

Abstract
The ability to monitor RNAs of interest in living cells is crucial to understanding the function, dynamics, and regulation of this important class of molecules. In recent years, numerous strategies have been developed with the goal of imaging individual RNAs of interest in living cells, each with their own advantages and limitations. This chapter provides an overview of current methods of live-cell RNA imaging, including a detailed discussion of genetically encoded strategies for labeling RNAs in mammalian cells. This chapter then focuses on the development and use of “RNA mimics of GFP” or Spinach technology for tagging mammalian RNAs, and includes a detailed protocol for imaging 5S and CGG60 RNA with the recently described Spinach2 tag.
1. Introduction
Our knowledge of the diverse roles of RNA molecules in every aspect of gene expression has expanded tremendously in recent years. To understand better the abundance, dynamics, and localization of individual RNAs, tools for labeling and imaging RNAs are necessary to study their spatiotemporal regulation. Recently, several major independent advances in imaging technology have allowed for RNAs to be monitored in living cells.
Many powerful RNA imaging techniques involve the use of exogenously added probes, such as labeled antisense probes (Molenaar et al., 2001; Molenaar et al., 2004) and molecular beacons (Bao et al., 2004; Bratu et al., 2003; Nitin et al., 2004; Tyagi & Alsmadi, 2004). These techniques have the advantage of targeting native, non-engineered RNAs. However, they are limited by the ability to deliver probe into cells without perturbing the biological system (Santangelo et al., 2012). However, several fully genetically encoded systems for studying RNA in living cells have been developed. These include the MS2-GFP imaging system, the Pumilio-split FP imaging system, and the use of “RNA mimics of GFP” to tag RNAs of interest. For the purposes of this chapter, these genetically encoded systems will be discussed in detail.
The MS2-GFP system for imaging tagged RNAs in living cells provided the first genetically encoded system for labeling endogenous RNAs (Brodsky & Silver, 2002; Fusco et al., 2003). In this system, an RNA of interest is tagged with up to 24 copies of the MS2 RNA hairpin and coexpressed with the MS2 coat protein tagged with GFP (Figure 1A) (Fusco et al., 2003; Shav-Tal et al., 2004). Because MS2-GFP binds to the MS2 hairpin as a high-affinity dimer, the tagged RNAs become labeled with up to 48 GFP molecules, allowing for imaging of even individual mRNAs in living cells. This technique was recently used to study the trafficking of individual β-actin mRNAs in the neuronal cells of a living mouse (Park et al., 2014).
Figure 1. Genetically encoded strategies for labeling RNA in living cells.

A) The MS2-GFP imaging system involves labeling an mRNA of interest (black line) with up to 24 copies of the MS2 RNA hairpin in the 3′ UTR (gray line). When this tagged RNA is coexpressed with the MS2 coat protein fused to GFP, these hairpins are bound by MS2-GFP as dimers, leading to tagging by up to 48 MS2-GFP molecules. B) The PUM-HD split-FP imaging system involves engineering two PUM-HDs to distinct 8-nt regions in an RNA of interest. Each PUM-HD is then fused to half of a split fluorescent protein. When both PUM-HDs are bound to the target RNA, fluorescence complementation occurs and the RNA is labeled with a single fluorescent protein. C) The MS2/PP7 split FP imaging system involves tagging an RNA with both the MS2 RNA hairpin (gray hairpin) and the PP7 RNA hairpin (blue hairpin) in alternation. MS2 and PP7 coat proteins are each tagged with complementary halves of an FP. When both are bound, complementation occurs, leading to labeling of RNAs. D) Imaging an RNA of interest by Spinach tagging involves fusing Spinach to either the 5′ or 3′ end of an RNA and incubating cells with a dye that is nonfluorescent in solution. Only when the dye is bound to the tagged RNA does the Spinach-dye complex become fluorescent, specifically labeling the tagged RNA.
Although powerful, this technique has several drawbacks. First, the MS2-GFP fusion protein is fluorescent throughout the cell, which can lead to high background. To mitigate this problem, MS2-GFP can be fused with a nuclear localization signal, so that the excess unbound MS2-GFP concentrates in the nucleus, lowering cytoplasmic background fluorescence. However, this prevents nuclear and cytoplasmic RNAs from being imaged in a single experiment. In addition, the presence of up to 48 nuclear localization signals tethered to the tagged RNA can lead to mislocalization and confound the interpretation of trafficking behaviors of tagged RNAs (Tyagi, 2009).
To bypass some of these problems, a similar system was developed using split fluorescent proteins fused to Pumilio homology domains (PUM-HDs) (Ozawa et al., 2007). PUM-HDs are site-specific RNA binding proteins that can be engineered to bind an 8-nt target of interest. In this case, the authors fused two halves of a fluorescent protein to two different PUM-HDs that were targeted to adjacent 8-nt sites on an RNA. Only when both proteins bound were the two halves of the fluorescent protein brought into close proximity to allow for bimolecular fluorescence complementation and fluorescence signal (Figure 1B). This technique has several advantages. The first is that unbound proteins are nonfluorescent, reducing background fluorescence. The second is that PUM-HDs can be engineered to target native RNAs. A primary disadvantage is that each RNA of interest is only labeled with a single fluorescent protein, which does not allow imaging of individual RNA molecules. A second disadvantage is that PUM-HDs must be engineered every time a new RNA is targeted.
