

Abstract
Determining the relative contribution of amyloid plaques and neurofibrillary tangles to brain dysfunction in Alzheimer disease is critical for therapeutic approaches, but until recently could only be assessed at autopsy. We report a patient with posterior cortical atrophy (visual variant of Alzheimer disease) who was studied using the novel tau tracer [18F]AV-1451 in conjunction with [11C]Pittsburgh compound B (PIB; amyloid) and [18F]fluorodeoxyglucose (FDG) positron emission tomography. Whereas [11C]PIB bound throughout association neocortex, [18F]AV-1451 was selectively retained in posterior brain regions that were affected clinically and showed markedly reduced [18F]FDG uptake. This provides preliminary in vivo evidence that tau is more closely linked to hypometabolism and symptomatology than amyloid.
Among the most striking neuroimaging findings in Alzheimer disease (AD) is the dissociation between the distribution of amyloid-β (Aβ) and patterns of neurode-generation. Across AD phenotypes, Aβ is deposited relatively symmetrically throughout the neocortex,1–3 whereas atrophy and hypometabolism are more focal and mirror clinical symptoms.1,3–6 Based on the close association found at autopsy between neurofibrillary tangles, neuronal injury,7 and cognitive status,8 tau pathology could be the missing link that ties Aβ to neurodegeneration and symptomatology. The study of relationships between tau pathology and other pathogenic processes in AD has been hindered by the lack of tau-specific imaging biomarkers that could demonstrate the distribution of neurofibrillary tangles during life. The recent advent of a novel positron emission tomography (PET) tracer ([18F]AV-1451, formerly called [18F]T807) allows in vivo assessment of regional tau load.9,10 In this study, we compared patterns of [18F]AV-1451, [11C]Pittsburgh compound B (PIB; measure of fibrillar Aβ), and [18F]fluorodeoxyglucose (FDG; glucose metabolism) retention in a patient with posterior cortical atrophy (PCA). PCA is a clinical–radiological syndrome generally associated with AD pathology11 that affects occipital, parietal, and occipitotemporal cortices, resulting in progressive visuospatial and visuoperceptive deficits.12 We hypothesized that [11C]PIB would show binding throughout the neocortex, whereas [18F]AV-1451 (increased) and [18F]FDG (decreased) would show more restricted uptake patterns in clinically affected posterior brain regions.
Patient and Methods
Subject
A 56-year-old right-handed man presented to the University of California, San Francisco (UCSF) Memory and Aging Center with a 3.5-year history of visual loss and cognitive decline. Early symptoms included marked visuospatial difficulties in the absence of memory, language, and executive function problems. His clinical presentation at first evaluation (Mini-Mental State Examination = 19 of 30, Clinical Dementia Rating = 1) included primary visuoperceptual deficits (left homonymous hemianopia), ventral and dorsal visual stream dysfunction, alexia, and elements of Gerstmann and Bálint syndromes. Although not an early symptom by history, mild-to-moderate memory deficits were evident on formal neuropsychological testing, whereas verbally mediated language and executive functions remained relatively preserved (Table). Elemental neurological examination was unremarkable. The patient is a homozygous apolipoprotein E ε4 allele carrier and has a strong family history of AD, with both parents affected, at around age 50 years (father) and 70 years (mother). Screening results for autosomal dominant gene mutations are not available. Temporoparietal and occipital brain atrophy on magnetic resonance imaging (MRI) supported the clinical diagnosis of PCA, with suspected underlying AD pathology. At 9-months clinical follow-up, the patient showed progression of existing symptoms and newly acquired prosopagnosia, trouble judging distance, and visual illusions.
