The specificity of these anti-AR antibodies was confirmed by analyzing total AR expression in AR-positive and AR-negative cell lines; AR-positive LNCaP and C4-2 demonstrated high signal for AR fluorescence, whereas the AR-negative PC3 and LHS, as well as COS7 cells, had left-shifted peaks for AR fluorescence that were set as background fluorescence (Supplemental Figure S1). Overall, our flow cytometry analysis revealed that AR levels do not change during the cell cycle.
AR localization is different in interphase and mitotic cells
AR localization in prostate cancer cells was assessed by confocal microscopy in cells enriched in mitosis. AR demonstrated predominantly nuclear staining in interphase cells. In mitotic cells marked by the characteristic DNA condensation of condensed chromosomes, a substantial portion of the AR was excluded from chromosomes. LNCaP and C4-2 cells had similar patterns of AR staining in interphase and mitotic cells (Figure 3A, 3B). AR-negative prostate epithelial cells, RWPE-1, did not stain for AR further confirming the specificity of the AR antibody (Figure 3C).
Fig. 3. AR protein localization in interphase and mitotic prostate cancer cells by confocal microscopy.

Cells were grown on cover slips, enriched in G2M by nocodazole (50 ng/ml) treatment for 16 h, fixed, and processed for indirect immunofluorescence. AR localization was analyzed using AR21 (red). DNA was stained with DAPI. Images were acquired using confocal microscopy (40× objective). (A) LNCaP, (B) C4-2, and (C) RWPE-1 cells (negative control for AR staining). Mitotic cells are marked by asterisks.
To further validate the change in AR localization in mitotic cells observed by confocal microscopy and quantify this observation, we analyzed AR localization during the cell cycle in LNCaP and C4-2 cells using the ImageStream imaging flow cytometer. This technology combines advantages of flow cytometry and epifluorescence microscopy enabling quantitative analysis of protein localization. In asynchronous LNCaP cells less than 1% of cells are found in mitosis (Figure 7D). To enrich cells in mitosis without the confounding variable of chemical arrest, cells were synchronized in S phase with aphidicolin and then released so that the cells were free of chemical modifiers when progressing through mitosis (Figure 4A). Similar to what we observed in our confocal studies, AR staining was predominantly nuclear in interphase cells (Figure 4B, 4C) and was largely excluded from mitotic chromosomes (Figure 4D). The specificity of the AR staining was confirmed by the absence of staining in the AR-negative cell line, LHS (Supplemental Figure S2A).
Fig. 7. AR is phosphorylated on S308 in mitosis in prostate cancer cell lines.

LNCaP and C4-2 cells were enriched in G2/M by aphidicolin/nocodazole treatment processed for flow cytometry. (A) and (H), DNA profiles for asynchronous and chemically enriched G2/M LNCaP cells, respectively. (B), (E) and (I), dual fluorescence analyses of S308 phosphorylation and DNA in asynchronous LNCaP, asynchronous C4-2, and G2/M enriched LNCaP cells, respectively. Histograms show overlaid DNA profiles of phospho-S308 positive (red) and phospho-S308 negative (blue) populations for asynchronous LNCaP (C), asynchronous C4-2 (F), and G2/M enriched LNCaP (J) cells. Dual fluorescence analysis of S308 phosphorylation and the mitotic marker phospho-histone H3 staining in asynchronous LNCaP (D), asynchronous C4-2 (G), and G2/M enriched LNCaP cells, respectively.
Fig. 4. AR localization is different in interphase and mitotic prostate cancer cells.
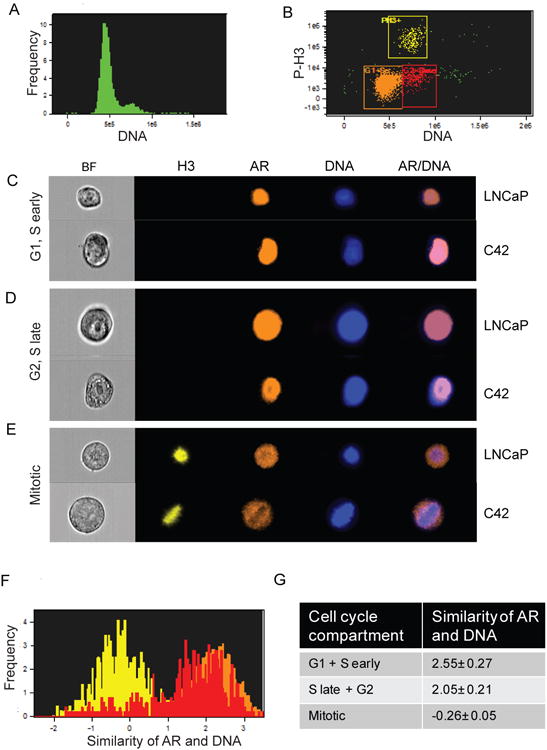
LNCaP cells were enriched in mitosis by aphidicolin block-release: cells were treated with aphidicolin for 24 h, washed, and released for 12 h. Cells were fixed, stained for total AR, phospho-histone-H3, and DNA, and analyzed by using ImageStream. (A) Histogram shows cell cycle profile for LNCaP cells subjected to analysis. (B) Dual-parameter fluorescence analysis of phospho-histone H3 and DNA shows gating strategy for isolating cell cycle populations based on DNA content and presence of the mitotic marker. Yellow: phospho-histone-H3-positive mitotic cells; red and orange: phospho-histone-H3-negative cells; orange: G1 and S early; and red: S late and G2M. (C, D, and E) Representative images of LNCaP and C4-2 cells in each cell cycle compartment are shown. Note that only mitotic cells are positive for phospho histone-H3. (F) Representative graphical illustration of AR and DNA similarity scores in 3 populations: mitotic, G1 and S early, and S late and G2 cells. Color-coding is as in B. (G) Table shows similarity scores of AR and DNA in cell cycle compartments. Values shown are Mean ± SEM from four independent experiments. The similarity scores are statistically different for G1 + S early and Mitotic populations (p = 0.0002) and S late + G2 and Mitotic populations (p = 0.0002). Similarity scores for G1 + S early and S late + G2 are not different (p = 0.24). Post hoc Tukey test was used
To quantitatively assess AR localization with mitotic chromosomes, we examined similarity scores, which reflect the extent to which AR staining coincides with DNA staining. A positive similarity score reflects high co-localization, whereas a low or negative similarity score indicates that the AR does not co-localize with mitotic chromosomes. For example, the similarity score for phospho-histone H3 and DNA is 2.8 ± 0.02 (Supplemental Figure S2B). This score provides an example of a high co-localization value. The beta2-adrenergic receptor is a G-protein coupled receptor localized on the cell surface in the absence of beta-agonist (Koryakina et al. 2012). Thus, this protein provides an example of a negative co-localization with DNA and a low similarity score; the similarity score for the beta2-adrenergic receptor and DNA is -0.43 ± 0.01 (Supplemental Figure S2C, S2D).
