

Summary
Variant surface antigens play an important role in the pathogenesis of Plasmodium falciparum malaria. To date, intensive work has mainly focused on the role in parasite virulence of the P. falciparum Erythrocyte Membrane Protein 1 (PfEMP1) encoded by the var multigene family. Two other multigene families coding for STEVOR and RIFIN have recently also been shown to be expressed in the invasive merozoite as well as on the surface of the infected erythrocyte, implicating them as potential parasite virulence factors. Here we report that STEVOR is an erythrocyte-binding protein recognizing Glycophorin C on the red blood cell (RBC) surface. STEVOR expression on the RBC leads to PfEMP1-independent rosette formation, while antibodies targeting STEVOR in the merozoite can effectively inhibit invasion. Our results suggest a novel role of STEVOR in enabling infected erythrocytes at the schizont stage to bind uninfected erythrocytes to form rosettes, thereby protecting released merozoites from immune detection.
Introduction
The disease manifestations of Plasmodium malaria are caused by the cyclical rounds of RBC infection by the parasite. Following invasion of an RBC, the asexual infectious cycle proceeds through ring, trophozoite and schizont stages, at which point rupture of the RBC membrane releases the merozoite progeny into the blood stream for a new round of invasion. The ability of the parasite to escape host immunity and establish long lasting chronic infections is thought to include the presentation of immunogenic variant surface antigens (VSA) at the surface of the infected RBC (iRBC) (Craig and Scherf, 2001; Kyes et al., 2001). P. falciparum is responsible for the most severe form of the disease in humans. Analysis of the P. falciparum genome (Gardner et al., 2002) has identified three major families of variant genes: the var genes encoding Erythrocyte Membrane Protein 1 (PfEMP1); the repetitive interspersed family (rif) encoding the RIFIN proteins; and the subtelomeric variant open reading frame (stevor) genes encoding the STEVOR proteins. The subsequent publication of the genome sequences of other malaria parasite species has shown that all appear to contain large multigene families coding for small variant genes similar to rif and stevor, while var appears to be a unique feature of P. falciparum (Carlton et al., 2008; Carlton et al., 2002; Janssen et al., 2004; Pain et al., 2008). In P. falciparum, the expression of VSA, besides enabling the parasite to escape host immunity by means of antigenic variation (Craig and Scherf, 2001), is thought to allow deep-tissue sequestration of iRBCs containing the trophozoite and schizont stages, thereby preventing their removal via the spleen (Newbold et al., 1999; Saul, 1999; Sherman et al., 2003) while at the same time retaining them in a favorable microenvironment promoting rapid asexual multiplication (Saul, 1999). Rosetting, another binding phenomenon by which iRBCs bind uninfected RBCs has been linked to disease severity (Carlson et al., 1990a; Doumbo et al., 2009; Rowe et al., 2002). Rosetting has been hypothesized to shield iRBCs from destruction by the immune system and enhance invasion of RBCs (Cockburn et al., 2002), though the latter is challenged by data suggesting that rosetting neither targets parasites to uninfected RBCs (Clough et al., 1998) nor protects the merozoites from invasion-inhibitory antibodies (Clough et al., 1998; Deans and Rowe, 2006) reflecting the fact that it may be difficult to directly ascertain the role of rosetting using in vitro culture techniques.
PfEMP1 proteins are proposed to be one of the main targets for naturally acquired immunity to malaria as well as being the main mediator for sequestration and rosetting. The intensive works carried out on var genes (Borst et al., 1995; Craig and Scherf, 2001; Ferreira et al., 2004; Flick and Chen, 2004; Scherf et al., 2008), have significantly enhanced our understanding on the biological role of PfEMP1, while at the same time somewhat detracting from the fact that this protein family is unique to P. falciparum and that other Plasmodium spp evade host immunity, rosette and sequester to a lesser extent in its absence suggesting evolvement of PfEMP1-independent mechanisms for immune evasion, sequestration and rosetting. The common feature for all Plasmodium spp is the presence of small VSA that could play an important role in these processes. In the case of P. falciparum, the expression of RIFIN and STEVOR on the iRBC surface (Fernandez et al., 1999; Kyes et al., 1999; Niang et al., 2009) implies that they confer an important survival advantage. The expression of STEVOR in multiple parasite stages including merozoites (Blythe et al., 2008; Khattab et al., 2008; Khattab and Meri, 2011; Petter et al., 2007), sporozoites and gametocytes (McRobert et al., 2004) suggests that STEVOR mediates multiple distinct functions (Blythe et al., 2004). The observation that some synthetic peptides based on a STEVOR sequence had some RBC binding activity (Garcia et al., 2005), suggested that STEVOR could function in the invasion process by enhancing initial interaction of merozoites with RBC. A direct role in parasite-induced pathogenesis is indicated by studies showing that STEVOR expression impacts the deformability of the erythrocyte membrane of both asexual (Sanyal et al., 2012) and sexual stages (Tiburcio et al., 2012) stages iRBC, probably facilitating parasite sequestration in deep tissue vasculature.
Here we show that STEVOR specifically binds to a chymotrypsin-resistant RBC receptor through its N-terminal semi-conserved region. Anti-STEVOR antibodies can inhibit merozoite invasion of RBC indicating a role of STEVOR during the invasion process. Using a range of experimental approaches, we provide clear evidence that STEVOR mediates rosetting independently of PfEMP1 through interaction with Glycophorin C (GPC) on the RBC surface and that STEVOR-mediated rosetting provides a relative growth advantage by protecting merozoites from invasion-blocking antibodies. Taken together, these findings demonstrate that, in addition to PfEMP1, STEVOR potentially contributes to parasite-mediated pathology by enabling iRBC to bind uninfected RBCs to form rosettes, thereby protecting released merozoites from immune detection and facilitating initial interactions of merozoite with RBCs through its expression in the merozoite.
RESULTS
Anti-STEVOR antibodies inhibit in vitro merozoite invasion
To study whether STEVOR is involved in merozoite invasion, we tested the previously described anti-STEVOR sera (anti-S1 and anti-S2) raised against the semi-conserved regions of two different members of STEVOR (Blythe et al., 2008; Niang et al., 2009) to specifically inhibit merozoite invasion of RBCs. We assessed effect of the anti-S1 and anti-S2 sera on five different parasite clones that had been shown to express distinct members of STEVOR (Niang et al., 2009). The 3D7 derived clone 5A expresses STEVOR that is recognized both anti-STEVOR sera while clones 3.2C and 5.2A express STEVOR variants recognized by only anti-S1 and -S2, respectively. Clone 5B does not express STEVOR that is recognized by either anti-sera while the laboratory clone A4 appears not to express any STEVOR at all (Blythe et al., 2008; Kyes et al., 1999). Both anti-S1 and -S2 sera significantly inhibited invasion of 5A parasites (Figure 1A) in a dose dependent manner with invasion inhibition being nearly as high as that observed with MSP119 antibody directed against the Merozoite Surface protein 1 (MSP1). No inhibition was observed with the pre-immune serum. In contrast anti-S1 is more efficient (31% at 1:10 dilution compared to 10% for anti-S2) in inhibiting invasion of parasite clone 3.2C (Figure 1B) while anti-S2 is more efficient (33.28% at 1:10 dilution as compared to 8.14% for anti-S1) in inhibiting clone 5.2A (Figure 1C), while neither sera showed any inhibition of either clone 5B or A4 (Figure 1D). A similar dose dependent invasion inhibitory effect was seen when using increasing concentrations of IgG purified from rabbit anti-S1 (Figure S1A) on the 5A parasite, while no invasion inhibition was observed when using identical concentrations of Transferrin that was obtained from the same rabbit sera (Figure 1E). All these results show that the invasion-inhibitory effect of the anti-STEVOR sera is variant specific and strongly suggest that variation in the expression of STEVOR in the merozoite may be an important immune evasion mechanism.