TABLE I. Neuropsychological Test Scores.
| Cognitive Domain/Task | Score, Total Correct/Total Possible Points |
|---|---|
| Verbal episodic memory | |
| CVLT-9 total learning | 19/36 |
| CVLT-9 delayed recall, 10-minute | 1/9 |
| CVLT-9 recognition | 7/9, 4 FP |
| Language | |
| Boston Naming Test | 9/15a |
| Peabody Picture Vocabulary Test | 15/16 |
| Verbal agility | 6/6 |
| Sentence comprehension | 4/5 |
| Visuospatial | |
| Intersecting pentagons | 0/1 |
| Benson figure copy | 3/17 |
| Calculations | 2/5 |
| VOSP spatial localization | 2/10 |
| Executive functions/attention | |
| Digit span forward | 7/8 |
| Digit span backward | 4/8 |
| Letter fluency, D | 12 |
Where applicable, total scores as well as total points possible for each test are provided.
Stimulus cues were administered for more than half the items on the task due to spatial misperceptions. However, due to significant visuospatial processing deficits, he still was unable to perceive the items adequately.
CVLT-9 = California Verbal Learning Test, 9-item version; FP = false positives; VOSP = Visual Object and Space Perception Battery.
MRI
The patient underwent MRI at the UCSF Neuroimaging Center on a 3T Siemens (Erlangen, Germany) Tim Trio. T1-weighted magnetization-prepared rapid gradient echo (MP-RAGE) was acquired as previously described.3 MP-RAGE sequences were processed using FreeSurfer 5.1 to define native space reference regions and cortical regions of interest (ROIs).
PET
PET scans were performed at Lawrence Berkeley National Laboratory (LBNL) on a Siemens Biograph 6 Truepoint PET/computed tomography (CT) scanner in 3-dimensional acquisition mode. A low-dose CT was performed for attenuation correction prior to all scans. [11C]PIB and [18F]FDG-PET were acquired on the same day as previously described.3 [18F]AV-1451 was acquired 46 days later. [18F]AV-1451 was synthesized and radiolabeled at LBNL's Biomedical Isotope Facility. One hundred minutes of dynamic data acquisition followed 9.5mCi of [18F]AV-1451 injected intravenously. PET data were reconstructed using an ordered subset expectation maximization algorithm with weighted attenuation. Images were smoothed with a 4mm Gaussian kernel with scatter correction and evaluated prior to analysis for patient motion and adequacy of statistical counts.
PET images were coregistered to the patient's MP-RAGE using SPM8. Dynamic 90-minute [11C]PIB data were analyzed using Logan graphical analysis with FreeSurfer-derived gray matter cerebellum as the reference region, yielding voxelwise distribution volume ratios (DVRs).13 [18F]FDG-PET images were summed, and standardized uptake value ratios (SUVrs) were calculated for the 30- to 60-minute postinjection interval using mean activity in the pons (defined in FreeSurfer and manually edited) as the reference region.14 Consistent with the initial report on human [18F]AV-1451/[18F]T807-PET,10 SUVr images at t = 80 to 100 minutes postinjection were created by normalizing summed activity from the realigned frames to mean activity in cerebellar gray matter, an area relatively spared of neurofibrillary tangle pathology even in advanced AD.15
Mean DVR/SUVr values were extracted for all tracers within 15 a priori selected FreeSurfer ROIs reflecting both clinically involved and spared cortical regions: bilateral occipital (calcarine, cuneus, and lateral occipital cortex), parietal (inferior and superior parietal, precuneus, isthmus, posterior cingulate, and supramarginal gyrus), temporal (superior, middle, and inferior gyri), and frontal (superior and middle gyri and orbitofrontal cortex). Pearson correlations were performed across these regions to assess the relationships between PET tracers.
Results
In accordance with the patient's clinical presentation, reduced [18F]FDG uptake was observed in primary visual and visual association cortices that included bilateral lateral occipital cortex and temporoparietal regions, more severe in the right hemisphere than the left (Fig 1). There was also mild hypometabolism in the right dorsolateral pre-frontal cortex. The [11C]PIB scan showed a typical AD-like pattern characterized by binding throughout the association neocortex. The occipital cortex showed low binding relative to the other lobes. In contrast, [18F]AV-1451 was selectively retained by posterior brain regions, in close correspondence with the regions that showed most prominent hypometabolism. Additionally, slightly elevated [18F]AV-1451 retention was observed in the right dorsolateral pre-frontal cortex, paralleling the mild hypometabolism on [18F]FDG-PET. Overall, the [18F]AV-1451 and [18F]FDG appeared strikingly as mirror images, with regions of high [18F]AV-1451 uptake corresponding to low [18F]FDG uptake and vice versa (see Fig 1).
