

Abstract
Aicardi–Goutières syndrome is an inflammatory disease occurring due to mutations in any of TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR or IFIH1. We report on 374 patients from 299 families with mutations in these seven genes. Most patients conformed to one of two fairly stereotyped clinical profiles; either exhibiting an in utero disease-onset (74 patients; 22.8% of all patients where data were available), or a post-natal presentation, usually within the first year of life (223 patients; 68.6%), characterized by a sub-acute encephalopathy and a loss of previously acquired skills. Other clinically distinct phenotypes were also observed; particularly, bilateral striatal necrosis (13 patients; 3.6%) and non-syndromic spastic paraparesis (12 patients; 3.4%). We recorded 69 deaths (19.3% of patients with follow-up data). Of 285 patients for whom data were available, 210 (73.7%) were profoundly disabled, with no useful motor, speech and intellectual function. Chilblains, glaucoma, hypothyroidism, cardiomyopathy, intracerebral vasculitis, peripheral neuropathy, bowel inflammation and systemic lupus erythematosus were seen frequently enough to be confirmed as real associations with the Aicardi-Goutieres syndrome phenotype. We observed a robust relationship between mutations in all seven genes with increased type I interferon activity in cerebrospinal fluid and serum, and the increased expression of interferon-stimulated gene transcripts in peripheral blood. We recorded a positive correlation between the level of cerebrospinal fluid interferon activity assayed within one year of disease presentation and the degree of subsequent disability. Interferon-stimulated gene transcripts remained high in most patients, indicating an ongoing disease process. On the basis of substantial morbidity and mortality, our data highlight the urgent need to define coherent treatment strategies for the phenotypes associated with mutations in the Aicardi–Goutières syndrome-related genes. Our findings also make it clear that a window of therapeutic opportunity exists relevant to the majority of affected patients and indicate that the assessment of type I interferon activity might serve as a useful biomarker in future clinical trials.
Keywords: Aicardi–Goutières syndrome, bilateral striatal necrosis, spastic paraparesis, type I interferon, interferon signature
INTRODUCTION
Aicardi–Goutières syndrome (AGS) is a rare genetic disorder most consistently affecting the brain and the skin. The diagnosis is usually made in the context of an early-onset encephalopathy characterized by basal ganglia calcification and white matter abnormalities. However, since the original description [Aicardi and Goutieres, 1984], a wider spectrum of disease presentation, progression and outcome has been recognized. In 2007 [Rice et al., 2007b], we reported a genotype-phenotype analysis of 98 cases with pathogenic variants in the four genes, TREX1 [Crow et al., 2006a], RNASEH2A, RNASEH2B, and RNASEH2C [Crow et al., 2006b], known to be associated with AGS at that time. Since then three further genes, SAMHD1 [Rice et al., 2009], ADAR [Rice et al., 2012] and IFIH1 [Rice et al., 2014], have been described as mutated in patients demonstrating a phenotype consistent with AGS, and the spectrum of disease resulting from mutations in the AGS-related genes has broadened, in part due to the advent of the new sequencing technologies.
These seven genes encode proteins, namely TREX1, the RNase H2 complex, SAMHD1, ADAR and IFIH1 (MDA5), each of which is involved in nucleic acid metabolism/signaling. Patients with AGS consistently demonstrate increased levels of interferon activity in the cerebrospinal fluid and serum [Lebon et al., 1988], and an increased expression of interferon-stimulated genes (ISGs) in peripheral blood [Rice et al., 2013a], a so-called interferon signature. These observations are important in identifying AGS as an inflammatory disorder associated with the induction of a type I interferon mediated innate immune response, likely driven by endogenously-derived nucleic acids [Crow and Rehwinkel, 2009].
Here we present genetic and clinical data on 374 mutation-positive patients from 299 families encompassing all seven known AGS-related genes. In doing so, we provide a comprehensive view of the associated disease spectrum, natural history, and genotype-phenotype correlations, information which is prerequisite for the assessment of outcome in future clinical trials.
MATERIALS AND METHODS
Patient data
Subjects were ascertained through our own clinical practice and through contact with international collaborators. Patients were included where we observed either biallelic mutations in one of TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 and ADAR, a recognized heterozygous disease-causing mutation in TREX1 (p.Asp18Asn, p.Asp18His or p.Asp200Asn) or ADAR (p. Gly1007Arg), or a dominant mutation in IFIH1 (see Supplementary Table VII for cDNA mutations). We also collected data on patients with a characteristic phenotype of AGS who were heterozygous for otherwise presumed recessive mutations in these genes. Variants were considered to be pathogenic on the basis of a combination of criteria including multiple ascertainment in affected patients, appropriate segregation within families, de novo occurrence, the output of pathogenicity prediction packages, evolutionary conservation, frequency in publically available sequencing databases, and the results of published or previously unpublished functional assays and structural studies. Mutations are recorded according to Human Genome Variation Society (HGVS) nomenclature and the following transcripts: TREX1, NM_033629.4; RNASEH2A, NM_006397.2; RNASEH2B, NM_024570.3; RNASEH2C, NM_032193.3; SAMHD1, NM_015474.3; ADAR, NM_001111.4; IFIH1, NM_022168.2. A multiplex ligation-dependent probe amplification (MLPA) assay was used to look for copy number variants in TREX1, RNASEH2A, RNASEH2B, RNASEH2C and SAMHD1 (MRC-Holland).
Clinical and laboratory data were obtained through direct clinical contact and/or from medical records, recorded in a REDCap database [Harris et al., 2009] and reviewed by either Y.J.C. (304 patients), S.O. (42 patients) or A.V. (28 patients). Information about every clinical characteristic was not available for all patients. Assessments of the gross motor function, manual ability and communication status of patients over the age of 1 year were made using the Gross Motor Function Classification System (GMFCS) [Palisano et al., 1997], the Manual Ability Classification System (MACS) [Eliasson et al., 2006] and the Communication Function Classification System (CFCS) [Hidecker et al., 2011], respectively.
The study was approved by a U.K. Multicentre Research Ethics Committee (reference number 04:MRE00/19), the Mondino Ethics Committee (3549/2009, September 30, 2009 and December 11, 2009) and the Children’s National Medical Center Institutional Review Board.
RESULTS
Mutation data
The mutations observed by gene, and the number of times (by family) that they were seen, are given in Figure 1, Supplementary Figure 1 (A–G) and Supplementary Tables I–VII.