Figure 1. In vitro inhibition of P. falciparum merozoite invasion by anti-STEVOR antibodies.
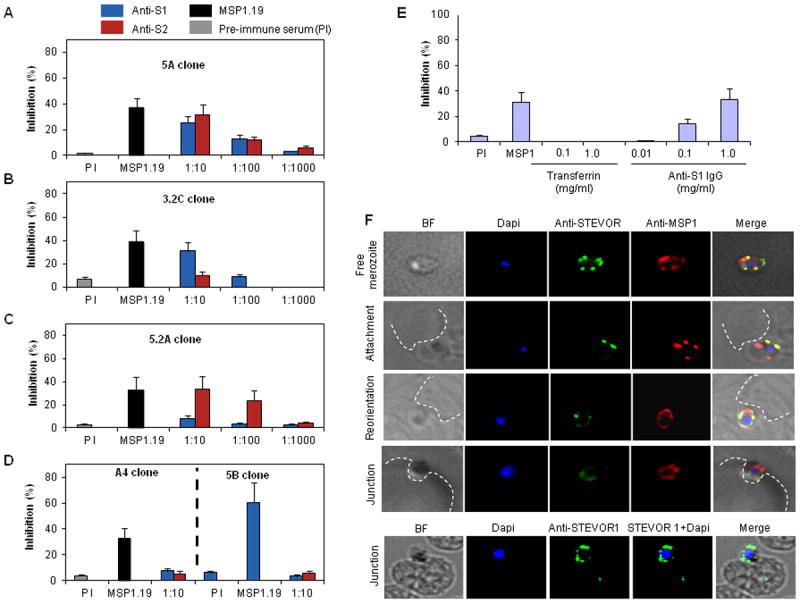
Invasion of 5A (A), 3.2C (B), 5.2A (C) 5B and A4 (D) P. falciparum parasite clones in the presence of different dilutions of anti-S1 and -S2 sera. Rabbit anti-PfMSP1.19 antibody (1:10 dilution) and pre-immune serum (PI, 1:10) were used as positive and negative controls respectively. Data are presented as the percentage of inhibition normalized to control without antiserum and represent the average of three independent experiments. Error bars denote two Standard Deviations (SD). E- Specificity- and concentration-dependent inhibition of 5A merozoite invasion into RBCs by IgG purified from rabbit anti-S1. Purified Transferrin protein that co-precipitated with the IgG fraction (see also Figure S1A) did not inhibit merozoite invasion. F- Dual life IFA staining of 5A-merozoite with anti-S2 (green) and anti-MSP1.19 (red) showing constant expression and location of STEVOR on the merozoite surface at different steps of the invasion process (free merozoite, attachment, reorientation and junction). Fixed IFA confirming STEVOR location on the merozoite surface in cytochalasin D-junction arrested 5A merozoites (junction). The fluorescent images (individual stains and merged images) and the bright field (BF) are shown. The dotted white lines delineate the uninfected RBC membrane.
Investigation of the effect of anti-STEVOR antibodies on merozoite invasion using live video microscopy shows that it inhibits invasion at an early step (movie S1). Time-lapse image sequences of invading merozoites show that many merozoites contacted but failed to invade RBCs and these merozoites detached after a few seconds suggesting that the antibodies interfere with the initial binding of merozoites to RBCs (movie S1 and Figure S1D). In contrast, no inhibition was observed in the absence of STEVOR sera (movie S2).
Live IFA on 5A merozoites using anti-S2 and anti-MSP1 antibodies showed clear localization of STEVOR along with MSP1 on the surface of live merozoite (Figure 1F, free merozoite) consistent with antibody recognition of STEVOR on the merozoite surface interfering with invasion. Furthermore, STEVOR consistently co-localized with MSP1 at the surface of the merozoite at different steps of the invasion process and no evidence was obtained that STEVOR is involved in the tight junction during invasion as illustrated by STEVOR expression on the merozoite surface by live and fixed IFA of cytochalasin-D-junction-arrested merozoites (Figure 1F).
STEVOR binds to human red blood cells through its N-terminal semi-conserved region
Based on the observation that STEVOR is exposed on the iRBC surface and the results reported here of its role in merozoite invasion, we investigate whether STEVOR binds to RBC. We expressed the semi-conserved (SC), hypervariable (HVR) and full length (FL) regions of three different stevor genes (Figure 2A) on the surface of COS-7 cells and tested their ability to bind to RBC. A GFP construct expressing the P. vivax Duffy binding protein region II (PEGFP-PvDBPII) (Chitnis and Miller, 1994; Gao et al., 2008), was used as a positive control in these assays. The SC regions (ST1C, ST2C, ST3C) of the three stevor genes bind human RBCs efficiently compared to very limited binding of the HVRs (ST1H, ST2H, ST3H) (Figure 2B). Binding activity of ST3C was the highest and comparable to binding of the PvDBPII control (Figure 2B and Figure S2). Expression of full-length stevor (ST3Full) did not lead to an increase of RBC binding as compared to the SC region alone (Figure 2B) demonstrating that the RBC binding domain is present in the SC region. No binding was observed with both controls pDisplay vector alone (pDispl) and non-transfected (NT) COS-7 (Figure 2B and Figure S2A) demonstrating that the observed RBC binding was dependent on STEVOR expression.
Figure 2. Erythrocyte binding of COS7 cells expressing different STEVOR regions.