The Singer laboratory recently developed a new technology that weds the advantages of the MS2-GFP system and this PUM-HD system, in which two different RNA binding proteins (the MS2 coat protein and the PP7 coat protein) are each tagged with a different half of a fluorescent protein (Wu et al., 2014). Then, these two fusion proteins are coexpressed with an RNA containing both MS2 and PP7 binding sites in the 3′ UTR. Only when both fusion proteins have bound do the two fluorescent protein halves interact to become fluorescent, nearly eliminating background fluorescence (Figure 1C). However, this technique still requires the large insertion of sequence into an RNA which may alter folding, function, or regulation.
A different approach for imaging RNA in living cells is based on appending RNA tags that directly confer fluorescence to RNAs of interest (Paige et al., 2011; Strack et al., 2013). Here, “RNA mimics of GFP” are used to tag RNAs for live-cell imaging (Figure 1D). A detailed discussion of the development of Spinach and its derivative Spinach2 follows, along with a detailed protocol for imaging RNA in mammalian cells.
2. Developing Spinach, an RNA mimic of GFP
In order to image RNA dynamics in living cells, we sought to develop an RNA tag that would be analogous to the GFP imaging system for protein localization in live cells. RNA aptamers had previously been described that can bind fluorescent dyes, which could potentially be used for labeling RNAs (Holeman et al.,1998). However, the fluorescence of these dyes was high regardless of whether they were bound to RNA, leading to high cellular background fluorescence. One exception was an aptamer that could conditionally activate a dye called malachite green (Babendure et al., 2003). In solution, malachite green is nonfluorescent, but when bound to a cognate aptamer RNA, it became brightly fluorescent. However, malachite green binds to cellular components which also induce its fluorescence, resulting in high background fluorescence levels (Paige et al., 2011).
To harness the power of an activatable dye, it is essential to use a dye that does not get activated by the diverse cellular constituents in the cell. Previous studies on GFP revealed that under protein denaturing conditions the chromophore was absorbant but not fluorescent, indicating that the folded protein modifies the structure of the chromophore to allow for fluorescence (Ward & Bokman, 1982). For this reason, we synthesized the GFP chromophore and several derivatives (Paige et al., 2011). We found these to be strongly absorbant, but essentially nonfluorescent in solution. Moreover, when cells were treated with these dyes, nominal cell-induced fluorescence was observed (Paige et al., 2011).
SELEX was then carried out against these dyes immobilized on agarose beads. Following SELEX, eluted RNAs were screened for their ability to induce fluorescence. The brightest RNA-dye pair was an RNA that binds 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI), a fluorophore that resembles the GFP fluorophore but contains two aromatic fluorine atoms. This RNA, designated Spinach, is approximately 80% as bright as wild-type GFP and half as bright as EGFP when bound to DFHBI (Figure 2) (Paige et al., 2011).
Figure 2. Schematic representations of the aptamers and dyes.

A) The mFOLD-predicted secondary structures of Spinach (left) and Spinach2 (right) are shown. The green highlighted region shows the regions that contain mutations. B) DFHBI (left) and DFHBI-1T (right).
Spinach-DFHBI fluorescence was then tested in bacterial cells. When E. coli cells overexpressing Spinach were incubated in media containing DFHBI, the cells were brightly fluorescent, indicating that Spinach was properly folded in this context and that DFHBI was able to enter the bacterial cells (Paige et al., 2011). Next, the utility of Spinach in tagging a mammalian RNA was examined. 5S RNA was tagged at the 3′ end with Spinach and expressed from its native promoter in HEK293-T cells. Here, diffuse cytoplasmic signal was observed for 5S-Spinach that was relocalized to stress granules after sucrose treatment, as expected for untagged 5S (Paige et al., 2011). These experiments demonstrated that Spinach-DFHBI could be used for live-cell imaging.
3. Imaging with Spinach2, a superfolding variant of Spinach
Although Spinach could be used to tag 5S RNA, the exposure times necessary for imaging revealed that 5S-Spinach was substantially dimmer than expected, suggesting that Spinach might not perform well as a tag in mammalian systems. Moreover, experiments tagging other RNAs demonstrated that Spinach was not bright enough to be imaged, although tagged RNA was readily detected by fluorescence in situ hybridization (Strack et al., 2013).
We reasoned that several factors could affect the fluorescence of Spinach in cells, including ion concentration, temperature, the overall percentage folded, and the amount that is folded in the context of tagged RNA. Testing these parameters showed that even under ideal in vitro conditions, only ~30% of Spinach is folded (Strack et al., 2013). Melting curve analysis also showed that more than 50% of Spinach is unfolded at 37°C, the temperature routinely used for imaging. Based on these results, we carried out systematic mutagenesis of Spinach to improve folding and thermostability. The result was Spinach2, which folds twice as well as Spinach in vitro and is substantially brighter than Spinach at 37°C (Strack et al., 2013) (Figure 2). Moreover, Spinach2 retains a high percent folding rate in diverse sequence contexts, unlike Spinach. These mutations seem to affect only the percentage of Spinach2 that folds properly, as the extinction coefficient, quantum yield, and KD for dye binding were unchanged (Strack et al., 2013).