FIG. 1.

Numbers and percentages of families with Aicardi–Goutières syndrome (AGS) with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR and IFIH1. D: denotes dominant mutation. One child with a neurological phenotype and a single heterozygous mutation in TREX1, and three children with single heterozygous mutations in RNASEH2B were also identified. In addition, four families demonstrating autosomal dominant segregation of familial chilblain lupus (FCL) with mutations in either TREX1 (two families) or SAMHD1 (one family) were ascertained. Mutations in RNASEH2B and TREX1 represent more than half of our cohort. Considering their relatively recent identification, it is possible that the proportion of patients with mutations in ADAR and IFIH1 may increase.
Biallelic mutations were recorded in TREX1 (65 families: 22%), RNASEH2A (14 families: 5%), RNASEH2B (104 families: 36%), RNASEH2C (35 families: 12%), SAMHD1 (38 families: 13%) and ADAR (18 families: 6%). Monoallelic, dominant, mutations of IFIH1 were identified in nine families. We ascertained four patients with a neurological phenotype to have either a single p.Asp18Asn (two patients), a p.Asp18His (one patient) or p.Asp200Asn (one patient) mutation in TREX1, and five patients to harbor the dominant p.Gly1007Arg mutation in ADAR. All of these dominant mutations arose de novo, except in one family where an unaffected father transmitted the ADAR p.Gly1007Arg mutation to two daughters by two different partners. We identified three patients with a combination of three predicted deleterious variants in two genes (Supplementary Table VIII). Three families demonstrating autosomal dominant segregation of an exclusively skin phenotype termed familial chilblain lupus (FCL), with either a p.Asp18Asn mutation in TREX1 (one family) or a p.Ile201Asn mutation in SAMHD1 (one family), together with a single family segregating FCL apparently due to a p.Gly126Trpfs*2 mutation in TREX1 (plus a p.Phe17Ser variant of uncertain significance) were also ascertained. These FCL cases are not discussed further.
One of two recently described synonymous RNASEH2A variants, c.69G>A (Val23Val) and c.75C>T (Arg25Arg), considered to be pathogenic as a result of altered splicing, were identified in five families [Rice et al., 2013b]. We also recorded two intronic variants in RNASEH2B (c.65–13G>A, three families; c.322–17A>G, one family) which appear to affect mRNA splicing and are likely to be disease causing (Supplementary Fig. 2, Supplementary Table III).
There were four children from four families with a convincing clinical diagnosis of AGS in whom, after screening all seven AGS-related genes, we could identify only a single, presumed recessive, mutation (TREX1 p.Arg114His, one case; RNASEH2B p.Cys125-Tyr, p.Leu52Trp and c.136+1del, one case each)(Supplementary Tables I, III). These mutations were present in an unaffected parent in every family. In all other neurologically affected individuals we were able to identify biallelic gene mutations (except relating to patients with known dominant mutations of TREX1, ADAR and IFIH1). Apart from a recurrent deletion (5′ and including exon 1) of SAMHD1 seen in 10 patients of Ashkenazi Jewish ancestry (Supplementary Table V), we observed only one large deletion of RNASEH2B (Supplementary Table III), and a single complex deletion/duplication in SAMHD1 (Supplementary Table V).
Details of recurrent mutations are given in Supplementary Table IX. The p.Arg114His mutation in TREX1 was seen in 35 of 70 TREX1-related families. Although most were of northern European ancestry, we also observed this variant in the homozygous state in a single family each of Turkish and Pakistani background. We recorded a TREX1 p.Glu20Glyfs*82 mutation in eight families of south Asian ethnicity (six in the homozygous state). As previously described [Crow et al., 2003; Crow et al., 2006a], a founder mutation, p.Arg164*, in TREX1 segregates in patients from the Cree Indian population. Remarkably, in a pan-ethnic cohort of 107 families with mutations in RNASEH2B, 97 harbored the p. Ala177Thr substitution (48 homozygotes; 49 heterozygotes). Twenty-four families of south Asian origin were homozygous for a p.Arg69Trp mutation in RNASEH2C, likely indicative of a founder mutation. As stated above, we recorded a recurrent deletion of SAMHD1 in 10 Ashkenazim families. A p.Arg145* mutation in SAMHD1, occurring on a shared ancestral haplotype, was seen in five families, four of whom were known to be Maltese. A p. Pro193Ala mutation was seen, always in the heterozygous state, in 13 of 22 ADAR mutation-positive families, mainly of northern European descent.
Clinical data
Thirty-seven patients (11.4%) (25 TREX1; two RNASEH2A; one RNASEH2B; three RNASEH2C; three SAMHD1; one ADAR; two IFIH1) presented at birth with a congenital infection-like syndrome comprising abnormal neurological signs (e.g., poor feeding, irritability, abnormal tone, abnormal movements and seizures) with thrombocytopenia and hepatosplenomegaly, thus indicating a prenatal onset of disease (Fig. 2). A further 37 patients (11.4%) (13 TREX1; one RNASEH2A; nine RNASEH2B; seven RNASEH2C; six SAMHD1; one ADAR) demonstrated neurological features at birth in the absence of obvious systemic features. Although precisely dating the onset of disease was difficult in most cases presenting beyond the neonatal period, the majority (223 of 325; 68.6%) of patients experienced obvious neurological dysfunction within the first year of life (Fig. 2). In those cases where the clinical history was unequivocal, 65 children (18.0%) demonstrated normal development up until the time of the onset of symptoms (Fig. 3), with the likelihood of exhibiting normal development prior to presentation being 63%, 57% and 21% in relation to mutations in IFIH1, ADAR and RNASEH2B, respectively, Twenty-eight patients (8.6%) presented after the age of 1 year, with 35% and 30% of patients with mutations in ADAR and IFIH1, respectively, demonstrating the onset of disease after this age. The latest age at presentation known to us was a child with a p.Gly1007Arg mutation in ADAR who developed features of a subacute dystonia beginning at age 5.
FIG. 2.