Similarity scores of AR and DNA were determined for three prostate cancer cell cycle populations: mitotic cells, and two interphase populations: G1+S-early, and S-late+G2 cells. For interphase populations, gating was based on DNA content (Figure 4B). In interphase cells the AR shows nuclear staining (Figure 4C, 4D) and high co-localization with DNA. The two interphase populations had similarity scores of 2.55±0.27 and 2.05±0.21 for G1+S-early and S-late+G2, respectively (Figure 4F, 4G). These values are close to the similarity score of the positive control, phospho-histone H3 (2.8 ± 0.02). For the G1+S-early population, 98.8% of the cells showed AR co-localization with DNA while the S-late+G2 population showed 88.8%; this reduced percentage is likely due to the redistribution of the AR from chromatin occurring in late G2 as cells are entering mitosis. In mitotic cells the similarity score of the AR and DNA was strongly left-shifted in 95.6% of the cells, yielding a negative similarity score of -0.26 ± 0.05 (Figure 4F and G). This similarity score of the AR and mitotic chromosomes was similar to the negative control, beta2-adrenergic receptor (Supplemental Figure S2C). Similar data were obtained with C4-2 cells (data not shown). These data demonstrate that AR localization changes in mitosis when compared to interphase cells. In interphase cells, the AR is nuclear and co-localized with DNA whereas in mitotic cells the AR is predominately excluded from chromosomes (Figure 4).
We next quantified the AR fluorescence intensity in each stage of mitosis to examine AR levels. The variation in AR fluorescence intensity for each stage of mitosis was only 5% from the AR fluorescence intensity of all mitotic cells. This indicates that AR levels do not change during mitosis and validates our previous observations that AR levels do not change during the cell cycle (Figure 2 and Supplemental Figure S1).
Hormone does not affect AR localization in mitosis
The effect of hormone on AR localization during mitosis was assessed. LNCaP and C4-2 cells were treated with 1nM DHT for 2hrs and analyzed by ImageStream. In both cell lines, DHT treatment for 2hrs did not significantly affect AR localization relative to DNA in mitotic cells. All p values were >0.05 for all comparisons of AR and DNA co-localization in untreated vs hormone treated populations; p=0.56 for (-) vs (+) DHT in Mitotic LNCaP; p=0.86 for (-) vs (+) DHT in Mitotic C4-2 (Supplemental Figure S3).
AR is excluded from chromosomes in all phases of mitosis
Our data above suggests that AR protein levels are preserved during mitosis and that AR localization changes leading to AR exclusion from DNA. To analyze this further we examined AR localization in all phases of mitosis. To isolate cells in specific phases of mitosis we used an algorithm that analyzes the shape of condensed chromosomes (Güttinger et al. 2009). Cells in different mitotic phases: prophase, metaphase, anaphase, and telophase, were isolated based on the relationship between the width of chromosomes and the aspect ratio (ratio of width to length) of the phospho-histone H3 signal (Figure 5A). Although the majority of mitotic cells were either in prophase or metaphase, we were able to isolate cells in all mitotic phases, including anaphase and telophase (Figure 5B-E). In all mitotic phases the AR was present and largely excluded from DNA. These data further demonstrate that during mitosis the AR is predominately excluded from mitotic chromosomes.
Fig. 5. AR localization during mitosis.
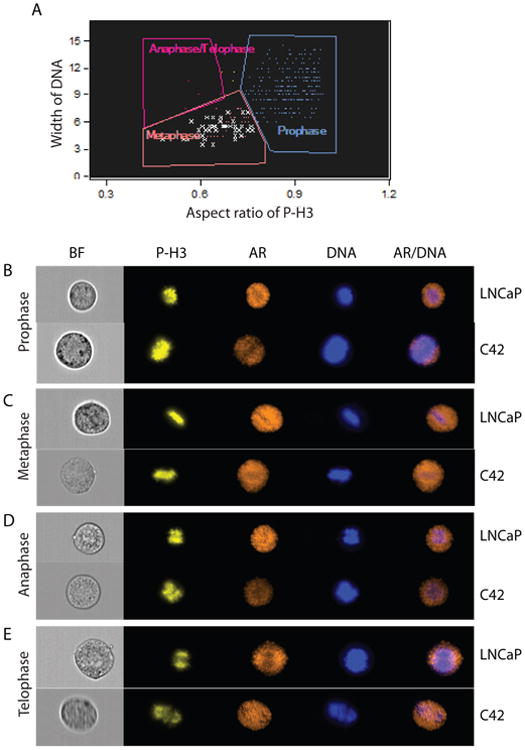
LNCaP and C4-2 were enriched in mitosis as previously described. Cells in different mitotic phases were isolated for analysis based on the Aspect Ratio of the phospho-histone H3 signal and width of DNA as shown in (A). Representative images of LNCaP and C4-2 cells in prophase (B), metaphase (C), anaphase (D), and telophase (E) are shown.