Spinach2 was then used to tag the noncoding RNAs 5S and 7SK. In both cases, Spinach2 was markedly brighter than Spinach (Strack et al., 2013). In addition, Spinach2 was used to label a “toxic” model RNA associated with Fragile X tremor/ataxia syndrome (FXTAS). When the 5′ UTR of FMR1 contains 55–200 CGG repeats, the resulting RNA forms aggregates (Dombrowski et al., 2002; Fu et al., 1991; Hagerman et al., 2001; Tassone et al., 2004) that are thought to cause toxicity by sequestration of nuclear proteins involved with essential cellular processes such as microRNA biogenesis (Sellier et al., 2013) and splicing (Sellier et al., 2010). For our work, Spinach2 was used to label a model transcript containing 60 CGG repeats in order to image the dynamic nature of these RNAs in living cells.
Although Spinach2 provides a simple and convenient approach to image RNA in living cells, the technology is currently limited to labeling highly abundant RNAs, and is not yet suited for imaging individual RNAs. The remainder of this chapter will provide a detailed protocol for imaging 5S-Spinach2 and CGG60-Spinach2 in mammalian cells, and will provide a general framework for tagging any RNA of interest with Spinach2.
3.1 Tagging an RNA of interest with Spinach2
Our work tagging RNAs with Spinach and Spinach2 has revealed that the presence of an RNA scaffold sequence greatly enhances imaging in cells (Strack et al., 2013). For this reason, we routinely tag RNAs with Spinach2 in the context of a modified human tRNALys scaffold (Ponchon & Dardel, 2007), in which Spinach2 is inserted into a truncated anticodon stem loop. This scaffold has been shown to enhance the folding of Spinach, and was essential for its stability in bacterial cells (Paige et al., 2011). Both 5S and CGG60 were also successfully imaged in cells when fused to tRNA-Spinach2.
The folding of both the target RNA and the Spinach2 tag may be affected by the position of insertion. For this reason, we typically generate RNAs with both 5′ and 3′ tRNA-Spinach2 tags and test them for fluorescence in vitro as well as in cells. Spinach2-tagged RNAs that retain the fluorescence of Spinach2 in vitro are then tested for performance in live cells.
3.2 Testing a tagged RNA for Spinach2 fluorescence in vitro
Design primers to amplify your tagged RNA of interest by PCR. The forward promoter should contain the T7 promoter sequence and two additional Gs (5′-TAATACGACTCACTATAGGG-3′) directly upstream of the target RNA sequence.
Carry out PCR with these primers using your tagged constructs as template. Also amplify tRNA-Spinach2 as a positive control and untagged target RNA as a negative control. Run the products on an agarose gel and gel purify the PCR products to ensure the correct band is used to template transcription.
Carry out in vitro transcription using the Ampliscribe transcription kit (Ambion) with a total transcription volume of 10 μl. Incubate the reaction at 37°C for 4–16 h. Following incubation, treat reaction with 1 μl DNase included in the kit to remove any residual template at 37°C for 1 h.
Purify RNA using ammonium acetate precipitation. Add 1 μl GlycoBlue copreciptant (Life Technologies) and 10 μl 5 M ammonium acetate to each 10 μl reaction. Incubate on ice for 15–30 min. Spin at 20,000 × g at 4°C for 15 minutes. Remove supernatant. Carefully wash pellet with ice-cold 70% ethanol. Spin at 20,000 × g at 4°C for 5 minutes. Remove supernatant carefully and do not disturb the pellets. Allow the tubes to sit open on the bench for 10–20 minutes or until the pellet is dry. Resuspend pellet in 50 μl RNase-free water. Store samples on ice when in use and at −20°C for long term stability.
Measure the RNA concentration using an absorbance spectrophotometer such as a NanoDrop. To convert the concentration from ng/μl to molarity, simply divide the value in ng/μl by the molecular weight (MW) in kDa. For example, if 300 ng/μl Spinach2 RNA is measured, this value is divided by the MW of Spinach2, which is 31.3 kDa to obtain a concentration of 9.6 μM.
Measure fluorescence of RNA-fluorophore complex. Prepare DFHBI (or DFHBI-1T) as a 40 mM stock in DMSO (10 mg/ml), and dilute to 100 μM in H2O immediately before use. The 40 mM stock can be stored indefinitely at 4°C in the dark. Prepare a 200 μl solution containing 1 μM RNA and 10 μM DFHBI (Lucerna) in a buffer with a final concentration of 40 mM K-HEPES pH 7.4, 100 mM KCl, and 1 mM MgCl2. Smaller or larger volumes may be needed based on the sample volume appropriate for the fluorimeter being used. Sample can be measured immediately after mixing in a fluorimeter. To measure fluorescence, use an excitation wavelength of 470 nm light and an emission wavelength between 500–520 nm.