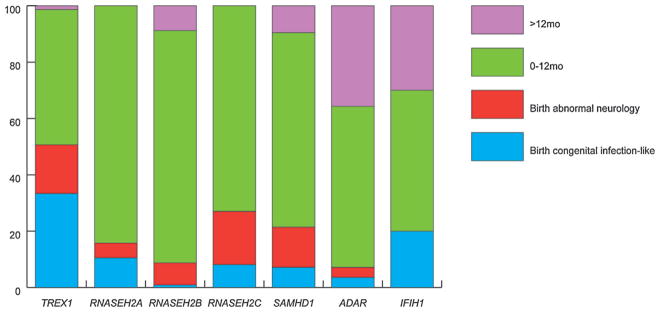
Age at presentation by genotype. Percentage of patients with either biallelic mutations or a recognized dominant mutation in one of the known AGS-related genes, in families where at least one individual has a neurological phenotype, i.e., excluding families with only FCL. Congenital infection-like describes patients with a neurological phenotype at birth plus thrombocytopenia and hepatosplenomegaly. Most patients present within the first year of life. Mutations in TREX1 were most frequently associated with a congenital infection-like presentation, while children presenting after the age of one year were most likely to harbor mutations in ADAR or IFIH1.
FIG. 3.
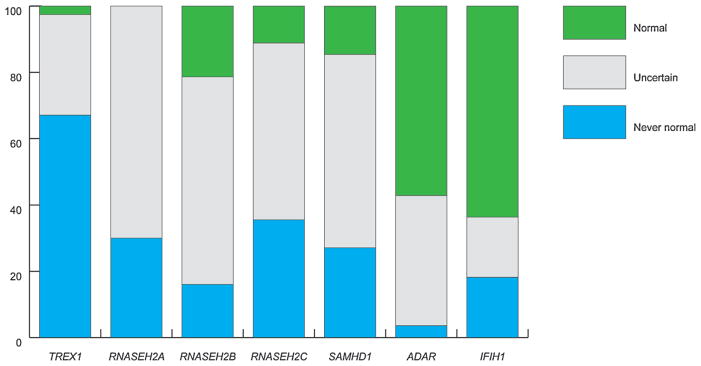
Development prior to presentation according to mutated gene. Percentage of patients with either biallelic mutations or a recognized dominant mutation in one of the known AGS genes, in families where at least one individual has a neurological phenotype, i.e., excluding families only with FCL. Never normal: presentation at or after birth without a period of normal development. Uncertain: presentation after birth where developmental status prior to presentation was uncertain. Normal: presentation after birth with definitely normal development prior to disease onset. Patients presenting with a period of normal development were more likely to harbor mutations in ADAR or IFIH1.
Most patients were considered to conform to the relatively stereotyped clinical profile previously described in the context of AGS, characterized by severe neurological dysfunction at birth or with onset in the first year of life, variably manifesting as spasticity, dystonia, seizures (140 of 362 patients, 39%), cortical blindness (111 of 362 patients, 31%) sometimes with pale optic discs, progressive microcephaly and psychomotor retardation. However, we also observed 13 children (3.6%) with the acute or sub-acute onset of severe dystonia and features of bilateral striatal necrosis on neuroimaging, in the absence of other features of AGS, all of whom carried mutations in ADAR (Supplementary Table X [Livingston et al., 2014]). Furthermore, we identified 12 patients (3.4%) (six RNASEH2B; three SAMHD1; two ADAR; one IFIH1) with a pure spastic paraparesis phenotype in the presence of normal neuroimaging, or non-specific changes in cerebral white matter, and preserved intellect [Crow et al., 2014b].
An assessment of gross motor function, manual ability and communication status at last contact was made in a total of 294, 291, and 285 patients, respectively (Supplementary Figs. 3–5). Of the latter, 210 (74%) patients were recorded to have none of any purposeful gross motor, hand and communication function. Only 14 of 294 patients (four RNASEH2B; one RNASEH2C; four SAMHD1; two ADAR; three IFHI1) were able to walk with no/ minimal support. Patients with mutations in RNASEH2B, SAMHD1, ADAR and IFIH1 were more likely to retain some useful, albeit often still limited, motor and communication abilities, i.e., they scored better than V, V and V on the GMFCS, MACS, and CFCS rating scales (three TREX1; two RNASEH2A; 32 RNASEH2B; three RNASEH2C; 19 SAMHD1; 10 ADAR; six IFIH1) (Fig. 4). A marked discrepancy in the severity of neurological outcome was observed between siblings in a small number of families. For example, an older sister to a severely neurologically affected female child was identified to have a homozygous p.Arg69Trp mutation in RNASEH2C and a history of chilblains in the absence of any other features [Vogt et al., 2013].
FIG. 4.
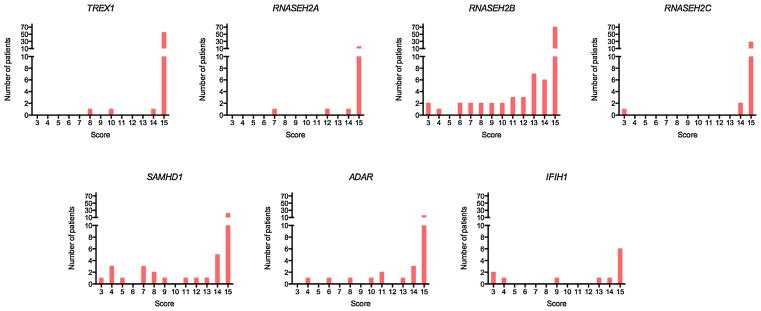
Degree of disability by mutated gene. Numbers represent the sum of GMFCS (Gross Motor Function Classification System for Cerebral Palsy), MACS (Manual Ability Classification System), and CFCS (Communication Function Classification System) score at time of last contact/ death for each patient where three is normal and 15 is profoundly disabled. Number of patients with either biallelic mutations or a recognized dominant mutation in one of the known AGS-related genes, in families where at least one individual has a neurological phenotype, i.e., excluding families with only FCL. Although most patients (74%) are severely neurologically damaged, this was more likely to be the case in children with mutations in TREX1, RNASEH2A or RNASEH2C.
We recorded data relating to status at last contact/known age at death in 357 patients (Fig. 5, Supplementary Table XI). Sixty-nine cases (19.3%) were ascertained to have died, with 37 of these deaths occurring in the first 5 years after birth, and mutations in TREX1 being associated with the highest number of deaths (26; 33.3% of all patients with mutations in TREX1). Sixty-eight patients (19.0%) were known to have lived beyond the age of 15 years, and we are aware of eight patients still alive at more than 30 years of age.
FIG. 5.