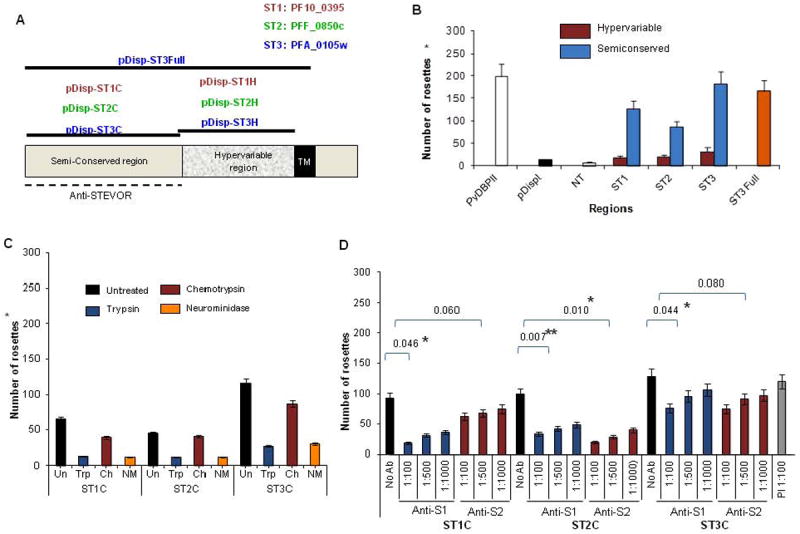
A- Chimeric constructs for the expression of different STEVOR regions on the surface of COS-7 cells (see details in supplemental methods). The generation of ST1Full and ST2Full was unsuccessful and therefore removed from the figure. The 1TM model of STEVOR is depicted here based on recent findings (Joannin et al., 2011; Niang et al., 2009). Dashed line delineates the regions served for generation of anti-S1 and -S2. B- Erythrocyte binding of COS-7 cells expressing the SC and HVR regions of the three stevor genes. The P. vivax Duffy Binding Protein II (PvDBPII) and the empty pDisplay vector (pDispl) and/or non-transfected COS-7 (NT) were used as controls. Binding of the ST3Full is also shown.C- Binding of the three SC regions to untreated and enzyme-treated erythrocytes showing significant reduction of binding following trypsin and neuraminidase treatments.D- Pattern of inhibition of the binding of the three SC regions by anti-STEVOR antibodies. Preimmune serum (PI) at 1:100 dilution was used as control. Error bars represent standard deviations (SD) of 3 independent experiments. Effects of anti-S1 and anti-S2 were statistically compared to control (no antibody), significant differences are shown by asterisk (* indicates p<0.05, ** indicates P<0.01).
The specificity of the SC binding was assessed using chymotrypsin-, trypsin- or neuraminidase-pretreated RBCs. These enzymes cleave specific receptors from the RBC and have been a useful tool for studying the interaction of malaria proteins with RBCs (Duraisingh et al., 2003; Perkins and Holt, 1988). The efficiency of the enzyme treatment was assessed by flow cytometry quantification using monoclonal antibodies against Glycophorin C (GPC) and Complement Receptor 1 (CR1) RBC surface proteins whose sensitivities to these enzymes are well established (Dolan et al., 1994; Maier et al., 2003; Sim, 1985). The assay confirmed the GPC sensitivity to trypsin and neuraminidase treatments while being chymotrypsin-resistant (Figure S2B) (Maier et al., 2003) while CR1 is neuraminidase-resistant and trypsin- and chymotrypsin-sensitive (Figure S2B) (Sim, 1985; Spadafora et al., 2010).
Chymotrypsin treatment had limited or no impact on RBC binding of the three different SC regions analyzed whereas trypsin and neuraminidase treatment significantly reduced the binding (Figure 2C) suggesting that STEVOR binds to a chymotrypsin-resistant receptor on the RBC surface. Reduction of binding to RBCs treated with neuraminidase suggests a surface glycoprotein receptor similar to glycophorin A or C.
Anti-STEVOR antibodies specifically inhibit erythrocyte binding
The anti-S1 and -S2 sera are able to inhibit merozoite invasion possibly by directly interfering with the binding of STEVOR to the RBC. We tested the ability of the two sera to block the binding of the SC regions of the different STEVORs. Anti-S1 and anti-S2 sera were raised against the semi-conserved regions of PF10_0395 and PFF_0850c genes respectively which served for the generation of ST1 and ST2 constructs.
Anti-S1 antibody efficiently inhibited RBC binding of ST1C, while the inhibitory effect of anti-S2 binding was low (Figure 2D). The reverse was seen with the ST2C construct that was more inhibited by anti-S2 antibody as compared to anti-S1 (Figure 2D). In both cases specific inhibition of the binding by the anti-sera’s was dose dependent (Figure 2D). While a low level of cross-reactivity was observed with anti-S1 and -S2, respectively, on ST2C and ST1C RBC binding, both had a reduced ability to block the binding of the ST3C region (Figure 2D) consistent with variant specific recognition by the antibodies. Pre-immune serum did not inhibit binding (Figure 2D) and anti-STEVOR sera was unable to inhibit RBC binding of PvDBPII (data not shown), showing that the inhibition of RBC binding was STEVOR specific.
STEVOR plays a role in the rosetting of RBC infected with P. falciparum
Our finding that STEVOR can bind to a RBC receptor led us to investigate whether STEVOR could mediate rosetting. We enriched cultures for rosetting-positive (R+) iRBCs (Figure 3A, i) from 5A, 5.2A and 3.2C STEVOR-expressing parasite clones. Various sizes of rosettes were observed, with rosettes of up to 8 uninfected RBCs (Figure 3A, ii-v). Western blot analysis of schizont extracts of 5A-R+ and 5A-R- parasites using anti-S2 and anti-GPC (loading control) antibodies showed significantly higher STEVOR expression with 5A-R+ (Figure 3B and Figure S3A). A similar increase in STEVOR expression was observed with 3.2C-R+ and 5.2A-R+ parasites (Figure 3C) suggesting that STEVOR may be important for rosetting of P. falciparum-iRBCs.
Figure 3. Rosetting enrichment and STEVOR expression of 5A, 3.2C and 5.2A (R+) and (R-) parasites.