Calculate the fluorescence emission intensity of the tagged samples relative to Spinach2 control.
This method of measuring the fluorescence of tagged constructs in vitro ensures that Spinach2 is folded properly in the context of the tagged RNA. Using this method, we were able to observe that the original Spinach exhibits misfolding depending on the identity of the flanking sequences, while Spinach2 generally folds robustly regardless of the identity of adjacent sequences. In our experience, tagged RNAs that exhibit less than 50% of the brightness of Spinach2 alone, on a mole per mole basis, are typically not likely to exhibit fluorescence in in vivo imaging experiments. If this occurs, changing the position of the tRNA-Spinach2 tag within the RNA may improve signal and should be attempted before further imaging is pursued. For 5S-Spinach2 and CGG60-Spinach2, the signals were at least 70% as high as untagged Spinach2.
3.3 Expressing 5S RNA in HEK 293-T cells
Obtain HEK 293-T cells (ATCC #CRL-11268) and grow to 80–90% confluence in Dulbecco’s modified Eagle medium (DMEM) + 10% fetal bovine serum (FBS) + penicillin/streptomycin.
Prior to splitting cells for imaging, treat two wells of a 24-well glass-bottom dish (Mat Tek Corp # P24G-1.5-13F) with 300 μL of poly-L-lysine (PLL) per well for 2 h at 37°C. Remove PLL and wash wells with 1 mL ddH2O followed by UV sterilization for 5 min. Plating cells directly onto coated glass coverslips allows for direct detection of live cells on an inverted widefield microscope. PLL coating followed by treatment with laminin (next step) provides a substrate that HEK 293-T cells can adhere to more readily than uncoated glass.
Add 100 μl of 10 μg/mL laminin (Trevigen Inc, Cat # 3400-010-01) to each well and incubate for 2 h at 37°C. Remove laminin and wash with 1 mL sterile ddH2O.
Remove media from 10 cm dish of HEK 293-T cells by aspiration. Replace with 10 mL prewarmed, 37°C DMEM + 10% FBS without antibiotics and create a cell suspension by repeated pipetting. For imaging, we routinely use DMEM that contains high glucose, L-glutamine, and sodium pyruvate for robust cell growth (Life technologies, Cat # 11995-065). Media may contain or lack phenol red. Prior to transfection, cells are grown without additional antibiotic, as specified in the FugeneHD manual.
Seed both wells in the 24-well coated dish with 40,000 HEK 293-T cells in 500 μL DMEM + 10% FBS without antibiotic. Allow cells to adhere and grow for 24 h. After 24 h, cells should be ~25% confluent.
Transfect cells using a 3:1 ratio of DNA to FugeneHD (Promega, Cat # E2311). In the first well, transfect cells with pAV5S-lambda (negative control). In the second well, transfect pAV5S-Spinach2. For transfections, add 25 μL water to each of two tubes. Next, add 600 μg of appropriate plasmid to each tube and mix. Next, add 1.65 μL FugeneHD to each tube and mix immediately by vigorously flicking the tube. Collect liquid at the bottom by brief centrifugation and incubate for 5–10 minutes at room temperature.
Add 25 μL of transfection mixture in droplets over the appropriate well of cells. Mix by gently pipetting up and down 5 times using a 1000 μL pipette set on 400 μL pipetting volume, being careful not to detach cells.
12–24 h post-transfection, replace media with 500 μL DMEM + 10% FBS + penicillin/streptomycin and allow cells to grow for 24 h.
Image cells 48 h post-transfection (see below for details).
This protocol should ensure robust expression of 5S-Spinach2 in HEK 293-T cells. The relatively low confluence of cells at the time of transfection is important for optimal imaging conditions, because after 48 h of growth following transfection, they reach only 50–80% confluence. At this cell density, individual cells that are spread out nicely along the glass bottom are readily imaged, and cellular death and autofluorescence are relatively low. Parallel experiments using a control plasmid expressing a fluorescent protein can be used to control for transfection efficiency.
5S-Spinach2 can, in principle, be imaged in any mammalian cell type that is readily transfected. However, we routinely image this construct in HEK293-T cells due to their robust expression levels. Highest expression will be observed in cell lines that express the T antigen, as the pAV5S vector uses the SV40 origin of replication. 5S-Spinach expression can also be further increased by induction with sucrose, as described previously, which enhances the transcription of 5S and relocalizes the 5S-Spinach2 signal to punctate stress granules (Paige et al., 2011).
3.4 Expressing CGG60-Spinach2 in COS-7 cells
-
1
Grow COS-7 cells (ATCC CRL-1651) to 80–90% confluence in Dulbecco’s modified Eagle medium (DMEM) + 10% fetal bovine serum (FBS) + penicillin/streptomycin. Ideally, the COS-7 cells used for imaging will be no more than 5–15 passages.
-
2
Resuspend COS-7 cells. Incubate COS-7 cells with 2 ml TrypLE (Life Technologies, Cat # 12604-013) at 37°C for 3–5 min. Resuspend cells in 8 mL prewarmed, 37°C DMEM + 10% FBS without antibiotics and create a cell suspension by trituration.