(A) Known status of AGS patients by age at last contact/age at death. (B) Known status of AGS patients by mutated gene. Number of patients with either biallelic mutations or a recognized dominant mutation in one of the known AGS-related genes, in families where at least one individual has a neurological phenotype i.e., excluding families with FCL only. Although there is a significant mortality associated with mutations in the AGS-related genes, a number of patients have been recorded to survive into adulthood. Mutations in TREX1 were associated with a greater risk of death than mutations in the other AGS-related genes.
Chilblains [Tolmie et al., 1995] were reported in 113 patients (31.2%) and were seen in association with mutations in all of the AGS-related genes, although only one patient with ADAR-related disease was reported to exhibit such lesions (in contrast to 26 of 48 [54.2%] patients with mutations in SAMHD1) (Fig. 6, Supplementary Table XII). The next most frequently described association was with glaucoma [Crow et al., 2004], which was recorded in 23 patients (6.3%) (10 of 48 [20.8%] patients with SAMHD1 mutations; no patients with mutations in ADAR or IFIH1). Most cases of glaucoma presented in the first 6 months of life, but one patient was diagnosed with bilateral disease requiring treatment at the age of 6 years. Intracerebral large vessel disease [Ramesh et al., 2010], usually identified retrospectively after a catastrophic intracerebral accident, was confirmed in nine patients, all with mutations in SAMHD1. A patient with mutations in TREX1 suffered a life-threatening intracerebral hemorrhage at age of 3 years, but without prior imaging evidence of a vascular anomaly. A further patient with TREX1 mutations [Olivieri et al., 2013] was noted to have a porencephalic lesion at the level of the left caudate nucleus due to an ischemic event in the territory of the perforating vessels.
FIG. 6.
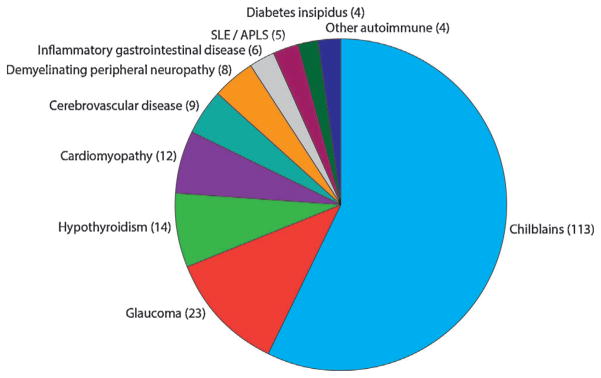
Frequency of associated phenotypes in AGS patients. Number of patients with either biallelic mutations or a recognized dominant mutation in one of the known AGS-related genes, in families where at least one individual has a neurological phenotype, i.e., excluding families with FCL only. SLE/APLS: Systemic lupus erythematosus/antiphospholipid syndrome. Inflammatory gastrointestinal disease: Crohns disease, atrophic gastritis, coeliac disease, autoimmune hepatitis, non-specific colitis. Other autoimmune: one diabetes mellitus, one hyperparathyroidism, one growth hormone deficiency, one adrenal insufficiency.
Hypothyroidism requiring replacement therapy was reported in 14 patients (3.9%) (six TREX1). Twelve cases (3.3%), nine with mutations in TREX1, were diagnosed with an infantile-onset hypertrophic cardiomyopathy. Eight patients were recorded to have a demyelinating peripheral neuropathy. Four patients were diagnosed with central diabetes insipidus (three TREX1), one with diabetes mellitus, one with hyperparathyroidism, one with growth hormone deficiency and one with both autoimmune gastritis and adrenal insufficiency. Six patients experienced inflammatory gastrointestinal problems (variably diagnosed as Crohn disease, atrophic gastritis, coeliac disease, autoimmune hepatitis and non-specific colitis). Four patients (two ADAR; two IFIH1) received a formal diagnosis of systemic lupus erythematosus (SLE), and one (TREX1) case developed antiphospholipid syndrome [Olivieri et al., 2013]. Three patients (two SAMHD1; one TREX1) demonstrated a panniculitis which in one case necessitated the use of high-dose immunosuppressive therapy. As previously described [Abe et al., 2014; Rice et al., 2007a], particularly widespread involvement of the skin was seen in three patients with an AGS phenotype due to dominant mutations in TREX1, with one of these patients experiencing a severe dactylitis showing limited responsive to high-dose immunosuppression. Two patients with SAMHD1-related disease developed a significant non-destructive arthropathy [Dale et al., 2010]. One affected individual with a homozygous splice-site mutation (c.1609–1G>C) in SAMHD1, and an additional predicted pathogenic heterozygous lesion in ADAR (p. Ala562Thr), developed chronic lymphocytic leukemia at the age of 24 years [Clifford et al., 2014].
Cerebrospinal fluid (CSF) and serum interferon activity, interferon-stimulated gene transcripts (ISGs) in blood, and CSF pterin levels
We have recently provided a detailed assessment of the level of interferon activity in CSF and serum measured using a cytopathic cell assay [Lebon et al., 1988; Lebon et al., 2002], and of the expression of a panel of ISGs in peripheral blood assessed by quantitative PCR [Rice et al., 2013a]. Summarizing data across the complete cohort described here, interferon activity in CSF and serum was consistently raised in mutation-positive patients, was negatively correlated with age (CSF, r =−0·601; serum, r = −0·274), and was higher in CSF than in serum in 93 of 134 paired samples (Supplementary Figs. 6–8, Supplementary Table XIII). Additionally, we recorded the level of pterins (in particular, neopterin) in CSF to be elevated in 48 patients sampled on 43 of 52 occasions, and to be negatively correlated with age (r = −0.617) (Supplementary Fig. 9, Supplementary Table XIII). We collated 233 CSF white cell count readings (from 158 patients: 167 of 233 results abnormal) (Supplementary Table XIII), which were also negatively correlated with age (r = −0.5559) (Supplementary Fig. 10). We derived an interferon score in peripheral blood for 100 patients measured on 146 occasions. A positive score (>2.4) was recorded in 67 of 68 (98%) patients with mutations in any of TREX1, RNA-SEH2A, RNASEH2C, SAMHD1, ADAR and IFIH1. In contrast, 10 of 32 (31%) patients with mutations in RNASEH2B demonstrated a normal interferon score (<2.4, i.e., within +2 SD of the control population) (Fig. 7, Supplementary Fig. 11, Supplementary Table XIII). While 77% (160 of 207) and 73% (115 of 158) of, respectively, CSF and serum interferon activity measurements were made before the age 24 months, only 15 of 146 (10%) ISG readings were made before this age (with the majority, 80% — 118 of 146, taken after the age of 4 years). The median value of CSF interferon activity recorded within 1 year of disease onset was significantly lower in patients with a combined score across the GMFCS, MACS and CFCS rating scales of <15, and demonstrated a positive correlation with disability in patients with mutations in all genes (Fig. 8).