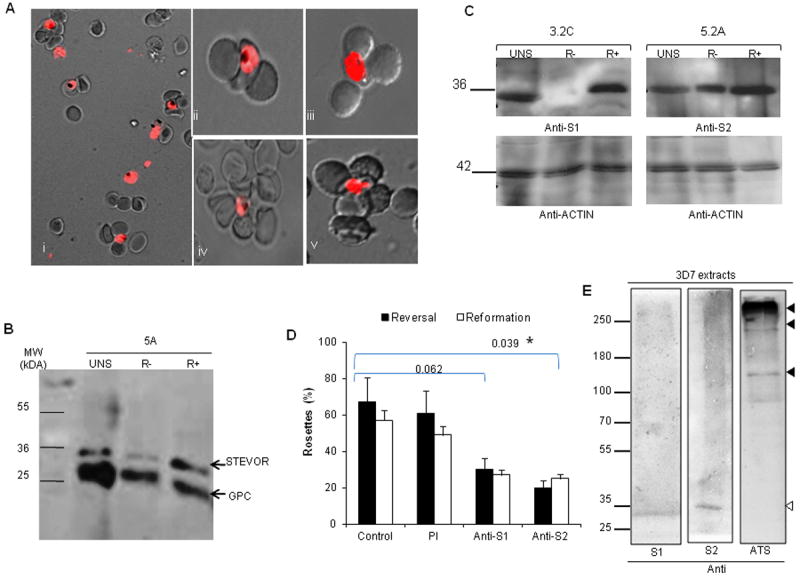
A- Live wet-preparation images of 5A rosetting parasites in whole field (i) and at single iRBC level (ii-v) viewed under fluorescent and direct light microscope at 100X magnification. Infected RBCs were stained with ethidium bromide. B-C Western blot of schizont extracts from unselected (UNS), R+ and R- 5A (B), 3.2C and 5.2A (C) clones with rabbit anti-S1 or -S2 showing higher STEVOR expression in R+ parasites. Mouse anti-GPC and rabbit anti-actin were used to show equal loading. D- Disruption (reversal) and blockage (reformation) of 5A rosetting by anti-S1 and -S2. Effects of anti-S1 and anti-S2 were statistically compared to control. Asterisk shows significant difference (p<0.05). E- Western blot of 3D7 schizont extract performed with a low percentage gel showing the specific recognition of the lower molecular STEVOR by anti-STEVOR sera (white arrow) and the absence of cross-reactivity with PfEMP1 (black arrows). PfEMP1 was only detected by anti-ATS antibody (black arrows). Data are expressed as percentage of rosettes in 200 counts. Error bars represent SD of two independent experiments.
Both anti-S1 and -S2 sera significantly disrupted and prevented rosetting of 5A-R+ parasites in rosette disruption (reversal) and reformation assays as compared to the untreated and pre-immune (PI) serum-treated controls (Figure 3D). While the pre-incubation of purified 5A-R+-iRBCs with anti-S1 or -S2 sera significantly blocked rosette reformation with only 27% and 25% parasite forming rosettes respectively as compared to 57% and 49% seen in the untreated and PI-treated controls respectively (Figure 3D), pre-incubation of uninfected erythrocytes with anti-STEVOR sera had no impact on rosetting (data not shown).
Western blot of 3D7 schizont extract probed with anti-STEVOR antibodies and antibody against the acidic terminal segment (ATS) of PfEMP1 showed specific recognition of STEVOR by both anti-STEVOR sera (Figure 3E) with no cross-reactivity with PfEMP1 (Figure 3E). The anti-ATS was generated and validated by IFA showing that it recognizes PfEMP1 on the iRBC surface of wild type 3D7 iRBCs (Figure S3C). In contrast, anti-ATS antibody specifically recognized the high molecular weight PfEMP1 bands (Figure 3E, black arrows) with no cross-reactivity with the lower molecular weight STEVOR protein (Figure 3E, anti-ATS). This strongly rules out that cross reactivity to PfEMP1 is responsible for the rosettes disruption seen here.
While these results suggest that STEVOR may play an important role in rosette formation it was important to establish whether rosette formation also occur in parasites that do not express STEVOR. We therefore tested for rosetting the A4 parasites clone that was previously shown to neither transcribe, nor express STEVOR (Blythe et al., 2008; Kyes et al., 1999). The A4 clone had been previously shown not to rosette and furthermore the predominant PfEMP1 variant expressed, encoded by the A4var gene, does not bind RBC although others have been able to select the A4 clone for rosetting through extensive rounds of selection (Albrecht et al., 2011). Our attempts using the same selection protocol as used for all the other parasite lines in this study failed to enrich A4 for rosetting (data not shown).
STEVOR is a key requirement for rosetting of A4 parasites
In order to functionally investigate the requirement of STEVOR for rosetting of A4, we stably transfected the A4 parasites with a full length STEVOR-GFP fusion protein using a modified version of the previously described pARL-STEVOR80 vector (Przyborski et al., 2005) resulting in the A4-pARL-STEVOR1-GFP parasite line (Figure 4A). Correct expression of the ~63 kDa STEVOR-GFP fusion protein was confirmed by western blot of schizont extract using anti-GFP antibody (Figure 4B, top panel). The same membrane was re-probed with anti-Glycophorin A antibody to show equal loading (Figure 4B, bottom panel). Life IFA using anti-S1 antibody showed that STEVOR is expressed on the surface of A4-pARL-STEVOR1-iRBC. Most STEVOR remains within the iRBC as shown by the GFP expression (Figure 4C), in line with previous work showing that most of the endogenous STEVOR remains within the Maurer’s Clefts (Przyborski et al., 2005). The A4-pARL-STEVOR1 parasite line was successfully enriched for rosetting (Figure 4D) supporting an important role of STEVOR in rosetting of this parasite. While we observed reduced rosetting frequency in this transfected line compared to the 5A-R+ parasites, there was no difference in the size of rosettes formed (Figure 4D). Critically, anti-S1 raised against the transfected STEVOR1 significantly disrupted rosetting of the A4-pARL-STEVOR1-R+ parasites compared to the low inhibitory effect of anti-S2 (Figure 4E). These results convincingly demonstrate that STEVOR can be a key requirement for rosetting of iRBC.
Figure 4. Functional investigation of STEVOR requirement for rosetting of A4 parasites.
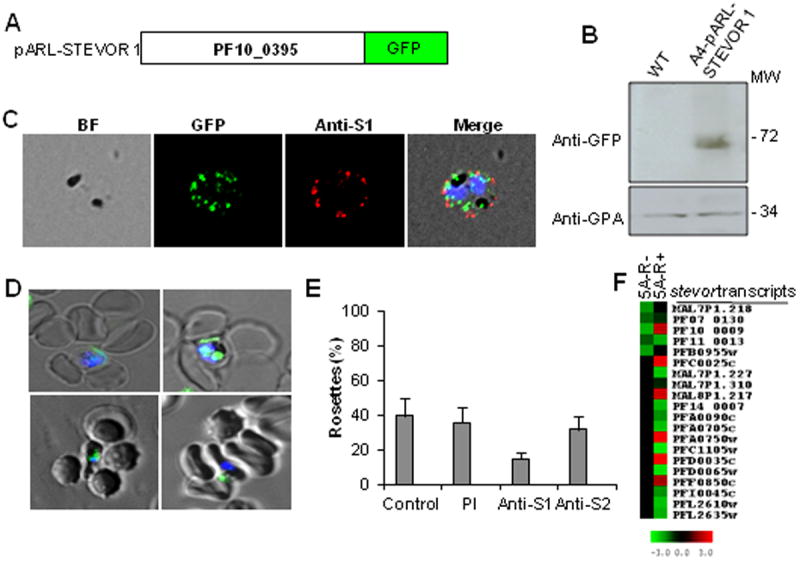
A- Generation of pARL-STEVOR1-GFP construct encoding a full length stevor gene (PF10_0395) driven by the cg4 promoter obtained from modification of the pARL-STEVOR80 vector. B- Western blot validation of the correct GFP fusion protein expression with anti-GFP antibody and anti-GPA antibody (loading control). C- Live IFA showing correct surface trafficking of the GFP fusion protein using anti-S1 antibody. From left to right are shown bright field (BF), nuclei staining (DAPI), GFP, anti-S1 staining and merged images. D- Live wet-preparation images of A4-pARL-STEVOR1-R+ iRBCs viewed under fluorescent and direct light microscope at 100X magnification. Parasite nuclei were stained with DAPI (2 μg/ml); GFP expression is shown as green dots within the iRBCs. E- Disruption of A4-pARL-STEVOR1 rosetting by anti-STEVOR sera showing 62.5% inhibition by anti-S1 compared to 20% for anti-S2. Data represent average of two independent experiments. Error bars denote two standard deviation.F- Microarray analysis of stevor transcript levels of 5A-R+ and 5A-R-. Red and green indicate upregulated and downregulated genes, respectively, in relation to a pool of RNA.