AR is phosphorylated on S308 in mitosis
The AR is phosphorylated at multiple sites with different functional consequences including regulation of AR localization (Koryakina et al. 2014). CDK1 is activated in mitosis, associates with the AR (Chen et al. 2006; Gordon et al. 2010) and regulates AR S81 phosphorylation (Chen et al. 2006, 2012). In addition to S81, the AR sequence contains multiple CDK1 consensus phosphorylation sites including a full site at S308. To directly test whether CDK1 can phosphorylate the AR on S308, we performed an in vitro kinase assay. CDK1, but not CDK5 or CDK9, phosphorylates AR S308 (Figure 6A). When S308 phosphorylation is examined in asynchronous LNCaP cells sorted into G1, S-early, S-late, and G2/M, peak S308 phosphorylation is observed in G2/M, consistent with peak CDK1 activity and an increase in Cyclin B levels (Figure 6B).
Fig. 6. CDK1 phosphorylates the AR on S308, which is increased in G2/M.
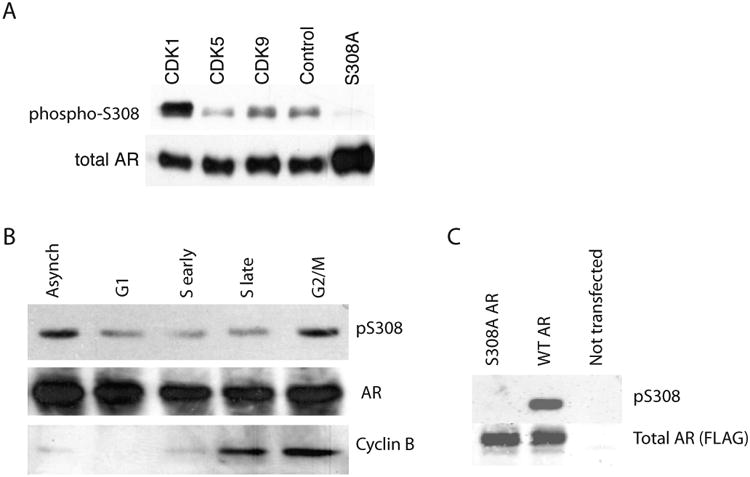
(A) Kinase assays were performed using purified CDKs and AR. Phosphorylation on S308 was assessed using phospho-S308 specific antibodies. Control represents no kinase control. S308A represents unphosphorylatable mutant. (B) LNCaP cells were stained with a DNA dye and sorted into cell cycle compartments by flow cytometry based on DNA content. AR was immunoprecipitated from 1×106 cells in each cell cycle compartment and immunoblotted for total and phospho-S308 AR. Phospho-S308 signal is increased in G2/M when Cyclin B1 protein levels from lysate are increased. (C) Phospho-S308 antibody specificity: COS7 cells transiently expressing either WT or S308A AR were treated with 1 nM DHT for 16hrs. AR was immunoprecipitated with anti-Flag antibody and blotted with anti-phospho-S308 (top panel) or anti-Flag antibody (lower panel).
To further explore the cell cycle regulation of AR S308 a series of flow cytometry experiments in asynchronous and G2/M enriched LNCaP and C4-2 prostate cancer cells was performed. The specificity of the phospho-specific S308 antibody was confirmed by analyzing AR negative prostate cell lines and COS7 cells transfected with Flag-tagged WT and S308A mutant AR (Supplemental Figure S4). Strong signal was observed in cells expressing WT AR, whereas no signal was seen in cells expressing S308A in COS7 cells expressing equivalent amounts of WT and S308A. AR negative cell lines, LHS and PC3, did not have a phospho-S308 signal further confirming phospho-S308 antibody specificity.
The DNA profile for asynchronous LNCaP cells is shown in Figure 7A. A small fraction of asynchronous LNCaP cells expressed AR S308 phosphorylation (Figure 7B). All cells positive for phospho-S308 are in G2/M, whereas phospho-S308 negative cells are found in all compartments of the cell cycle (Figure 7B, C, D). Similar results were obtained in C4-2 cells (Figure 7 E, F, G). To confirm that S308 phosphorylation occurs in G2/M, we analyzed LNCaP cells enriched in G2/M. This led to 60% of LNCaP cells enriched in G2/M (Figure 7H) and a 40% increase in the phospho-S308 signal (Figure 7I). All phospho-S308 positive cells were localized in G2/M (Figure 7J, 7K). To control for any potential confounding effects of chemical enrichment, we utilized an alternative approach where the dynamics of S308 phosphorylation was analyzed in cells synchronized in S-phase by aphidicolin and then released to progress through G2/M. As cells move through G2/M, the fraction of cells staining for phospho-S308 increased, peaking at 12hrs after release (Supplemental Figure S5). These data confirm that the AR is phosphorylated on S308 in G2/M.
Interestingly, when the phospho-S308 fraction was analyzed specifically in G2/M by gating on DNA content, approximately 14% of asynchronous LNCaP cells were positive for phospho-S308, whereas in cells enriched in G2/M by nocodazole, approximately 85% of cells were positive for phospho-S308 (Supplemental Figure S6). Nocodazole treatment causes arrest of cells in prometaphase by interfering with microtubule polymerization. It is interesting to note that the proportion of the phospho-S308 positive cells was higher in cells treated with nocodazole, suggesting that AR is likely phosphorylated on S308 in early mitosis. To further delineate the cell cycle compartment where S308 phosphorylation takes place, cells were analyzed for the expression of the mitotic marker phospho-histone H3. Dual fluorescence analysis of phospho-S308 and phospho-histone H3 in LNCaP and C4-2 cells revealed that all phospho-S308 positive cells expressed the mitotic marker phospho-histone H3 (Figure 7D, 7G, 7K). Thus, our flow cytometry data indicate that the AR is phosphorylated on S308 in mitosis. Importantly, we found that AR S308 phosphorylation is hormone independent. To assess the effect of hormone on S308 phosphorylation, LNCaP cells were treated with a range of doses of DHT, 0.01-10 nm, in steroid-free media for 2hrs and 16hrs and phospho-S308 signal was analyzed by using flow cytometry. As shown in Table 1, hormone treatment did not affect S308 phosphorylation.