FIG. 7.

(A) Quantitative reverse transcription PCR (qPCR) showing the interferon score derived from a panel of six interferon stimulated genes (ISGs) measured in whole blood in 100 AGS patients and 29 controls. The median fold change of the six probes combined was calculated to given an interferon score for each individual. Red bars show the median RQ value for each probe in each group. Samples colored red have a positive interferon score (>2.4) whereas samples colored blue have a normal interferon score (within +2 SD of the median for the control population). For subjects with repeat samples, the median combined measurement is shown. RQ is equal to 2−ΔΔCt, i.e., the normalized fold change relative to a control. One way ANOVA with Dunnett’s multiple comparison test. Almost all patients demonstrate a positive interferon score compared to controls, except for individuals with mutations in RNASEH2B, where 31% of patients demonstrated a normal interferon signature. (B) ISG RQ by mutated gene compared to controls. Red bars show the median RQ value for each probe in each group. One way ANOVA with Dunnett’s multiple comparison test. These data sets include some measurements published previously [Rice et al., 2013a]. These data indicate a clear upregulation of the expression of the six interferon stimulated genes assayed in patients compared to controls, with lower median values in patients with mutations in RNASEH2B.
FIG. 8.

CSF interferon measurements in patients assayed within one year of disease onset, plotted against disability score. (A) CSF interferon measurements in patients with a combined GMCSF, MACS, and CFCS score of 15 compared to patients with a score less than 15. Red bars show the median CSF interferon. Unpaired t-test of log transformed data. (B) CSF interferon measurements plotted against the combined disability score. In patients with serial measurements only the first measurement is shown. These data sets include some measurements published previously [Lebon et al., 1988, Lebon et al., 2002, Rice et al., 2013a]. There is a possible association between interferon activity in the cerebrospinal fluid measured in the first year of life and disability outcome.
DISCUSSION
Here, we present the mutational and phenotypic spectrum across seven genes known to be associated with a clinical diagnosis of AGS. Several points of note arise from these molecular and clinical data, which we discuss below.
Firstly, homozygous or compound heterozygous null mutations in TREX1 and in SAMHD1 are seen frequently, consistent with a complete loss of protein activity. In contrast, we have never observed biallelic null mutations in any of RNASEH2A, RNA-SEH2B, RNASEH2C or ADAR, indicating that such a state is either incompatible with life or is associated with phenotypes not ascertained here. We identified one child to have a maternally inherited C terminus frameshift mutation (p.Leu287Cysfs*11) in TREX1 (in combination with a second mutation), a molecular lesion previously considered to be exclusively relevant to the clinically distinct disorder retinal vasculopathy with cerebral leukodystrophy (RVCL) [Richards et al., 2007]. Whether the child and his mother are at risk of developing RVCL is unclear, but this result indicates that such mutations can be associated with the AGS phenotype.
Although AGS is most frequently inherited as an autosomal recessive trait, mutations in IFIH1 are all heterozygous gain-of-function [Rice et al., 2014], while the p.Gly1007Arg mutation in ADAR, seen in five patients, as well as the p.Asp18Asn, p.Asp18His and p.Asp200Asn mutations in TREX1, likely act as heterozygous dominant-negative alleles. To our knowledge, dominant mutations associated with a neurological phenotype have not been conclusively documented in RNASEH2A, RNASEH2B, RNASEH2C or SAMHD1.
Except in those patients with a previously recognized dominant mutation, we were able to define two likely pathogenic variants in all but four patients. These data indicate that pathogenic variants in non-coding regions relevant to gene regulation are rare in the clinical context that we have ascertained. Whether or not these four variants are contributory to the phenotype, or represent a chance association, is unclear. Possibilities include the presence of a cryptic second mutation, or non-penetrance in the transmitting parent. The significance of our finding of three patients with a combination of three predicted deleterious mutations in two genes is also uncertain. In this regard, we note that very few patients have been sequenced for mutations in all seven AGS-related genes.
We observed several founder mutations which may aid in screening of discrete populations, most obviously in the Ashkenazim where a carrier frequency of 1/138 was recorded for the recurrent SAMHD1 deletion (http://www.ashg.org/2013meeting/abstracts/fulltext/f130121959.htm). We also note that the p.Pro193Ala mutation in ADAR and the p.Ala177Thr substitution in RNASEH2B are associated with a non-negligible carrier frequency in the general population. In particular, the p.Pro193Ala has been seen on 32 and nine alleles in 4300 European Americans and 2203 African Americans, respectively (http://evs.gs.washington.edu/EVS/).
Although it was difficult to precisely date the onset of disease in many cases, in 65 patients, it was clear that the affected child demonstrated an initial period of normal development, with 28 children presenting after the age of 1 year. How the AGS-associated disease process is induced is uncertain, but could relate to an environmental trigger or genetic background. At least in the case of the ADAR-associated bilateral striatal necrosis phenotype, several parents gave a clear history of the onset of disease shortly after an infectious episode [Livingston et al., 2014].
The non-prospective nature of our data collection, with incomplete follow-up information and probable under-ascertainment of certain disease features, means that we are not able to derive formal mortality rates or risk statistics. However, it is clear from our results that AGS is a severe disease, with 74% of our cohort left with a profound combined deficit of motor and communication activity (we note that the CFCS does not assess intellectual function, and that some patients retained useful intellectual ability in the face of a major disturbance of communication skills). Mutations in TREX1 were frequently associated with a neonatal presentation, implying an in utero onset of disease, and with a high number of deaths. Mutations in ADAR, and IFIH1 were more likely to be seen in patients presenting after a definite period of normal development, and in patients presenting after the age of 1 year. Patients with mutations in these same two genes, as well as in RNASEH2B and SAMHD1, could also demonstrate some preservation of manual ability and communication skills. As well as clinically important differences in outcome between genes, we observed the same mutations in association with clinically distinct phenotypes (for example, mutations in ADAR can cause ‘classical’ AGS, ‘uncomplicated’ spastic paraparesis and bilateral striatal necrosis). There is no definite explanation for this variability in phenotypic expression and clinical severity, ranging from complete non-penetrance (including two IFIH1 mutation-positive individuals demonstrating a robust and sustained interferon signature who remain clinically asymptomatic at the ages of 48 and 79 years) [Rice et al., 2014], through isolated skin disease, to a severe neurological phenotype. Such variation, albeit apparently rare [Vogt et al., 2013], must be taken into account when interpreting the outcome of future clinical trials.