While we show here that STEVOR expression is essential for rosette formation in the A4 clone, the data does not exclude a role for PfEMP1 as well. We therefore analyzed the transcription of stevor, var and rif families in 5A-R+ and 5A-R- parasites by microarray using highly synchronized trophozoite stage cultures (22-28h post invasion). The analysis revealed downregulation of all stevor transcripts in the 5A-R- parasites while several upregulated stevor transcripts were detected in the 5A-R+ (Figure 4F). A single dominant var (PFL2665c) and no upregulated rif were seen in the 5A-R+ parasites (Figure S4A). While PFL2665c has not previously been implicated in rosetting it raises the possibility that both PfEMP1 and STEVOR are important for rosetting in 5A-iRBCs.
STEVOR can mediate rosetting of P. falciparum-iRBC independently of PfEMP1
To address whether rosetting can occur independently of PfEMP1 we carried out the rosette formation assay using 3D7ΔMAHRP1-iRBCs. In this parasite line, the disruption of the MAHRP1 protein has been shown to compromise trafficking of PfEMP1 to the iRBC surface (Spycher et al., 2008) suggesting that any adhesion or binding phenotype of this parasite line can be attributed to parasite ligands other than PfEMP1. We first showed that 3D7ΔMAHRP1 did not have impaired trafficking of STEVOR to the iRBC surface as both anti-S1 and -S2 were able to react with STEVOR antigens on the surface of live iRBCs (Figure 5A, top panel).
Figure 5. Analysis of STEVOR expression in the 3D7ΔMAHRP1 parasites.
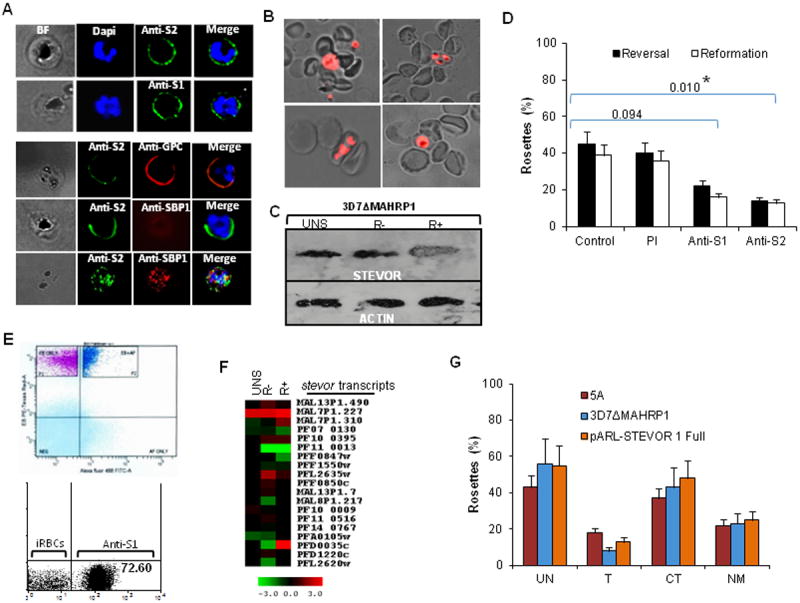
A- Detection of STEVOR on live 3D7ΔMAHRP1-R+ -iRBCs surface by anti-S1 and -S2 (top panel). Dual staining of live 3D7ΔMAHRP1-R+ -iRBCs with anti-S2 and anti-GPC antibodies (bottom panel, top row) or anti-SBP1 (bottom panel, middle row) illustrating the external location of both STEVOR and GPC and lack of surface staining of 3D7ΔMAHRP1-R+ iRBCs with anti-SBP1, respectively. Confirmation of SBP1 internal location is revealed by IFA on fixed parasites (bottom panel, bottom row). B- Live wet preparation images showing successful rosetting enrichment of 3D7ΔMAHRP1 line. C- Immunoblotting with rabbit anti-S2 showing increased STEVOR expression in 3D7ΔMAHRP1-R+. Rabbit anti-actin was used to show equal loading. D- Statistically compared inhibition of 3D7ΔMAHRP1 rosetting by anti-S1 and -S2 on rosetting reversal and reformation assays. Significant difference (P<0.05) is shown by an asterisk. E- Isolation and establishment of monovariant 3D7ΔMAHRP1 cultures expressing abundant STEVOR at the cell surface (dark blue subpopulation) by sorting and re-culturing the anti-S1 and -S2 (See also Figure S3E) surface positive iRBCs subpopulations (top panel) and re-evaluation for surface-positive STEVOR (see also Figure S3E) by flow cytometry after 5 weeks of weekly rosetting enrichment. F- Microarray analysis comparing stevor mRNA level in unselected (UNS)-, R- and R+ 3D7ΔMAHRP1-iRBCs; red and green indicate upregulated and downregulated genes respectively in relation to a pool of RNA. G- Rosette reformation of purified 5A-, 3D7ΔMAHRP1- and pARL-STEVOR1-R+ parasites with normal, trypsin-, chymotrypsin- and neuraminidase-treated RBCs revealing the chymotrypsin-resistant phenotype of all tested lines.
Dual live IFA of 3D7ΔMAHRP1-R+ iRBCs using anti-S2 antibody and antibody against GPC showed co-localization of STEVOR and GPC on the surface of parasites (Figure 5A, bottom panel). Only surface-expressed STEVOR was detected by live IFA of iRBCs and staining with antibody against the Maurer’s clefts protein SBP1 was negative, consistent with the internal location of this protein (Figure 5A, bottom panel). Expression of SBP1 was confirmed by dual IFA on fixed parasites showing double labeling of STEVOR and SBP1 (Figure 5A, bottom panel).