Table 1. The effect of Hormone on S308 phosphorylation.
LNCaP cells were grown in T-Media supplemented with 5% FBS. Prior to hormone treatment, media were switched to RPMI supplemented with 5% CSS. Cells were treated with vehicle or DHT ranging 0.01 – 10 nm for 2 h and 16 h. Cells were fixed and processed for flow cytometry. S308 phosphorylation was assessed using phospho-S308 specific antibodies. Listed are the mean signal ± SEM. Hormone treatment did not affect S308 phosphorylation: p = 0.979 for 2 h and p = 0.992 for 16 h using one way variance analysis.
| DHT treatment | Phospho-S308 Positive, 2 h | Phospho-S308 Positive, 16 h |
|---|---|---|
| Vehicle | 1.35 ± 0.19 | 1.68 ± 0.42 |
| 0.01 nm | 1.45 ± 0.48 | 1.66 ± 0.29 |
| 0.1 nm | 1.47 ± 0.40 | 1.57 ± 0.33 |
| 1 nm | 1.75 ± 0.61 | 1.80 ± 0.41 |
| 10 nm | 1.53 ± 0.57 | 1.81 ± 0.53 |
Pharmacologic inhibition of CDK1 activity decreases S308 phosphorylation
To address further whether S308 phosphorylation was mediated by CDK1, LNCaP cells were treated with either vehicle or the CDK1 selective inhibitor RO-3306 for 2hrs (Vassilev et al. 2006). This relatively short time of treatment was chosen to minimize the affects of CDK1 inhibition on the cell cycle. Figure 8 shows that treatment led to a dramatic inhibition of S308 phosphorylation (Figure 8). In vehicle-treated cells, approximately 10.8% of cells were positive for phospho-S308 in G2/M (Figure 8B, 8F), whereas in RO-3306-treated cells the phospho-S308 positive population decreased to 2.3% (Figure 8D, 8F). Treatment with the CDK1 inhibitor for 2hrs did not change the cell cycle profile (Figure 8E). These data, together with the in vitro kinase assay and the data showing an increase in S308 phosphorylation in phospho-histone H3 positive cells, suggest that CDK1 phosphorylates the AR on S308 in mitosis.
Fig. 8. Pharmacological inhibition of CDK1 results in decrease in S308 phosphorylation.
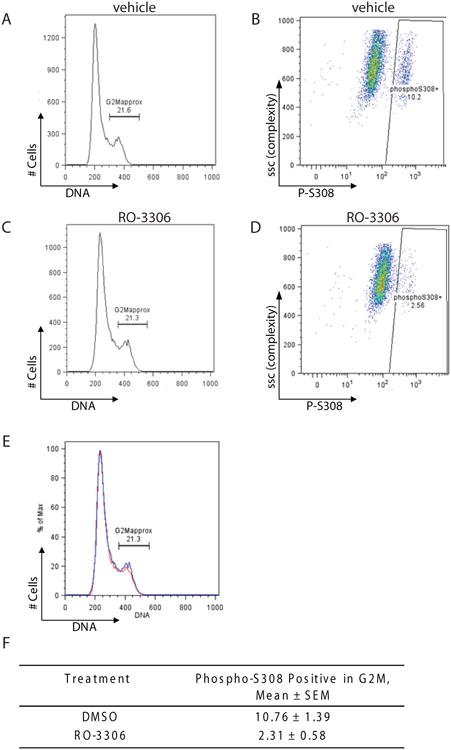
LNCaP cells were treated with either vehicle (A and B) or RO-3306 (C and D) for 2 h and phospho-S308 signal was analyzed by flow cytometry. Histograms (A) and (C) show DNA profiles for vehicle- (A) and RO-3306-treated (C) cells; gating on G2M phase is shown. B and D show phospho-S308 signal in G2M cells in vehicle and RO-3306-treated cells, respectively. (E) shows overlaid DNA profiles of RO-3306-treated (red) and vehicle-treated (blue) cells. (F) Table shows quantitation of S308 phosphorylation in RO-3306-treated and vehicle-treated cells, n=3.
AR S308 phosphorylation regulates AR localization in mitosis
Our confocal and ImageStream data demonstrate that AR localization changes during mitosis. We hypothesized that CDK1 phosphorylation of S308 in mitosis regulates this change in AR localization. To address this hypothesis, AR localization during the cell cycle was analyzed using a combination of phospho-S308 specific and three different anti-total AR antibodies using confocal microscopy. One of the anti-total AR antibodies we used, M441, is a mouse monoclonal developed against AR amino acids 299-315 and thus contains the phospho-site S308. Importantly, M441 only recognizes non-phosphorylated S308 AR; phosphorylation on S308 blocks M441 binding (Supplemental Figure S7). The anti-total AR antibody AR21 is a rabbit polyclonal raised against AR amino acids 1-21. AR21 recognizes both phosphorylated and non-phosphorylated S308 AR (Supplemental Figure S7). The immunostaining pattern of AR21 was identical to the pattern of a third anti-total AR antibody (Supplemental Figure S8). Thus, these anti-total AR antibodies can be used in conjunction with the phospho-S308 specific antibody to assess the localization of phospho-S308 and non-phospho-S308 AR during interphase and mitosis.