-
5
Plate four wells in the 24-well dish with 80,000 COS-7 cells in 500 μL DMEM + 10% FBS without antibiotic. Allow cells to adhere and grow for 24 h. After 24 h, cells should be ~40% confluent.
-
6
Transfect cells using a 3:1 ratio of DNA to FugeneHD. In well 1, transfect cells with pCGG60 (negative control). In well 2, transfect pCGG60-Spinach2. In well 3, cotransfect pCGG60 and pCDNA-m Cherry-Sam68. In well 4, cotransfect pCGG60-Spinach2 and pCDNA-m Cherry-Sam68. For transfections, add 25 μL water to each of two tubes. Next, add 600 μg of appropriate plasmid to each tube and mix. For the cotransfections, add 400 μg of the appropriate pCGG60 vector and 200 μg of pCDNA-mCherry-Sam68. Next, add 1.65 μL FugeneHD to each tube and mix immediately by vigorously flicking the tube. Collect liquid at the bottom by brief centrifugation and incubate for 5–10 minutes at room temperature.
-
7
Add 25 μL of transfection mixture in droplets over the appropriate well of cells. Mix by gently pipetting up and down 5 times using a 1000 μL pipette set on 400 μL pipetting volume, being careful not to resuspend cells.
-
8
12–24 h post-transfection, replace media with 500 μL DMEM + 10% FBS + penicillin/streptomycin and allow cells to grow for 12–24 h.
-
9
Image cells 24–48 h post-transfection (see below for details).
This protocol should allow for robust expression of CGG60-Spinach2 in ~10–40% of COS-7 cells. It is important to note that most cell types do not form aggregates after expression of CGG60 RNA(Sellier et al., 2010). CGG60 aggregates can be seen in COS-7 and differentiated PC12 cells, but not other common cell lines such as HeLa and HEK293-T cells. This indicates that CGG60 RNA aggregation is regulated by cellular pathways. Coexpression of mCherry-Sam68 serves as a positive control for CGG60 RNA expression and aggregate formation (Sellier et al., 2010; Strack et al., 2013). Typically, if the CGG60 RNA has aggregated, then the mCherry-Sam68 signal will be predominantly localized to intranuclear aggregates. However, if the CGG60 RNA is not expressed or is not aggregated, then the mCherry-Sam68 signal is diffusely nucleoplasmic, and not in foci. In these cells, CGG aggregate formation has not occurred and therefore will not be imaged.
CGG60 aggregates have been shown to grow larger over time (Sellier et al., 2010). This was also observed for CGG60-Spinach2 aggregates (Strack et al., 2013). At 24 h post transfection, cells are highly heterogeneous, yet typically contain 10–20 small-medium CGG60-Spinach2 foci per nucleus. By 72 h post-transfection, cells may contain fewer than 5 very large foci. It is important to note that CGG60 aggregates appear to be cytotoxic to COS-7 cells, which can reduce the number of CGG60-Spinach2-expressing cells after transfection. Additionally, passage of these cells is extremely difficult, with almost no positively transfected cells being observed after passaging.
4. Fluorescence imaging of Spinach2-tagged RNAs
All imaging of Spinach2 was carried out using a conventional widefield microscope (Nikon TE2000 epifluorescence microscope) with a CoolSnap HQ2 CCD camera through a 60X oil objective (Plan Apo 1.4 NA) housed entirely in an environmental chamber that regulates temperature and CO2 levels. Spinach2 fluorescence is captured through a FITC/EGFP filter set (480/40 ex and 535/50 em) and mCherry fluorescence is captured through a Texas Red filter set (560/55 ex and 645/75 em).
Spinach2 is non-fluorescent in the absence of dye. Until recently, the best dye for imaging was DFHBI (Lucerna) (Paige et al., 2011; Strack et al., 2013). However, testing of novel derivatives of DFHBI revealed that another dye, DFHBI-1T (Lucerna) (Figure 2), is optimally suited for mammalian imaging (Song et al., 2014). DFHBI-1T has three primary advantages. The first is that Spinach2-DFHBI-1T is nearly twice as bright as Spinach2-DFHBI in vitro. The second is that Spinach2-DFHBI-1T is red-shifted relative to Spinach2-DFHBI, allowing for a better spectral overlap with the GFP filter sets, which enhances the overall observed fluorescence signal. The third advantage is that DFHBI-1T itself, when incubated with cells, shows even lower background signals than DFHBI, allowing for greater sensitivity when imaging Spinach2-DFHBI-1T. When quantified in living cells, the signal for Spinach2-DFHBI-1T was greater than 1.6-fold higher than that of Spinach2-DFHBI (Song et al., 2014) (Figure 3). For these reasons, DFHBI-1T can be useful to improve the detection limits when imaging Spinach2-tagged RNAs in both bacterial and mammalian cells.
Figure 3. Spinach2-DFHBI-1T is brighter than Spinach2-DFHBI in live cells.