Enrichment of 3D7ΔMAHRP1 for rosetting shows that the parasite is able to form rosettes with similar efficiency as the 3D7 clones as there was no difference in the size of rosettes between 5A-R+ and 3D7ΔMAHRP1-R+ parasites though slightly more rosetting iRBCs were observed with the 5A-R+ parasites (Figure S3A and S3B). Similar to the 5A, 3.2C and 5.2A parasites, STEVOR expression was increased in 3D7ΔMAHRP1-R+ (Figure 5C). As observed with 5A-R+, anti-STEVOR sera were able to disrupt rosetting of 3D7ΔMAHRP1-R+ and block rosette reformation (Figure 5D).
To establish whether enrichment of 3D7ΔMAHRP1 for rosetting led to any surface expression of PfEMP1, anti-ATS antibody was used to detect a trypsin-cleaved ATS fragment in both wild type 3D7, unselected and 3D7ΔMAHRP1-R+ parasites. While the anti-ATS was able to detect a cleaved fragment in the WT 3D7 following trypsin treatment (Figure S3D, white arrow), no band was detected in 3D7ΔMAHRP1 parasites before and after rosetting enrichment (Figure S3C) consistent with the absence of PfEMP1 surface expression in 3D7ΔMAHRP1 line.
The contribution of STEVOR in 3D7ΔMAHRP1 rosetting was further estimated by establishing cultures expressing abundant STEVOR at the cell surface (Figure 5E, top left panel and Figure S3E) by sorting and re-culturing the anti-S1 and -S2 surface-positive iRBC sub-populations. Quantification of the level of surface-expressed STEVOR after 5 weeks of weekly rosetting enrichment showed a significant increase in the number of anti-S1 and -S2 surface-positive iRBC (72.60% and 83.79% respectively) along with 54% and 61% rosetting frequency respectively (Figure 5E, bottom left and right panels and Figure S3E). Microarray analysis of stevor, var and rif transcription in unselected, R- and R+ 3D7ΔMAHRP1 showed no enrichment of var and rif transcripts in unselected, R- and R+ parasites (Figure S4B) while several stevor transcripts were upregulated following rosetting enrichment (Figure 5F). In particular, PFD0035c showed increased mRNA levels in both 5A-R+ and 3D7ΔMAHRP1-R+ (Figure 4F and 5F). Taken together, this data convincingly demonstrates an important role of STEVOR in rosette formation of 3D7ΔMAHRP1 parasites in the absence of PfEMP1 surface expression.
Additional evidence supporting STEVOR-mediated rosetting came from the investigation of the ability of 5A, A4-pARLSTEVOR1 and 3D7ΔMAHRP1 parasites to form rosettes with trypsin-, chymotrypsin- and neuraminidase-treated RBCs. This assay revealed that rosettes formed by all three lines are resistant to chymotrypsin treatment, while being sensitive to trypsin and neuraminidase (Figure 5G). This shows a perfect match between the enzyme sensitivity of the rosetting lines with that of RBC binding by STEVOR recombinant proteins (Figure 2D). This finding also rules out PfEMP1, since PfEMP1-mediated rosettes are unaffected by enzymatic treatment (Rowe et al., 1994; Udomsangpetch et al., 1989).
STEVOR binding to RBC correlates with the level of GPC on the RBC surface
To gain more insight into the nature of the specific receptors(s) mediating STEVOR binding to RBC, we first tested the ability of monoclonal antibodies against Glycophorin A (GPA), GPC, CD36 and CR1 to block rosette reformation of 5A parasites. Pre-incubation of RBC with 50 μg/ml of GPA, GPC, CD36 and CR1 monoclonal antibodies prior to rosette formation with purified 5A-R+ -iRBCs or binding of COS7 cells expressing ST3C showed no effect of CD36 and GPA (Figure 6A) whereas some inhibition was seen for CR1 and a stronger inhibition for GPC (Figure 6A and data not shown). This suggested that GPC is a potential receptor for STEVOR.
Figure 6. STEVOR binding to RBC correlates with the level of GPC on the RBC surface.
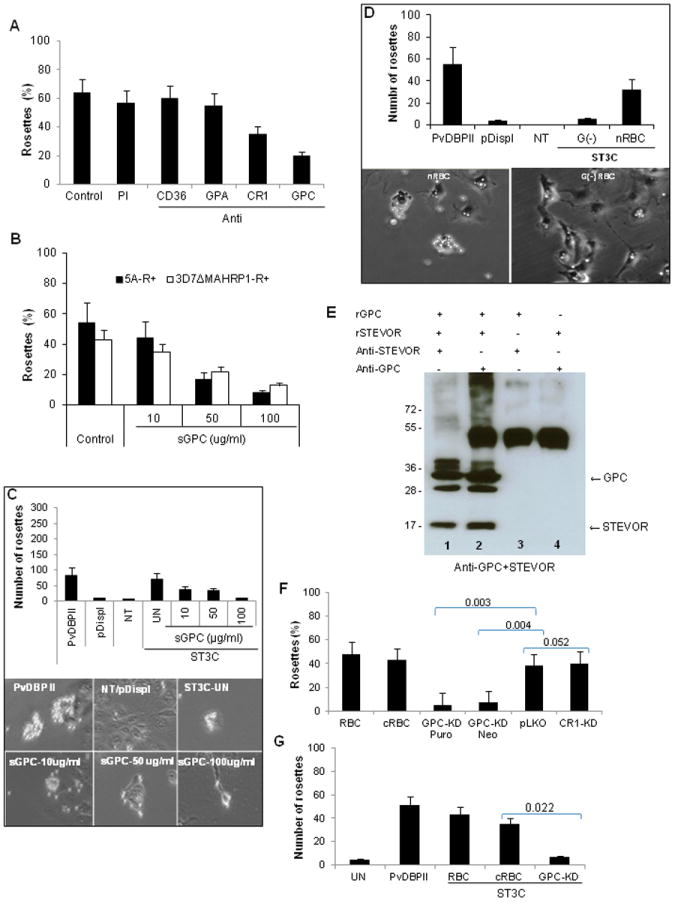
A- Effects of RBC treatment with 50 μg/ml of monoclonal antibodies against CD36, GPA, CR1 and GPC on rosette reformation of purified 5A-R+ iRBCs. Untreated RBCs (control) or RBCs treated with preimmune serum (PI) were used as controls. Data represent the average of two independent experiments. B and C- Concentration-dependent inhibition of rosetting of purified 5A-R+ and 3D7ΔMAHRP1-R+ iRBCs by soluble GPC (B) and binding of COS7 expressing ST3C construct (C). Soluble GPC reduces both the number of ST3C-expressing COS7 cells binding to RBC (C, top panel graph) as well the size of rosettes formed (C, bottom panel images) in a concentration-dependent manner compared to control (ST3C-UN). PvDBPII and untransfected (NT) and/or pDisplay vector-transfected (pDispl) were used as positive and negative controls for binding, respectively.D- Impairment (top panel graph) of binding of COS7 expressing ST3C to Gerbich negative (G(-)) RBC compared to normal RBC (nRBC) as illustrated by micrograph images (bottom panel images). PvDBPII and untransfected (NT) and/or pDisplay vector-transfected (pDispl) were used as positive and negative control for binding respectively. Data are presented as average of two independent experiments, each performed in duplicate. E- Pull down assay with recombinant STEVOR (rSTEVOR) and Glycophorin C (rGPC) in the presence (+) or absence (-) of anti-STEVOR or anti-GPC antibodies showing specific interactions of STEVOR and GPC (lanes 1 and 2). F and G- Impact of GPC knock down (KD) on rosetting of 5A-iRBCs (F) and ST3C binding (G). Rosetting was greatly reduced when performed with GPC knock down (KD) RBCs following puromycin (Puro, 97% KD compared to pLKO control, see also Figure S5) and Neomycin (Neo, 89% KD compared to pLKO control, Figure S5) selection compared to pLKO.In vitro generated-RBC (cRBC) and donor RBC (RBC) were used as controls. CR1 KD (27% KD compared to pLKO control, see also Figure S5) has no impact on rosetting. For ST3C binding (G), only Puromycin- selected GPC-knock down RBCs were used due to the limited number of available cells. Statistical differences of GPC and CR1-KD in comparison to pLKO control are shown.