Only mitotic, and not interphase, LNCaP cells had a phospho-S308 signal (Figure 9A). The AR-negative PC3 cells did not show a phospho-S308 signal (Supplemental Figure 9A). Interphase LNCaP cells, co-stained with M441 and AR21 total AR antibodies, had very similar nuclear patterns of staining and both of these antibodies showed very high co-localization with DNA, 92% and 80% of co-localization with DNA, respectively (Figure 9B and 9C). In contrast, in mitotic cells these antibodies had very different staining patterns. M441 staining remained closely co-localized with chromosomes (86% of co-localization), whereas most of the AR signal was excluded from chromosomes when cells were stained with AR21, (only 31% of the AR21 staining co-localized with DNA). Taken together, these experiments suggest that two forms of the AR are present in mitosis: phosphorylated S308 that is excluded from chromosomes and non-phosphorylated S308 that preserves chromosome association during mitosis.
Fig 9. P-S308 signal is excluded from chromatin in LNCaP cells.
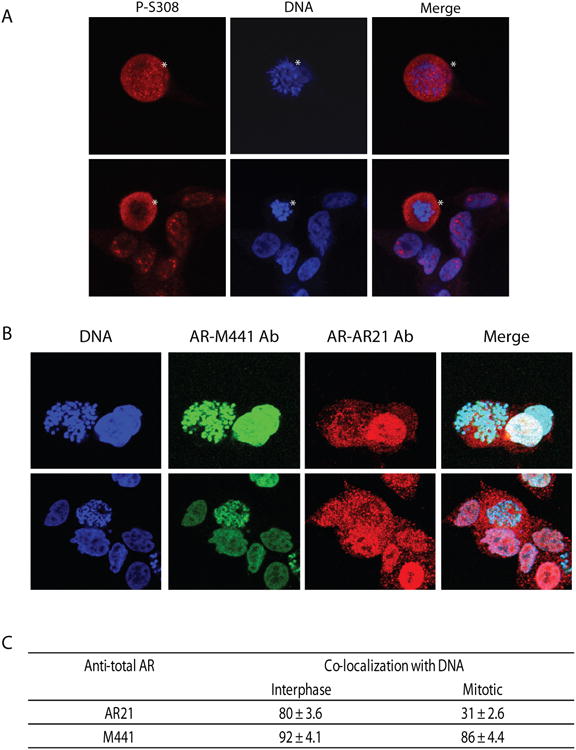
(A) LNCaP cells were enriched in mitosis, fixed and stained with phospho-S308 specific antibody. P-S308 signal is present only in mitotic cells (mitotic cells are marked by asterisks). (B) LNCaP cells were co-stained with two different anti-AR antibodies (M441 and AR21) targeting different epitopes. (C) Table shows quantitation of co-localization of the AR staining with DNA using the two different anti-AR antibodies in interphase and mitotic LNCaP cells.
Discussion
It is well established that the AR drives cell cycle progression by regulating transcription of genes that control the G1-S transition, with substantive lines of cross-communication between the AR and the cell cycle machinery (Balk & Knudsen 2008). However, little has been reported on the role of the AR in G2 or mitosis. In this study, we show data suggesting that AR phosphorylation on S308 by CDK1 regulates AR localization in mitosis and that this CDK1-mediated change in the AR localization correlates with changes in AR transcriptional activity.
Other nuclear receptors also are reported to regulate and be regulated by the cell cycle, although the specific mechanisms and modalities of those interactions are different from the AR. Previous studies on the Glucocorticoid Receptor (GR), Progesterone Receptor (PR), Estrogen Receptor (ER), and Thyroid Receptor (TR) have demonstrated that transcriptional activity is regulated as a function of the cell cycle (Hsu & DeFranco 1995; Maruvada et al. 2004; Narayanan et al. 2005; Dalvai & Bystricky 2010). Several studies have shown that GR-dependent transactivation is impaired in G2/M enriched nocodazole-treated cells when compared to asynchronous cells (Hsu et al. 1992; Hsu & DeFranco 1995; Matthews et al. 2011). However, others have shown that GR transcriptional activity was similar in asynchronous and G2-enriched cells, and repressed in mitosis due to chromatin condensation (Hu et al. 1994; Abel et al. 2002). GR hormone binding was elevated in S phase and G2 in Hela cells (Cidlowski & Cidlowski 1982). The progesterone receptor (PR) transcriptional activity peaks in S-phase through recruitment of cyclin/cdk2 to sites of PR transcription. Chemical arrest of MCF-7 breast cancer cells showed that several ERα target genes having higher expression in G1 compared to G2/M and that different selective estrogen modulators can either repress or stimulate expression of specific ERα target genes depending on the cell cycle phase (Dalvai & Bystricky 2010). TR expression and transactivation was essentially absent in G1, increased in late S and G2 and peaked in G2/M (Maruvada et al. 2004).
One previous study did address AR transcriptional activity during the cell cycle using reporter constructs in chemically arrested cells and 24 hour hormone treatment (Martinez & Danielsen 2002). Under those conditions, AR transcriptional activity was higher in G0 and S and reduced during the G1/S transition and these transcriptional effects paralleled changes in AR expression; however, the G2/M cell cycle compartment was not examined. In our study, we used a brief 2-hour change to charcoal-stripped media and androgen stimulus to assess androgen induced transcription of endogenous AR target genes SGK, SNAI2, and TMPRSS2 in different cell cycle compartments by FACs and found that androgen-stimulated AR transactivation is highest in G1 and essentially abrogated in G2/M. We did not observe differences in AR expression during the cell cycle (see Figures 2 and 4). There are many differences between our study and Martinez et al, including our use of human cell lines, endogenous gene targets, and asynchronous sorted cells that may account for the different observations of AR levels during the cell cycle. Interestingly, when AR localization of exogenous GFP-tagged AR in response to hormone was examined, increased nuclear localization was observed in G2 cells (Szafran et al. 2008). This observation would suggest that AR transcriptional activity would be highest in G2. Our study only examined a small subset of AR target genes. To date, no published study has examined the global cell cycle dependent gene expression profile of the AR, or any other steroid hormone receptor. Thus, the increased hormone induced AR nuclear localization in G2 may lead to increased transactivation of G2 specific AR target genes.