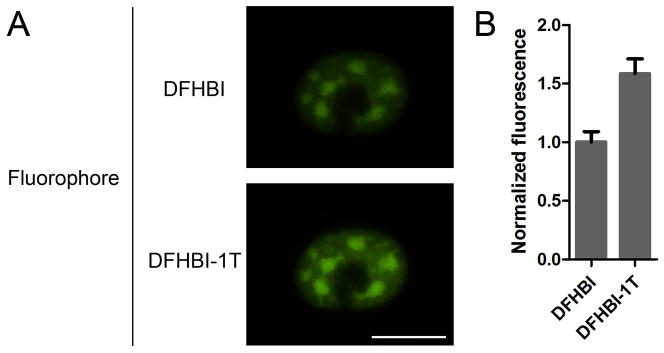
A) Shown is a COS-7 cell expressing CGG60-Spinach2. The cell was imaged using a widefield microscope with EGFP filter sets and a 100 msec exposure time. Cells were first incubated with 20 μM DFHBI and imaged. Media was then exchanged with media lacking dye for 30 min to remove DFHBI. This media was then supplemented with 20 μM DFHBI-1T for 30 min. Spinach2-DFHBI-1T images were collected following this incubation. B) Quantification of green fluorescence signal from Spinach2-DFHBI and Spinach2-DFHBI-1T in living cells. Scale bar, 20 μm. Images were used with permission from Song et al., 2014.
Spinach2-DFHBI is known to photobleach rapidly in some situations (Han et al). However, due to the nature of the RNA-dye complex, this photobleaching is reversible as the bleached dye molecule is unbound and replaced with an unbleached dye molecule. Thus, in time course imaging where images are taken at least ten sec apart, Spinach-DFHBI and Spinach2-DFHBI-1T signal is remarkably resistant to photobleaching.
4.1 Imaging 5S-Spinach2
Create a 10X stock of imaging solution containing 200 mM Na-HEPES pH 7.4, 50 mM MgSO4, and 200 μM DFHBI-1T.
Thirty minutes prior to imaging add 50 μL imaging solution directly into media of both wells and mix by gently swirling the plate. Incubate the plate at 37°C for at least 30 min to allow the dye to enter the cells.
Using your imaging software, take images of negative control cells transfected with pAV5S-lambda using 1 sec exposure times. Using linear adjustment, subtract background from these images such that any green autofluorescence or residual fluorescence from DFHBI-1T is eliminated. Save these background adjustment values.
Image cells expressing 5S-Spinach2 using 1 sec exposure times. Carry out background subtraction using the background adjustment values obtained for negative control cells in step 3. All resulting green fluorescence represents 5S-Spinach2.
This imaging protocol will produce images of 5S-Spinach2 in HEK293-T cells (Figure 4). However, it should be noted that 5S-Spinach2 signal when diffuse throughout the nucleus and cytoplasm is not easily observed by the naked eye. Indeed, optimal images are obtained using 0.5–1 sec exposure times and background subtraction. Cotransfection with a plasmid expressing mCherry can be used to determine which cells are transfected with pAV5S-Spinach2; however, the green fluorescence signal is noticeably lower in cotransfected cells. If cotransfection is used, it must also be carried out for the control plasmid to ensure proper background subtraction.
Figure 4. HEK 293T cells expressing 5S-Spinach2.
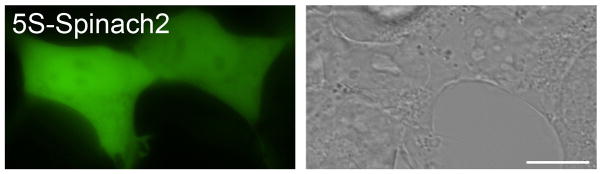
Cells were incubated with 20 μM DFHBI for 30 min prior to imaging. Shown are green fluorescence (left) and differential interference contrast (DIC, right) images. Fluorescence image was collected by widefield microscopy with EGFP filter sets with a 1 sec exposure time. Scale bar, 10 μm.
To ensure specificity of signal, cells can be imaged before and 30 minutes after addition of DFHBI-1T. In addition, after imaging with DFHBI-1T, imaging media can be replaced with media lacking dye. 30 minutes after dye removal, Spinach2 fluorescence will disappear. Finally, the 5S promoter is induced by treatment with 600 mM sucrose for 30 min and sucrose treatment causes 5S-Spinach to relocalize to stress granules (Paige et al., 2011). This relocalization to distinct foci can also be observed for 5S-Spinach2 and can provide additional confirmation that the fluorescent signal reflects Spinach2.
4.2 Imaging CGG60-Spinach2
Create a 10X stock of imaging solution containing 200 mM Na-HEPES pH 7.4, 50 mM MgSO4, and 200 μM DFHBI-1T.
Thirty minutes prior to imaging add 50 μL imaging solution directly into media of both wells and mix by gently swirling the plate. Incubate the plate at 37°C for 30 min to allow for dye to enter the cells.
Using your imaging software, take images of negative control cells transfected with pCGG60 using 100 msec exposure times. Using linear adjustment, subtract background from these images such that any green autofluorescence or residual fluorescence from DFHBI-1T is eliminated. Save these background adjustment values. Repeat for cells coexpressing CGG60 and mCherry-Sam68 to subtract any contribution of mCherry signal in the green channel.