Data represent the average of three independent experiments. Error bars denote standard deviation.
To further validate this finding, we tested the ability of soluble GPC (sGPC) to prevent rosetting and binding of STEVOR to RBC. Soluble GPC competitively blocked both rosette formation of 5A- and 3D7ΔMAHRP1-R+ parasites (Figure 6B) as well as RBC binding of COS7 expressing ST3C (Figure 6C) in a concentration-dependent manner. Increasing concentration of sGPC reduced the number of rosettes formed by COS7 cells expressing ST3C and the size of rosettes (Figure 6C). Additionally, the binding of COS7 cells expressing ST3C to normal and Gerbich negative (G(-)) RBCs was compared and only weak binding of ST3C to G(-) RBC was observed compared to normal RBCs (nRBC) (Figure 6D). These data along with the findings that GPC antibody (unlike other antibodies) and soluble GPC can compete with the binding of ST3C support that GPC is a receptor for STEVOR.
Direct support for a STEVOR-GPC interaction was obtained using immunoprecipitation. This approach showed that both GPC and STEVOR can pull down each other in the presence of both recombinant proteins and either anti-STEVOR (Figure 6E, column 1) or anti-GPC (Figure 6E, column 2), whereas no reactivity was seen in the absence of either recombinant STEVOR (Figure 6E, column 3) or recombinant GPC (Figure 6E, column 4).
We then used of the recently described in vitro RBC culture system combined with lentiviral transduction (Bei et al., 2010a) to knock down GPC. Stable targeted knockdown of gene expression was confirmed by flow cytometry measuring GPC RBC surface expression on day 18 of ex-vivo culture as described (Bei et al. 2010a). The mean level of GPC knock down (GPC-KD) was 97% and 89% respectively following puromycin and neomycin selection compared to control cultured RBC (cRBC) (Figure S5).
The binding frequency of the GPC-KD cells was compared to that of RBC transduced with scrambled pLKO and cRBC controls in both rosette reformation assay with purified 5A-R+ parasites and RBC binding assay to COS7 expressing ST3C. A significant reduction of rosette frequency was observed with the GPC-KD RBCs compared to cRBC and pLKO controls (Figure 6F) as well as in a COS7 experiment with ST3C (Figure 6G). No impact of GPA-KD (27% KD compared to pLKO) was observed on ST3C binding (Figure S5). These data convincingly demonstrate that STEVOR binding to RBC is mediated through interaction with GPC.
Invasion efficiency of R+ and R- parasites
Growth of R+ and R- parasites of the 5A clone under static and suspension conditions at 1% starting parasitemia showed that R+ parasites have a significantly increased parasite multiplication as compared to R- parasites (p<0.001) in both culture conditions (Figure 7A). Moreover, the enhanced parasite multiplication rates of R+ parasites are greater in suspension culture as compared to static cultures (p<0.001) (Figure S6), which is consistent with rosetting providing a particular growth advantage under flow conditions observed in vivo. To follow this in more detail, R+ and R- parasites of 5A, 3.2C and 5.2A were cultured under suspension conditions for three rounds of replication. Under these conditions, parasitemia increased steadily from an initial starting level of 0.1% to a level of around 12.5% and 8.7% in 5A- and 5.2A-R+, respectively, compared to 8.8% and 7.03% in R- parasites for the same clones (Fig 7A and 7B, D5). An enhanced parasite multiplication rate was also observed for the 3.2C-R+over the 3.2C-R- parasites (Figure 7C, D5), though the overall growth of this clone was much slower compared to 5A and 5.2A.
Figure 7. Targeting of invasion by R+ and R- of 5A parasites.
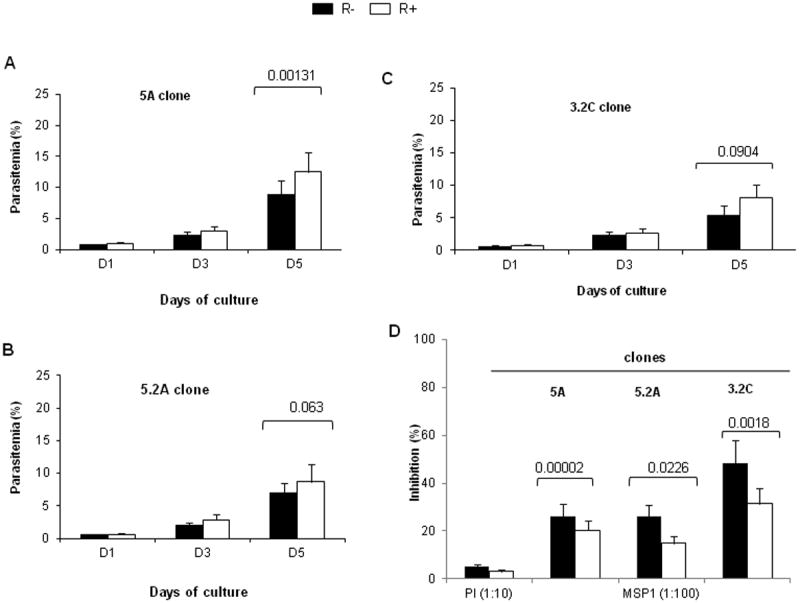
Comparison of invasion efficiency of R+ and R- parasites of 5A (A), 5.2A (B) and 3.2C (C) clones grown in suspension following three consecutive rounds of replication at a 0.1-0.2% starting parasitemia showing growth advantage of R+ over R- parasites. D- Invasion of 5A-, 5.2A- and 3.2C-R+ and -R- parasites in the presence of a high concentration (1:100 dilution) of MSP1.19 inhibitory antibody and non inhibitory pre-immune serum (PI, 1:10 dilution). Data is presented as percentage of inhibition compared to control without serum and represent the average of two independent experiments performed in triplicate each. Statistical comparisons are shown.