An earlier study analyzing the effect of the cell cycle on AR protein expression suggested that AR protein expression was lost in mitosis and that the AR functions as a mitotic licensing factor (Litvinov et al. 2006). However, this is in contrast to our experiments reported here as well as studies from other groups showing AR expression in mitosis (Kumar et al. 2008). In our study, we show AR expression throughout the cell cycle. In Kumar et al, overexpression of GFP-tagged AR in COS-1 cells led to agonist-mediated docking of AR onto mitotic chromatin (Kumar et al. 2008). AR association with chromatin was dependent on the method used for fixation/permeabilization, where one method provided evidence for endogenous hormone-bound AR associated with mitotic chromatin and others showed the majority of the AR excluded from chromatin. In our studies different fixation/permeabilization methods did not reveal differences in AR localization or phosphorylation (data not shown). In further support of AR expression during mitosis, studies have also shown that CDK1 activity, which peaks in G2/M, stabilizes the AR (Chen et al. 2006). One possible explanation for the discrepancy among these reports of AR expression in mitosis is that the AR is degraded late in mitosis. However, using ImageStream analysis we observed endogenous AR in all mitotic phases, including telophase, consistent with AR expression being maintained throughout mitosis. Another possibility is the method used to analyze the flow cytometry data (Litvinov et al. 2006), which could lead to alternative conclusions.
While uncommon, there is a precedent for transcription factor association and activity during mitosis even though chromatin favors a highly condensed state and global transcription during mitosis is silenced (Gottesfeld & Forbes 1997). Most basal transcription factors, RNA polymerases and sequence-specific DNA binding proteins and histone modifying enzymes are absent from condensed mitotic chromosomes. However, evidence suggests that certain transcription factors remain bound to mitotic condensed chromosomes (Burke et al. 2005; Saradhi et al. 2005; Yan et al. 2006; Young et al. 2007). For example, the zinc-finger protein CTCF and the lineage-specific transcription factor Runx2 are implicated in maintenance of epigenetic marks in mitosis (Burke et al. 2005; Young et al. 2007) and the forkhead FoxI1 transcription factor is implicated in organization of mitotic chromatin structure (Yan et al. 2006).
Our data are in general agreement with studies on the GR localization demonstrating GR nuclear localization throughout interphase and exclusion from the nucleus in mitosis (Hsu et al. 1992; Matthews et al. 2011). In these studies it was suggested that recycled GRs do not re-enter the nucleus in G2- synchronized cells (Hsu et al. 1992). GR localization in mitosis did not change with dexamethasone treatment, which is similar to our finding that hormone treatment did not alter AR localization during mitosis. Interestingly, dexamethasone-induced GR transactivation was blocked in G2 synchronized cells, but not in asynchronous cells, although the number of receptors remained the same in both populations. This latter finding is consistent with our observations of AR transcriptional activity decreasing in G2/M although AR protein levels remain unchanged throughout the cell cycle. Interestingly, the binding of a nuclear receptor family member pregnane and xenobiotic receptor (PXR) to mitotic chromatin during all mitotic phases was reported in an over-expression system, although the significance of that association was not elucidated (Saradhi et al. 2005).
Here we show that AR phosphorylated on S308 is excluded from chromosomes and non-phosphorylated S308 preserves chromosome association during mitosis in a hormone independent manner. AR phosphorylation on S308 is coincident with peak CDK1 activity. Previous work from our lab and others has collectively demonstrated that CDK1 interacts with the AR and can phosphorylate AR S81 (Chen et al. 2006, 2012; Gordon et al. 2010) and nocodazole arrested cells in G2/M have elevated AR S81 phosphorylation (Chen et al. 2006, 2012) although our work suggests that CDK9 is the major AR S81 kinase (Gordon et al. 2010) and it remains unclear under what biological conditions each of the reported CDKs phosphorylate the AR on S81. In experiments reported here we show that CDK1 phosphorylates the AR on S308 in vitro and that pharmacological inhibition of CDK1 activity dramatically decreases S308 phosphorylation in prostate cancer cells. Interestingly, even though we previously observed an increase in S308 phosphorylation in response to hormone (Gioeli et al. 2002), a thorough analysis of S308 phosphorylation in mitotic cells reported here indicates that S308 phosphorylation is hormone-independent. It is likely that the slight increase in AR S308 phosphorylation observed during phospho-peptide mapping studies were due to an increase in the percent of mitotic cells resulting from hormone stimulation. A previous study also showed that S308 is phosphorylated by CDK11p58/Cyclin D3 complex in G2/M leading to repression of AR transcriptional activity (Zong et al. 2007). This is in agreement with our earlier publication (Gioeli et al. 2002) and parallels our observations in this report where AR transcriptional activity decreases coincident with S308 phosphorylation. Interestingly, one study has suggested that S308 phosphorylation correlates with patient survival (McCall et al. 2013), which is consistent with the data suggesting that phosphorylation on this site may reduce AR transcriptional activity.
Interestingly, the GR is also phosphorylated in a cell cycle dependent manner (Matthews et al. 2011). Studies on the GR suggest that G2-synchronized cells have a distinct phosphorylation pattern compared to asynchronous cells (Hsu et al. 1992), although comparison of HPLC phosphopeptide maps revealed no phospho-site-specific differences in WCL2 cells synchronized in S-phase and G2/M (Hu et al. 1994). Overall GR phosphorylation is increased in G2/M compared to S-phase, whereas hormone-dependent induction is higher in S-phase and negligible in G2/M. GR phosphorylation on S203 and S211 occurred in a ligand-independent fashion in G2/M and ligand-independent GR transactivation required S211 phosphorylation (Matthews et al. 2011).
Our experiments provide evidence that the cell cycle effects AR function. Taken together, the present study proposes a model in which the AR S308 phosphorylation by CDK1 results in the AR exclusion from mitotic chromatin thereby negatively regulating AR dependent transcription. This study begins to integrate the complexity of cycling cells and signal transduction by kinases with AR biology.