FIGURE 1.
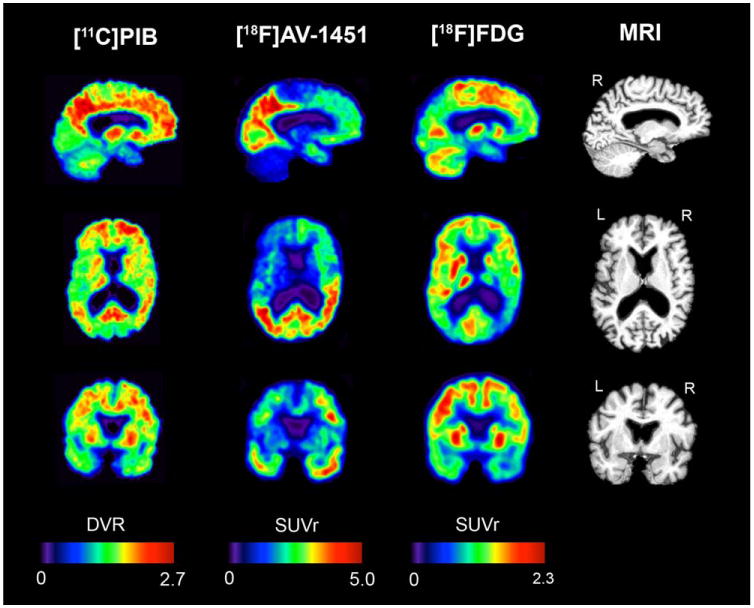
Sagittal, axial, and coronal slices of [11C]Pittsburgh compound B (PIB), [18F]AV-1451, and [18F]fluorodeoxyglucose (FDG) positron emission tomography alongside structural magnetic resonance imaging (MRI) scan. DVR = distribution volume ratio; L = left; R = right; SUVr = standardized uptake value ratio
Pearson correlations across 15 cortical ROIs confirmed the visual impression and showed a strong correlation between [18F]FDG and [18F]AV-1451 (r = −0.60, p<0.001, Fig 2A) in the absence of a correlation between [18F]FDG and [11C]PIB (r = 0.17, p = 0.38, see Fig 2B). [18F]AV-1451 and [11C]PIB did not correlate (r = 0.20, p = 0.29).
FIGURE 2.
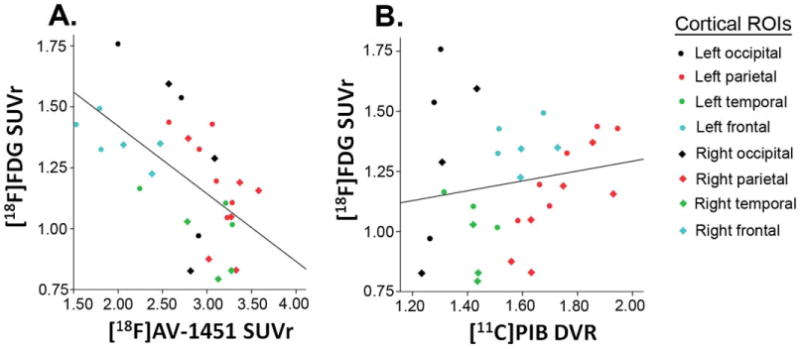
Pearson correlations between [18F]fluorodeoxyglucose (FDG) uptake and (A) [18F]AV-1451 retention (r = −0.60, p<0.001) and (B) [11C]Pittsburgh compound B (PIB) binding (r = 0.17, p = 0.38) across the neocortex suggest a stronger relationship with hypometabolism for tau than for amyloid pathology. See Patient and Methods section for detailed information on which brain regions were included within each cortical region of interest (ROI). DVR = distribution volume ratio; SUVr = standardized uptake value ratio
Discussion
The main finding of the present study was that in a patient with PCA the pattern of increased [18F]AV-1451 retention in posterior brain regions highly overlapped with regions that showed decreased [18F]FDG uptake, whereas [11C]PIB binding was distributed diffusely throughout association neocortex, consequently showing no correlation with [18F]FDG uptake. This provides preliminary in vivo evidence that hypometabolism and symptomatology are more closely linked to tau than to Aβ pathology.7,8
Aggregation of Aβ is an early pathophysiological event in the evolution of AD.16 The ability to measure Aβ in cerebrospinal fluid or by using PET has offered profound insights into preclinical and prodromal stages of AD17 and has shown great potential as a diagnostic add-on to the clinical workup of patients with cognitive deficits.18 on the other hand, biomarker studies have shown that the relationships between Aβ pathology and most downstream processes such as glucose hypometabolism, brain atrophy, disease severity, progression, and clinical presentation are modest at best.1,3 Furthermore, clinical trials applying anti-Aβ monoclonal antibodies have thus far failed to show a clinical benefit in AD dementia.19 These findings suggest that, at least at the dementia stage, non-Aβ processes are driving disease manifestation. Tau pathology is a likely candidate given its devastating effects on synaptic function,7 the strong correlation between the spatial distribution of tau and clinical evolution of the disease,20 and the close association between postmortem tau burden and cognitive performance during life.8 Tau imaging with PET provides a unique opportunity to study these molecular, anatomical, and clinical interactions in vivo.