As recently described, our data show an almost 100% correlation between a positive interferon score and the presence of disease-associated mutations in TREX1, RNASEH2A, RNASEH2C, SAMHD1, ADAR, and IFIH1 [Rice et al., 2013a]. In contrast, 31% of patients with mutations in RNASEH2B did not demonstrate an overexpression of ISG transcripts in blood. Since ISG sampling was usually performed many years after initial diagnosis, it remains possible that all patients demonstrate a positive interferon signature at the time of disease onset, and that levels fall more quickly in patients with RNASEH2B mutations. Of note, our data suggest a positive correlation between the levels of CSF interferon activity assayed within one year of disease presentation, and disability as measured using a combined score across the GMFCS, MACS, and CFCS rating scales.
Beyond an initial encephalopathic phase, generally lasting several months, continued neurological deterioration was not obvious in most patients; indeed, some parents reported a slow but steady acquisition of new skills over time (although we also note that it is difficult to assess a loss/gain of skills in a child who is already profoundly neurologically compromised, and that a few parents described possible further episodes of regression). This observation is consistent with the survival of some patients into the fourth decade of life, and a definite trend towards a decline in interferon activity in CSF and serum, as well as CSF levels of the inflammatory marker neopterin [Dale et al., 2009] and the CSF white cell count, over time. Since only a limited number of samples were collected during the early stage of the disease, our ISG data do not contradict this suggestion, although they clearly demonstrate that an interferon signature persists long term in most patients, indicative of an ongoing inflammatory process. Such persistence is reflected clinically by the high frequency of recurrent chilblains, most typically occurring in the winter months, and the intracranial large-vessel disease particularly seen in patients with mutations in SAMHD1. Why the AGS-associated clinical phenotype apparently ‘abates’ neurologically beyond the initial subacute encephalopathic phase, and whether or not patients are at risk of neurological disease progression, or ‘flares’, in later life, is still uncertain.
We note the consistent association of the AGS phenotype with glaucoma, hypothyroidism, cardiomyopathy and a demyelinating peripheral neuropathy. All might be overlooked in the case of a severely disabled individual unable to report symptoms, and so we would recommend that these states are searched for on a proactive basis. Empirically, and because of the possibility for treatment, we would suggest life-long surveillance, perhaps annually, for glaucoma and thyroid function. Our own work [Ramesh et al., 2010], and that of others [Thiele et al., 2010; Xin et al., 2011], shows that the risk of cerebrovascular accidents in the context of SAMHD1-related disease is high, and indicates a particular role for SAMHD1 in blood vessel integrity and homeostasis. Given the potential for intervention, individuals with mutations in SAMHD1 might benefit from screening for intracranial arteriopathy, although such a decision would need to take account of the overall clinical situation and continued uncertainty about management in the face of such lesions.
Patients with both AGS and SLE were first described over 14 years ago [Aicardi and Goutieres, 2000; Dale et al., 2000], and heterozygous mutations in TREX1 have been identified in non-syndromic lupus [Lee-Kirsch et al., 2007]. However, the frequency with which such mutations occur in SLE is unclear [Barizzone et al., 2013; Namjou et al., 2011]. Although we have not undertaken prospective testing of a large group of patients specifically addressing the point, our data indicate that the frequency of clinically diagnosed lupus in patients with AGS is low (only four cases in our series). More generally, following our description of progressive arthropathy with distal joint contractures and painful mouth ulcers in association with biallelic SAMHD1 mutations [Dale et al., 2010], and considering the associated high frequency of chilblains (54%), glaucoma (21%) and intracranial vasculopathy (18%), we would suggest that there is a need to consider mutation analysis of SAMHD1 (and possibly the other AGS-related genes), in a broad range of inflammatory phenotypes.
Our experience indicates that carriers of recessive mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 and ADAR do not normally manifest disease features. In particular, we are not aware of a proven increase in the incidence of cancer in these individuals, nor of cancer in affected patients. However, the documented role of the RNase H2 complex in removing mis-incorporated ribonucleotides from DNA [Reijns et al., 2012], and the observation of a patient with mutations in SAMHD1 developing chronic lymphocytic leukemia at the age of 24 years [Clifford et al., 2014], indicates the need for long-term observation of patients for features of malignancy.
We would not expect to be able to reverse neurological damage already accrued at the time of initiating treatment, a fact of particular relevance for patients affected in utero and displaying pathological signs at birth. However, the majority of children with AGS demonstrate the onset of disease at a variable time post-natally. This observation is important in suggesting that treatment in the early stages of the disease might result in an attenuation of the associated inflammation and consequent tissue injury. In certain cases, e.g., where chilblains are a particular problem, and in the context of the recognized later-presenting phenotypes described above, treatment beyond the sub-acute encephalopathic phase might be beneficial even in the presence of significant neurological dysfunction.
With the integration of new sequencing technologies into standard clinical practice, we predict that the spectrum of phenotypes associated with mutations in the AGS-related genes will broaden. These observations beg the question as to whether such cases should actually be referred to as AGS. Irrespective of nosology, it is probable that these phenotypes likely all relate to a common pathology, involving an upregulation of type I interferons stimulated by endogenous nucleic acids [Crow, 2011; Crow, 2015], and might therefore potentially benefit from similar anti-interferon/ anti-inflammatory therapeutic strategies [Crow et al., 2014a].
Supplementary Material
Acknowledgments
Grant sponsor: European Union’s Seventh Framework Programme; Grant number: GA 241779; Grant sponsor: European Research Council; Grant number: GA 309449; Grant sponsor: National Research Agency (France) under the “Investments for the Future” program; Grant number: ANR-10-IAHU-01; Grant sponsor: Parsons Family Foundation.
We sincerely thank the patients and their families included in this research. We thank the International Aicardi–Goutières syndrome Association (IAGSA) and all other clinicians who have contributed patients/data not included here. We thank Dr Anna Schuh and Dr Ruth Clifford for providing sequence data. This paper is dedicated to the memory of Dr. John L Tolmie.