Supplementary Material
Acknowledgments
We would like the thank Dr Huy Ta and Christopher McNair for critically reading the manuscript.
We would also like to thank Joanne Lannigan and Mike Solga of the UVA Flow Cytometry Core Facility for outstanding technical support.
Grant Support: National Cancer Institute (R01 CA124706 and R01 CA178338, DG), the Paul Mellon Urologic Cancer Institute (DG), the Farrow Fellowship (YK), and Prostate Cancer Foundation Challenge Award (KEK).
Footnotes
Declaration of interest: no conflict of interest that could be perceived as prejudicing the impartiality of the research is reported.
References
- Abel GA, Wochnik GM, Rüegg J, Rouyer A, Holsboer F, Rein T. Molecular Endocrinology. Vol. 16. Baltimore, Md: 2002. Activity of the GR in G2 and mitosis; pp. 1352–1366. [DOI] [PubMed] [Google Scholar]
- Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nuclear Receptor Signaling. 2008;6:e001. doi: 10.1621/nrs.06001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CJ, Petre CE, Morey LM, Wang Y, Revelo MP, Haiman CA, Lu S, Fenoglio-Preiser CM, Li J, Knudsen ES, et al. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci U S A. 2006;103:2190–2195. doi: 10.1073/pnas.0506281103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke LJ, Zhang R, Bartkuhn M, Tiwari VK, Tavoosidana G, Kurukuti S, Weth C, Leers J, Galjart N, Ohlsson R, et al. CTCF binding and higher order chromatin structure of the H19 locus are maintained in mitotic chromatin. The EMBO Journal. 2005;24:3291–3300. doi: 10.1038/sj.emboj.7600793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xu Y, Yuan X, Bubley GJ, Balk SP. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0604193103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Gulla S, Cai C, Balk SP. Androgen receptor serine 81 phosphorylation mediates chromatin binding and transcriptional activation. The Journal of Biological Chemistry. 2012;287:8571–8583. doi: 10.1074/jbc.M111.325290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cidlowski JA, Cidlowski NB. Glucocorticoid receptors and the cell cycle: evidence that the accumulation of glucocorticoid receptors during the S phase of the cell cycle is dependent on ribonucleic acid and protein synthesis. Endocrinology. 1982;110:1653–1662. doi: 10.1210/endo-110-5-1653. [DOI] [PubMed] [Google Scholar]
- Comstock CE, Knudsen KE. The complex role of AR signaling after cytotoxic insult: implications for cell-cycle-based chemotherapeutics. Cell Cycle. 2007;6:1307–1313. doi: 10.4161/cc.6.11.4353. [DOI] [PubMed] [Google Scholar]
- Dalvai M, Bystricky K. Cell cycle and anti-estrogen effects synergize to regulate cell proliferation and ER target gene expression. PloS One. 2010;5:e11011. doi: 10.1371/journal.pone.0011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George D, Moul JW. Emerging treatment options for patients with castration-resistant prostate cancer. The Prostate. 2011 doi: 10.1002/pros.21435. [DOI] [PubMed] [Google Scholar]
- Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, White FM, Christian RE, Settlage RE, Shabanowitz J, et al. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J Biol Chem. 2002;277:29304–29314. doi: 10.1074/jbc.M204131200. [DOI] [PubMed] [Google Scholar]
- Gordon V, Bhadel S, Wunderlich W, Zhang J, Ficarro SB, Mollah SA, Shabanowitz J, Hunt DF, Xenarios I, Hahn WC, et al. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Mol Endocrinol. 2010;24:2267–2280. doi: 10.1210/me.2010-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesfeld JM, Forbes DJ. Mitotic repression of the transcriptional machinery. Trends in Biochemical Sciences. 1997;22:197–202. doi: 10.1016/s0968-0004(97)01045-1. [DOI] [PubMed] [Google Scholar]
- Güttinger S, Laurell E, Kutay U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nature Reviews Molecular Cell Biology. 2009;10:178–191. doi: 10.1038/nrm2641. [DOI] [PubMed] [Google Scholar]
- Van Hooser A, Goodrich DW, Allis CD, Brinkley BR, Mancini MA. Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. Journal of Cell Science. 1998;111(Pt 23):3497–3506. doi: 10.1242/jcs.111.23.3497. [DOI] [PubMed] [Google Scholar]
- Hsu SC, DeFranco DB. Selectivity of cell cycle regulation of glucocorticoid receptor function. The Journal of Biological Chemistry. 1995;270:3359–3364. doi: 10.1074/jbc.270.7.3359. [DOI] [PubMed] [Google Scholar]
- Hsu SC, Qi M, DeFranco DB. Cell cycle regulation of glucocorticoid receptor function. The EMBO Journal. 1992;11:3457–3468. doi: 10.1002/j.1460-2075.1992.tb05425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JM, Bodwell JE, Munck A. Molecular Endocrinology. Vol. 8. Baltimore, Md: 1994. Cell cycle-dependent glucocorticoid receptor phosphorylation and activity; pp. 1709–1713. [DOI] [PubMed] [Google Scholar]
- Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discovery. 2013;3:1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- Knudsen KE. The cyclin D1b splice variant: an old oncogene learns new tricks. Cell Div. 2006;1:15. doi: 10.1186/1747-1028-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen KE, Arden KC, Cavenee WK. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J Biol Chem. 1998;273:20213–20222. doi: 10.1074/jbc.273.32.20213. [DOI] [PubMed] [Google Scholar]
- Knudsen KE, Cavenee WK, Arden KC. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Research. 1999;59:2297–2301. [PubMed] [Google Scholar]