Image cells expressing CGG60-Spinach2. Due to the brightness of the foci, imaging should be carried out using 100 msec exposure times. Carry out background subtraction using the background adjustment values obtained for negative control cells in step 3. All resulting green fluorescence represents CGG60-Spinach2.
For cells coexpressing mCherry-Sam68, image cells using the Texas Red filter set using 100–400 msec exposure times.
CGG60-Spinach2 will form large foci that colocalize with mCherry-Sam68 (Figure 5) in COS-7 cells. Unlike 5S-Spinach2, which is only faintly visible, CGG60-Spinach2 is clearly visible by the naked eye through the microscope, as ~150–200 copies of CGG60-Spinach2 are present in a typical aggregate (Strack et al., 2013). CGG60-Spinach2 aggregates are highly heterogeneous (Figure 6) and can change dramatically over time(Strack et al., 2013).
Figure 5. CGG60-Spinach2 colocalizes with mCherry-Sam68.
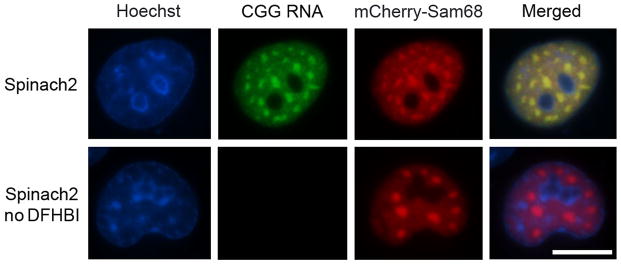
Shown are images of COS-7 nuclei containing CGG60 aggregates labeled with Spinach2-DFHBI or mCherry-Sam68. Spinach signal was collected by widefield microscopy with EGFP filter sets with a 100 msec exposure time. mCherry signal was collected using a Texas Red filter set and 200 msec exposure time. CGG60-Spinach2 and mCherry-Sam68 are shown with and without DFHBI. Scale bar, 20 μm.
Figure 6. Examples of COS-7 nuclei with CGG60-Spinach2 aggregates.
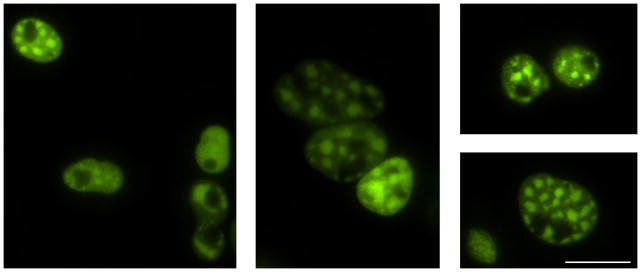
Aggregates are highly heterogeneous and range in both size and number. Some representatives are shown. Images were collected by widefield microscopy with EGFP filter sets with 50–200 msec exposure times.
5. Imaging other RNAs using Spinach2
Spinach2 can, in principle, be used to tag any RNA of interest for live-cell imaging. However, to date, the most successful imaging applications have involved tagging abundant and relatively long-lived RNAs (Strack et al., 2013). This is largely due to each RNA molecule being tagged by only a single Spinach2 molecule. Thus, with the current technology, the most likely RNAs to be tagged and imaged using Spinach2 include highly expressed non-coding RNAs, abundant viral RNAs, and other trinucleotide-repeat containing RNAs that form intranuclear aggregates.
The challenge of imaging rare or even individual RNAs using Spinach2 is currently being addressed. An obvious method for enhancing signal would be to attach “cassettes” of 2–20 Spinach2 molecules to an RNA of interest. Studies of this nature are currently underway in order to optimize the sequence, length, and number of Spinach2 tags necessary to image lower abundance RNAs in living cells.
In addition, new RNA-dye complexes with unique features, including brighter, better folding green fluorescent complexes as well as RNA-dye complexes in the yellow to far-red regions are being developed. Such studies will enhance RNA tagging in living cells and allow for imaging multiple RNAs in the same cell simultaneously.
Acknowledgments
We thank K.Y. Wu and J.S. Paige for their role in developing the original protocols for imaging Spinach in mammalian cells. This work was supported by US National Institutes of Health NINDS NS064516 (S.R.J.) and NIGMS F32 GM106683 (R.L.S.).