Footnotes
Conflict of interest: none.
Additional supporting information may be found in the online version of this article at the publisher’s website.
References
- Abe J, Nakamura K, Nishikomori R, Kato M, Mitsuiki N, Izawa K, Awaya T, Kawai T, Yasumi T, Toyoshima I, Hasegawa K, Ohshima Y, Hiragi T, Sasahara Y, Suzuki Y, Kikuchi M, Osaka H, Ohya T, Ninomiya S, Fujikawa S, Akasaka M, Iwata N, Kawakita A, Funatsuka M, Shintaku H, Ohara O, Ichinose H, Heike T. A nationwide survey of Aicardi-Goutieres syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology (Oxford) 2014;53:448–458. doi: 10.1093/rheumatology/ket372. [DOI] [PubMed] [Google Scholar]
- Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. doi: 10.1002/ana.410150109. [DOI] [PubMed] [Google Scholar]
- Aicardi J, Goutieres F. Systemic lupus erythematosus or Aicardi-Goutieres syndrome. Neuropediatrics. 2000;31:113. doi: 10.1055/s-2000-7533. [DOI] [PubMed] [Google Scholar]
- Barizzone N, Monti S, Mellone S, Godi M, Marchini M, Scorza R, Danieli MG, D’Alfonso S. Rare variants in the TREX1 gene and susceptibility to autoimmune diseases. Biomed Res Int. 2013;2013:471703. doi: 10.1155/2013/471703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford R, Louis T, Robbe P, Ackroyd S, Burns A, Timbs AT, Colopy Wright, Dreau G, Sigaux H, Judde F, Rotger JG, Telenti M, Lin A, Pasero YL, Maelfait P, Titsias J, Cohen M, Henderson DR, Ross SJ, Bentley MT, Hillmen D, Pettitt P, Rehwinkel A, Knight J, Taylor SJ, Crow JC, Benkirane YJ, Schuh M. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. 2014;123:1021–1031. doi: 10.1182/blood-2013-04-490847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ. Type I interferonopathies: A novel set of inborn errors of immunity. Ann N Y Acad Sci. 2011;1238:91–98. doi: 10.1111/j.1749-6632.2011.06220.x. [DOI] [PubMed] [Google Scholar]
- Crow YJ. Type I interferonopathies: Mendelian type I interferon up-regulation. Curr Opin Immunol. 2015;32:7–12. doi: 10.1016/j.coi.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, Michaud J, Roberts E, Stephenson JB, Woods CG, Lebon P. Cree encephalitis is allelic with Aicardi-Goutieres syndrome: Implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. 2003;40:183–187. doi: 10.1136/jmg.40.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006a;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006b;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Massey RF, Innes JR, Pairaudeau PW, Hill Rowland, Woods CA, Ali CG, Livingston M, Lebon JH, Nischall P, McEntagart K, Hindocha M, Winter N. Congenital glaucoma and brain stem atrophy as features of Aicardi-Goutieres syndrome. Am J Med Genet Part A. 2004;129A:303–307. doi: 10.1002/ajmg.a.30250. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18:R130–R136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Vanderver A, Orcesi S, Kuijpers TW, Rice GI. Therapies in Aicardi-Goutieres syndrome. Clin Exp Immunol. 2014a;175:1–8. doi: 10.1111/cei.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Zaki MS, Abdel-Hamid MS, Abdel-Salam GM, Boespflug-Tanguy O, Cordeiro NJV, Gleeson JG, Gowrinathan NR, Laugel V, Renaldo F, Rodriguez D, Livingston JH, Rice GI. Mutations in ADAR1, IFIH1 and RNASEH2B presenting as spastic paraplegia. Neuropediatrics. 2014b;45:386–391. doi: 10.1055/s-0034-1389161. [DOI] [PubMed] [Google Scholar]
- Dale RC, Brilot F, Fagan E, Earl J. Cerebrospinal fluid neopterin in paediatric neurology: A marker of active central nervous system inflammation. Dev Med Child Neurol. 2009;51:317–323. doi: 10.1111/j.1469-8749.2008.03225.x. [DOI] [PubMed] [Google Scholar]
- Dale RC, Gornall H, Singh-Grewal D, Alcausin M, Rice GI, Crow YJ. Familial Aicardi-Goutieres syndrome due to SAMHD1 mutations is associated with chronic arthropathy and contractures. Am J Med Genet A. 2010;152A:938–942. doi: 10.1002/ajmg.a.33359. [DOI] [PubMed] [Google Scholar]
- Dale RC, Tang SP, Heckmatt JZ, Tatnall FM. Familial systemic lupus erythematosus and congenital infection-like syndrome. Neuropediatrics. 2000;31:155–158. doi: 10.1055/s-2000-7492. [DOI] [PubMed] [Google Scholar]
- Eliasson AC, Krumlinde-Sundholm L, Rosblad B, Beckung E, Arner M, Ohrvall AM, Rosenbaum P. The Manual Ability Classification System (MACS) for children with cerebral palsy: Scale development and evidence of validity and reliability. Dev Med Child Neurol. 2006;48:549–554. doi: 10.1017/S0012162206001162. [DOI] [PubMed] [Google Scholar]
- Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap) – a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidecker MJ, Paneth N, Rosenbaum PL, Kent RD, Lillie J, Eulenberg JB, Chester K, Jr, Johnson B, Michalsen L, Evatt M, Taylor K. Developing and validating the Communication Function Classification System for individuals with cerebral palsy. Dev Med Child Neurol. 2011;53:704–710. doi: 10.1111/j.1469-8749.2011.03996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci. 1988;84:201–208. doi: 10.1016/0022-510x(88)90125-6. [DOI] [PubMed] [Google Scholar]
- Lebon P, Meritet JF, Krivine A, Rozenberg F. Interferon and Aicardi-Goutieres syndrome. Eur J Paediatr Neurol. 2002;6:A47–A53. doi: 10.1053/ejpn.2002.0574. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CG, Falconer A, Forte G, Jenkinson EM, Kasher PR, Szynkiewicz M, Rice GI, Crow YJ. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet. 2014;51:76–82. doi: 10.1136/jmedgenet-2013-102038. [DOI] [PubMed] [Google Scholar]
- Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, Alarcon-Riquelme ME, Gallant CJ, Boackle SA, Criswell LA, Kimberly RP, Brown E, Edberg J, Stevens AM, Jacob CO, Tsao BP, Gilkeson GS, Kamen DL, Merrill JT, Petri M, Goldman RR, Vila LM, Anaya JM, Niewold TB, Martin J, Pons-Estel BA, Sabio JM, Callejas JL, Vyse TJ, Bae SC, Perrino FW, Freedman BI, Scofield RH, Moser KL, Gaffney PM, James JA, Langefeld CD, Kaufman KM, Harley JB, Atkinson JP. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 2011;12:270–279. doi: 10.1038/gene.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri I, Cattalini M, Tonduti D, La Piana R, Uggetti C, Galli J, Meini A, Tincani A, Moratto D, Fazzi E, Balottin U, Orcesi S. Dysregulation of the immune system in Aicardi-Goutieres syndrome: Another example in a TREX1-mutated patient. Lupus. 2013;22:1064–1069. doi: 10.1177/0961203313498800. [DOI] [PubMed] [Google Scholar]
- Palisano R, Rosenbaum P, Walter S, Russell D, Wood E, Galuppi B. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol. 1997;39:214–223. doi: 10.1111/j.1469-8749.1997.tb07414.x. [DOI] [PubMed] [Google Scholar]
- Ramesh V, Bernardi B, Stafa A, Garone C, Franzoni E, Abinun M, Mitchell P, Mitra D, Friswell M, Nelson J, Shalev SA, Rice GI, Gornall H, Szynkiewicz M, Aymard F, Ganesan V, Prendiville J, Livingston JH, Crow YJ. Intracerebral large artery disease in Aicardi-Goutieres syndrome implicates SAMHD1 in vascular homeostasis. Dev Med Child Neurol. 2010;52:725–732. doi: 10.1111/j.1469-8749.2010.03727.x. [DOI] [PubMed] [Google Scholar]
- Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149:1008–1022. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O’Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007a;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen HJ, Corry PC, Cowan FM, Cox H, D’Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SG, Garcia-Cazorla A, Gener B, Goizet C, Goutieres F, Green AJ, Guet A, Hamel BC, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang YH, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EG, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard ML, Nischal KK, Ostergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Burgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JB, Tacke U, Tan TY, Till M, Tolmie JL, Tomlin P, Vagnarelli F, Valente EM, Van Coster RN, Van der Aa N, Vanderver A, Vles JS, Voit T, Wassmer E, Weschke B, Whiteford ML, Willemsen MA, Zankl A, Zuberi SM, Orcesi S, Fazzi E, Lebon P, Crow YJ. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007b;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, Crow YJ. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, Casarano M, Chouchane M, Cimaz R, Collins AE, Cordeiro NJ, Dale RC, Davidson JE, De Waele L, Desguerre I, Faivre L, Fazzi E, Isidor B, Lagae L, Latchman AR, Lebon P, Li C, Livingston JH, Lourenco CM, Mancardi MM, Masurel-Paulet A, McInnes IB, Menezes MP, Mignot C, O’Sullivan J, Orcesi S, Picco PP, Riva E, Robinson RA, Rodriguez D, Salvatici E, Scott C, Szybowska M, Tolmie JL, Vanderver A, Vanhulle C, Vieira JP, Webb K, Whitney RN, Williams SG, Wolfe LA, Zuberi SM, Hur S, Crow YJ. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, Barnerias C, Bernard G, Bodemer C, Botella MP, Cereda C, Chandler KE, Dabydeen L, Dale RC, De Laet C, De Goede CG, Del Toro M, Effat L, Enamorado NN, Fazzi E, Gener B, Haldre M, Lin JP, Livingston JH, Lourenco CM, Marques W, Jr, Oades P, Peterson P, Rasmussen M, Roubertie A, Schmidt JL, Shalev SA, Simon R, Spiegel R, Swoboda KJ, Temtamy SA, Vassallo G, Vilain CN, Vogt J, Wermenbol V, Whitehouse WP, Soler D, Olivieri I, Orcesi S, Aglan MS, Zaki MS, Abdel-Salam GM, Vanderver A, Kisand K, Rozenberg F, Lebon P, Crow YJ. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet Neurol. 2013a;12:1159–1169. doi: 10.1016/S1474-4422(13)70258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W, Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, Crow YJ. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Reijns MA, Coffin SR, Forte GM, Anderson BH, Szynkiewicz M, Gornall H, Gent D, Leitch A, Botella MP, Fazzi E, Gener B, Lagae L, Olivieri I, Orcesi S, Swoboda KJ, Perrino FW, Jackson AP, Crow YJ. Synonymous mutations in RNASEH2A create cryptic splice sites impairing RNase H2 enzyme function in Aicardi-Goutieres syndrome. Hum Mutat. 2013b;34:1066–1070. doi: 10.1002/humu.22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KR, de Vries B, Wan J, Kane MJ, Mamsa H, Schafer R, Stam AH, Haan J, de Jong PT, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068–1070. doi: 10.1038/ng2082. [DOI] [PubMed] [Google Scholar]
- Thiele H, du Moulin M, Barczyk K, George C, Schwindt W, Nurnberg G, Frosch M, Kurlemann G, Roth J, Nurnberg P, Rutsch F. Cerebral arterial stenoses and stroke: novel features of Aicardi-Goutieres syndrome caused by the Arg164X mutation in SAMHD1 are associated with altered cytokine expression. Hum Mutat. 2010;31:E1836–E1850. doi: 10.1002/humu.21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolmie JL, Shillito P, Hughes-Benzie R, Stephenson JB. The Aicardi-Goutieres syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis) J Med Genet. 1995;32:881–884. doi: 10.1136/jmg.32.11.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J, Agrawal S, Ibrahim Z, Southwood TR, Philip S, Macpherson L, Bhole MV, Crow YJ, Oley C. Striking intrafamilial phenotypic variability in Aicardi-Goutieres syndrome associated with the recurrent Asian founder mutation in RNASEH2C. Am J Med Genet Part A. 2013;161A:338–342. doi: 10.1002/ajmg.a.35712. [DOI] [PubMed] [Google Scholar]
- Xin B, Jones S, Puffenberger EG, Hinze C, Bright A, Tan H, Zhou A, Wu G, Vargus-Adams J, Agamanolis D, Wang H. Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc Natl Acad Sci USA. 2011;108:5372–5377. doi: 10.1073/pnas.1014265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.