- Koryakina Y, Jones SM, Cornett LE, Seely K, Brents L, Prather PL, Kofman A, Kurten RC. Effects of the β-agonist, isoprenaline, on the down-regulation, functional responsiveness and trafficking of β2-adrenergic receptors with N-terminal polymorphisms. Cell Biology International. 2012;36:1171–1183. doi: 10.1042/CBI20120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koryakina Y, Ta HQ, Gioeli D. Androgen receptor phosphorylation: biological context and functional consequences. Endocrine-Related Cancer. 2014;21:T131–T145. doi: 10.1530/ERC-13-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Chaturvedi NK, Kumar S, Tyagi RK. Agonist-mediated docking of androgen receptor onto the mitotic chromatin platform discriminates intrinsic mode of action of prostate cancer drugs. Biochimica et Biophysica Acta. 2008;1783:59–73. doi: 10.1016/j.bbamcr.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Lim JT, Mansukhani M, Weinstein IB. Cyclin-dependent kinase 6 associates with the androgen receptor and enhances its transcriptional activity in prostate cancer cells. Proc Natl Acad Sci U S A. 2005;102:5156–5161. doi: 10.1073/pnas.0501203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinov IV, Vander Griend DJ, Antony L, Dalrymple S, De Marzo AM, Drake CG, Isaacs JT. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc Natl Acad Sci U S A. 2006;103:15085–15090. doi: 10.1073/pnas.0603057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Liu M, Epner DE, Tsai SY, Tsai MJ. Androgen regulation of the cyclin-dependent kinase inhibitor p21 gene through an androgen response element in the proximal promoter. Mol Endocrinol. 1999;13:376–384. doi: 10.1210/mend.13.3.0254. [DOI] [PubMed] [Google Scholar]
- Martinez ED, Danielsen M. Loss of androgen receptor transcriptional activity at the G(1)/S transition. The Journal of Biological Chemistry. 2002;277:29719–29729. doi: 10.1074/jbc.M112134200. [DOI] [PubMed] [Google Scholar]
- Maruvada P, Dmitrieva NI, East-Palmer J, Yen PM. Cell cycle-dependent expression of thyroid hormone receptor-beta is a mechanism for variable hormone sensitivity. Molecular Biology of the Cell. 2004;15:1895–1903. doi: 10.1091/mbc.E03-09-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews L, Johnson J, Berry A, Trebble P, Cookson A, Spiller D, Rivers C, Norman M, White M, Ray D. Cell cycle phase regulates glucocorticoid receptor function. PloS One. 2011;6:e22289. doi: 10.1371/journal.pone.0022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall P, Adams CE, Willder JM, Bennett L, Qayyum T, Orange C, Underwood MA, Edwards J. Androgen receptor phosphorylation at serine 308 and serine 791 predicts enhanced survival in castrate resistant prostate cancer patients. International Journal of Molecular Sciences. 2013;14:16656–16671. doi: 10.3390/ijms140816656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Adigun AA, Edwards DP, Weigel NL. Cyclin-dependent kinase activity is required for progesterone receptor function: novel role for cyclin A/Cdk2 as a progesterone receptor coactivator. Molecular and Cellular Biology. 2005;25:264–277. doi: 10.1128/MCB.25.1.264-277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petre CE, Wetherill YB, Danielsen M, Knudsen KE. Cyclin D1: mechanism and consequence of androgen receptor co-repressor activity. J Biol Chem. 2002;277:2207–2215. doi: 10.1074/jbc.M106399200. [DOI] [PubMed] [Google Scholar]
- Reutens AT, Fu M, Wang C, Albanese C, McPhaul MJ, Sun Z, Balk SP, Janne OA, Palvimo JJ, Pestell RG. Cyclin D1 binds the androgen receptor and regulates hormone-dependent signaling in a p300/CBP-associated factor (P/CAF)-dependent manner. Molecular Endocrinology. 2001;15:797–811. doi: 10.1210/mend.15.5.0641. [DOI] [PubMed] [Google Scholar]
- Saradhi M, Sengupta A, Mukhopadhyay G, Tyagi RK. Pregnane and Xenobiotic Receptor (PXR/SXR) resides predominantly in the nuclear compartment of the interphase cell and associates with the condensed chromosomes during mitosis. Biochimica Et Biophysica Acta. 2005;1746:85–94. doi: 10.1016/j.bbamcr.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Szafran AT, Szwarc M, Marcelli M, Mancini MA. Androgen receptor functional analyses by high throughput imaging: determination of ligand, cell cycle, and mutation-specific effects. PloS One. 2008;3:e3605. doi: 10.1371/journal.pone.0003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A. 2006;103:10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth H, Bhadel S, Ivey M, Conaway M, Spencer A, Hernan R, Holemon H, Gioeli D. Identification of Kinases Regulating Prostate Cancer Cell Growth Using an RNAi Phenotypic Screen. PloS One. 2012;7:e38950. doi: 10.1371/journal.pone.0038950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783–7792. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Hashimoto Y, Kohri K, Ogata E, Kato S, Ikeda K, Nakanishi M. Cyclin E as a coactivator of the androgen receptor. J Cell Biol. 2000;150:873–880. doi: 10.1083/jcb.150.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Xu L, Crawford G, Wang Z, Burgess SM. The forkhead transcription factor FoxI1 remains bound to condensed mitotic chromosomes and stably remodels chromatin structure. Molecular and Cellular Biology. 2006;26:155–168. doi: 10.1128/MCB.26.1.155-168.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DW, Hassan MQ, Yang XQ, Galindo M, Javed A, Zaidi SK, Furcinitti P, Lapointe D, Montecino M, Lian JB, et al. Mitotic retention of gene expression patterns by the cell fate-determining transcription factor Runx2. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3189–3194. doi: 10.1073/pnas.0611419104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Chi Y, Wang Y, Yang Y, Zhang L, Chen H, Jiang J, Li Z, Hong Y, Wang H, et al. Cyclin D3/CDK11p58 complex is involved in the repression of androgen receptor. Mol Cell Biol. 2007;27:7125–7142. doi: 10.1128/MCB.01753-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.