Patients with atypical, nonamnestic clinical presentations of AD (ie, PCA, logopenic variant primary progressive aphasia, or behavioral/dysexecutive variant AD) share the plaques and tangles of typical amnestic AD, but show distinct neurodegenerative patterns and cognitive profiles. This study replicated previous amyloid PET findings in PCA that reported relatively diffuse [11C]PIB binding, with tracer retention in clinically affected posterior brain regions as well as in clinically and functionally spared frontal cortex.1,3 In contrast, [18F]AV-1451 was selectively retained in regions that are clinically affected in PCA, and show atrophy on MRI and hypometabolism on [18F]FDG-PET.3,4 This is in line with neuropathological studies showing that tau (in contrast to Aβ) is disproportionally elevated in brain regions that functionally and structurally deteriorate in PCA compared to relatively preserved brain regions.11
[18F]FDG and [18F]AV-1451 both showed excellent regional specificity in this PCA patient. However, tau radiotracers offer considerable potential added value over [18F]FDG in that they bind to a core element of AD pathology and hence could serve as a dual neurodegenerative and pathophysiological biomarker. Furthermore, the anticorrelation between [18F]FDG and [18F]AV-1451 was strong (r = −0.60) but not perfect. Current conceptual models of the AD pathophysiological cascade postulate that spread of tau may precede hypometabolism.16 There may also be differences in metabolic susceptibility to the neurotoxicity of tau across brain regions and individuals. Future studies with larger samples and longitudinal follow-up will be needed to explore the relationships between tau aggregation and brain metabolism.
A caveat of this study is the single-subject design; results will have to be replicated in larger samples. Furthermore, we did not correct PET data for partial volume effects. Opposing neurobiological directions of tau aggregation and glucose hypometabolism in AD, however, suggest that lack of partial volume correction, if any effect, may have underestimated the relationship between [18F]AV-1451 and [18F]FDG-PET. Finally, we acknowledge several methodological uncertainties that come with the novelty of [18F]AV-1451. The good correspondence between tracer uptake and the clinical picture, however, seems to support the use of [18F]AV-1451 PET in a clinical setting.
Acknowledgments
This research was funded by the Marie Curie FP7 International Outgoing Fellowship (628812; R.O.); donors of Alzheimer's Disease Research, a program of BrightFocus Foundation (R.O.); NIH National Institute on Aging (R01-AG045611, G.D.R.; R01-AG027859, W.J.J.; P50-AG023501, B.L.M.); Tau Consortium (G.D.R., W.J.J); John Douglas French Alzheimer's Foundation (G.D.R., B.L.M.); and State of California Department of Health Services Alzheimer's Disease Research Centre of California (04-33516, B.L.M). Avid Radiopharmaceuticals enabled use of the [18F]AV-1451 tracer, but did not provide direct funding and was not involved in data analysis or interpretation.