References
- Babendure JR, Adams SR, Tsien RY. Aptamers switch on fluorescence of triphenylmethane dyes. J Am Chem Soc. 2003;125:14716–14717. doi: 10.1021/ja037994o. [DOI] [PubMed] [Google Scholar]
- Bao G, Tsourkas A, Santangelo PJ. Engineering nanostructured probes for sensitive intracellular gene detection. Mech Chem Biosyst. 2004;1:23–36. [PubMed] [Google Scholar]
- Bratu DP, Cha BJ, Mhlanga MM, Kramer FR, Tyagi S. Visualizing the distribution and transport of mRNAs in living cells. Proc Natl Acad Sci U S A. 2003;100:13308–13313. doi: 10.1073/pnas.2233244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky AS, Silver PA. Identifying proteins that affect mRNA localization in living cells. Methods. 2002;26:151–155. doi: 10.1016/S1046-2023(02)00017-8. [DOI] [PubMed] [Google Scholar]
- Dombrowski C, Lévesque S, Morel ML, Rouillard P, Morgan K, Rousseau F. Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum Mol Genet. 2002;11:371–378. doi: 10.1093/hmg/11.4.371. [DOI] [PubMed] [Google Scholar]
- Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Warren ST. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- Fusco D, Accornero N, Lavoie B, Shenoy SM, Blanchard JM, Singer RH, Bertrand E. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr Biol. 2003;13:161–167. doi: 10.1016/s0960-9822(02)01436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- Han KY, Leslie BJ, Fei J, Zhang J, Ha T. Understanding the photophysics of the spinach-DFHBI RNA aptamer-fluorogen complex to improve live-cell RNA imaging. J Am Chem Soc. 2013;135:19033–19038. doi: 10.1021/ja411060p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holeman LA, Robinson SL, Szostak JW, Wilson C. Isolation and characterization of fluorophore-binding RNA aptamers. Fold Des. 1998;3:423–431. doi: 10.1016/S1359-0278(98)00059-5. [DOI] [PubMed] [Google Scholar]
- Molenaar C, Abdulle A, Gena A, Tanke HJ, Dirks RW. Poly(A)+ RNAs roam the cell nucleus and pass through speckle domains in transcriptionally active and inactive cells. J Cell Biol. 2004;165:191–202. doi: 10.1083/jcb.200310139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar C, Marras SA, Slats JC, Truffert JC, Lemaître M, Raap AK, Dirks RW, Tanke HJ. Linear 2′ O-Methyl RNA probes for the visualization of RNA in living cells. Nucleic Acids Res. 2001;29:E89–89. doi: 10.1093/nar/29.17.e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitin N, Santangelo PJ, Kim G, Nie S, Bao G. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nucleic Acids Res. 2004;32:e58. doi: 10.1093/nar/gnh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Natori Y, Sato M, Umezawa Y. Imaging dynamics of endogenous mitochondrial RNA in single living cells. Nat Methods. 2007;4:413–419. doi: 10.1038/nmeth1030. [DOI] [PubMed] [Google Scholar]
- Paige JS, Wu KY, Jaffrey SR. RNA mimics of green fluorescent protein. Science. 2011;333:642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, Lim H, Yoon YJ, Follenzi A, Nwokafor C, Lopez-Jones M, Meng X, Singer RH. Visualization of dynamics of single endogenous mRNA labeled in live mouse. Science. 2014;343:422–424. doi: 10.1126/science.1239200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponchon L, Dardel F. Recombinant RNA technology: the tRNA scaffold. Nat Methods. 2007;4:571–576. doi: 10.1038/nmeth1058. [DOI] [PubMed] [Google Scholar]
- Santangelo PJ, Alonas E, Jung J, Lifland AW, Zurla C. Probes for intracellular RNA imaging in live cells. Methods Enzymol. 2012;505:383–399. doi: 10.1016/B978-0-12-388448-0.00028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A, Tassone F, Willemsen R, Disney MD, Hagerman PJ, Todd PK, Charlet-Berguerand N. Sequestration of DROSHA and DGCR8 by Expanded CGG RNA Repeats Alters MicroRNA Processing in Fragile X-Associated Tremor/Ataxia Syndrome. Cell Rep. 2013;3:869–880. doi: 10.1016/j.celrep.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, Hagerman PJ, Charlet-Berguerand N. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29:1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shav-Tal Y, Darzacq X, Shenoy SM, Fusco D, Janicki SM, Spector DL, Singer RH. Dynamics of single mRNPs in nuclei of living cells. Science. 2004;304:1797–1800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Strack RL, Svensen N, Jaffrey SR. Plug-and-play fluorophores extend the spectral properties of Spinach. J Am Chem Soc. 2014;136:1198–1201. doi: 10.1021/ja410819x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack RL, Disney MD, Jaffrey SR. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat RNA. 2013 doi: 10.1038/nmeth.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- Tyagi S. Imaging intracellular RNA distribution and dynamics in living cells. Nat Methods. 2009;6:331–338. doi: 10.1038/nmeth.1321. [DOI] [PubMed] [Google Scholar]
- Tyagi S, Alsmadi O. Imaging native beta-actin mRNA in motile fibroblasts. Biophys J. 2004;87:4153–4162. doi: 10.1529/biophysj.104.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward WW, Bokman SH. Reversible denaturation of Aequorea green-fluorescent protein: physical separation and characterization of the renatured protein. Biochemistry. 1982;21:4535–4540. doi: 10.1021/bi00262a003. [DOI] [PubMed] [Google Scholar]
- Wu B, Chen J, Singer RH. Background free imaging of single mRNAs in live cells using split fluorescent proteins. Sci Rep. 2014;4:3615. doi: 10.1038/srep03615. [DOI] [PMC free article] [PubMed] [Google Scholar]