We thank J. Vogel, S. Lockhart, V. Shah, A. Fero, and K. Norton for their contributions in the acquisition and analysis of PET data.
Footnotes
Authorship: All authors designed the study. P.M.G., M.S., Z.A.M., B.M.B., and G.D.R. collected the data. R.O. and D.R.S. analyzed the data. R.O., D.R.S., W.J.J., and G.D.R. wrote the manuscript. S.L.B., J.P.O., M.J., P.M.G., M.S., Z.A.M., B.M.B., M.L.G.-T., and B.L.M. revised the manuscript.
Potential Conflicts of Interest: B.L.M.: Medical Director, John Douglas French Foundation; Scientific Director, Tau Consortium; Director/ Medical Advisory Board, Larry L. Hillblom Foundation; Scientific Advisory Board Member, National Institute for Health Research Cambridge Biomedical Research Centre and its subunit the Biomedical Research Unit in Dementia (UK). W.J.J.: personal fees, Genentech, Synarc, Hoffman LaRoche. G.D.R.: speaking fees, GE Healthcare, Medscape, Rockpointe.
References
- 1.Rabinovici GD, Furst AJ, Alkalay A, et al. Increased metabolic vulnerability in early-onset Alzheimer's disease is not related to amyloid burden. Brain. 2010;133:512–528. doi: 10.1093/brain/awp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ossenkoppele R, Zwan MD, Tolboom N, et al. Amyloid burden and metabolic function in early-onset Alzheimer's disease: parietal lobe involvement. Brain. 2012;135:2115–2125. doi: 10.1093/brain/aws113. [DOI] [PubMed] [Google Scholar]
- 3.Lehmann M, Ghosh PM, Madison C, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer's disease. Brain. 2013;136:844–858. doi: 10.1093/brain/aws327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ridgway GR, Lehmann M, Barnes J, et al. Early-onset Alzheimer disease clinical variants: multivariate analyses of cortical thickness. Neurology. 2012;79:80–84. doi: 10.1212/WNL.0b013e31825dce28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madhavan A, Whitwell JL, Weigand SD, et al. FDG PET and MRI in logopenic primary progressive aphasia versus dementia of the Alzheimer's type. PloS One. 2013;8:e62471. doi: 10.1371/journal.pone.0062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolk DA, Price JC, Madeira C, et al. Amyloid imaging in dementias with atypical presentation. Alzheimers Dement. 2012;8:389–398. doi: 10.1016/j.jalz.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron. 2014;82:756–771. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia CF, Arteaga J, Chen G, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer's disease. Alzheimers Dement. 2013;9:666–676. doi: 10.1016/j.jalz.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 10.Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34:457–468. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 11.Renner JA, Burns JM, Hou CE, et al. Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology. 2004;63:1175–1180. doi: 10.1212/01.wnl.0000140290.80962.bf. [DOI] [PubMed] [Google Scholar]
- 12.Crutch SJ, Lehmann M, Schott JM, et al. Posterior cortical atrophy. Lancet Neurol. 2012;11:170–178. doi: 10.1016/S1474-4422(11)70289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Logan J, Fowler JS, Volkow ND, et al. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 14.Minoshima S, Frey KA, Foster NL, Kuhl DE. Preserved pontine glucose metabolism in Alzheimer disease: a reference region for functional brain image (PET) analysis. J Comput Assist Tomogr. 1995;19:541–547. doi: 10.1097/00004728-199507000-00006. [DOI] [PubMed] [Google Scholar]
- 15.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropath. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 16.Jack CR, Jr, Holtzman DM. Biomarker modeling of Alzheimer's disease. Neuron. 2013;80:1347–1358. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the pre-clinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ossenkoppele R, Prins ND, Pijnenburg YA, et al. Impact of molecular imaging on the diagnostic process in a memory clinic. Alzheimers Dement. 2013;9:414–421. doi: 10.1016/j.jalz.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. New Engl J Med. 2014;370:322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]