We assessed whether STEVOR mediated rosetting was able to protect parasites from invasion-inhibitory antibodies. For this purpose R+ and R- parasites of 5A, 5.2A and 3.2C clones were grown in the presence of MSP1.19 invasion-inhibitory antibody. The data clearly shows that all R+ parasites are able to invade significantly better than R- parasites in the presence of a high concentration of anti-MSP1.19 antibody, suggesting protection against the invasion-inhibitory MSP1.19 antibody for the R+ selected population as compared to the R- population.
Taken together, these results strongly support that STEVOR-mediated rosetting provides the parasite with some growth advantage as it is able to protect merozoites from invasion-inhibitory antibodies.
DISCUSSION
Efficient adhesion of the invasive merozoite to the RBC, or the binding of iRBC to endothelial cells or uninfected RBC, are important factors contributing to P. falciparum-induced pathology. To date, numerous merozoite-specific adhesion molecules have been identified, while PfEMP1 has been proposed as the main mediator for rosetting and sequestration of the iRBC. In this study, we have shown that STEVOR is a RBC-binding protein that interacts with GPC and plays an important role in both merozoite invasion as well as rosette formation, making this protein family a key contributor to parasite-mediated pathology.
Our findings that STEVOR can mediate rosette formation on the iRBC surface independently of PfEMP1 as evidenced by an enzyme-sensitivity phenotype challenge the concept that the adhesion properties of P. falciparum iRBCs are exclusively mediated by PfEMP1. Rather, they support the possibility that others VSAs are important in ultimately defining iRBC adhesion. In P. falciparum, rosetting has been extensively linked to interactions of the major parasite ligand PfEMP1 with receptors on uninfected RBC (Carlson et al., 1990b; Handunnetti et al., 1992; Mercereau-Puijalon et al., 2008; Rowe et al., 1997). However, rosetting is observed in all human malaria species and has also been shown to occur in simian and rodent malaria parasites (Angus et al., 1996; Chotivanich et al., 1998; David et al., 1988; Mackinnon et al., 2002; Udomsangpetch et al., 1991). Moreover, within species, rosetting levels vary between parasite clones (Carlson et al., 1990a; Chotivanich et al., 1998; Rowe et al., 1995) consistent with this phenomenon being mediated by variant adhesins. The wide prevalence of rosetting suggests that it is a feature of both P. falciparum and non-falciparum parasites and therefore cannot be exclusively mediated by PfEMP1. Importantly, in P. falciparum rosetting had been initially associated with a group of low molecular mass proteins called rosettins (Helmby et al., 1993), with subsequent studies suggesting a relationship between rosettins and RIFINs (Fernandez et al., 1999; Kyes et al., 1999). These initial studies have been somewhat overshadowed by the finding that PfEMP1 can also mediate rosetting by interacting with CR1 (Rowe et al., 1997). The perceived notion of PfEMP1 being the sole contributor to sequestration as well as rosetting is inconsistent with current data showing that each iRBC expresses only a single member of PfEMP1, which again is linked to a specific adhesion phenotype (Baruch et al., 1997; Chen et al., 2000; Howell et al., 2008; Vogt et al., 2003). This exclusivity of adhesion phenotypes would suggest that each iRBC can only bind a single receptor, making it unlikely that sequestration and rosetting can be carried out by a single iRBC. However, co-expression of rosetting and cytoadherence receptors on the same iRBC demonstrates that an individual iRBC can express two surface ligands at the same time (Hasler et al., 1990). This observation was further supported by micrograph images indicating that sequestered iRBCs also bind to uninfected RBC (Kaul et al., 1991). The data presented here, suggests that STEVOR and PfEMP1 contribute to different iRBC adhesion phenotypes.
While a recent transcriptional analysis study (Claessens et al., 2011) did not identify stevor as a major differentially expressed component in the R+ lines tested, our data clearly showed upregulation of stevor transcription and expression in all lines tested. This discrepancy could be due to the use by Claessens et al. of very well characterized rosetting lines in which the dominant PfEMP1 variants mediating rosetting are well established.
Stronger RBC binding of ST3 as compared to the other two STEVOR variants tested indicated that not all STEVOR have equal capacity to form rosettes. In addition, the transcriptional data showed that only some stevor genes are upregulated, again consistent with the notion that only a subset of STEVOR family members mediates rosetting, and in line with the clonally variant nature of the rosetting phenotype.
The dual role of STEVOR in merozoite invasion and rosetting is conceptually attractive as it provides a direct link between rosetting and invasion. Not only would such a strategy provide protection of the released merozoites from immune attack, but at the same time localizes uninfected RBC for efficient merozoite attachment. Previous in vitro studies using either culture-adapted lines (Clough et al., 1998) or parasites collected from patients (Deans et al., 2006) showed no evidence for increased invasion efficiency of rosetting compared to non-rosetting parasites. In contrast, in vivo studies with a primate model strongly demonstrated an association between rosetting and high parasite multiplication efficiency (Le Scanf et al., 2008). Here, our data clearly shows that STEVOR-mediated rosetting not only confers a growth advantage but also provides protection against invasion-blocking antibodies, strongly supporting the proposed dual role of STEVOR. Importantly, our findings suggest that disruption of STEVOR-mediated rosettes could significantly enhance the efficiency of invasion-inhibitory antibodies in vivo. This is in line with previous reports suggesting that rosetting enhances parasite invasion into uninfected RBC (Le Scanf et al., 2008; Wahlgren et al., 1989) and/or protects merozoites from invasion-inhibitory antibodies (Ruangjirachuporn et al., 1992).
While we have only demonstrated binding of STEVOR to RBC, a recent study showing a lack of transcription of var, A-type rif and stevor multicopy gene families in parasites circulating in the peripheral blood of splenectomized patients is consistent with STEVOR also playing a role in sequestration of iRBCs to endothelial receptors (Bachmann et al., 2009). This is further supported by a recent study showing that STEVOR expression impacts on the deformability of the RBC membrane of both asexual and sexual stage iRBCs, probably facilitating parasite sequestration in deep tissue vasculature (Sanyal et al., 2011; Tiburcio et al., 2012). In P. falciparum, sequestration and rosetting now need to be seen as a multistep process that involves not only PfEMP1 but also STEVOR. In the broader context of Plasmodium species biology, expression of parasite adhesins on the iRBC surface has been linked to sequestration of the rodent parasites P. chabaudi and P. berghei (Franke-Fayard et al., 2005; Mackinnon et al., 2002) and possibly P. vivax (Anstey et al., 2007; Chotivanich et al., 1998). Common to all these parasites is the pir multigene family that codes for small variant antigens that have features similar to STEVOR and RIFIN (Cunningham et al., 2010; Janssen et al., 2002; Jemmely et al., 2010). This would suggest that PIR are adhesins that enable these parasites to rosette and sequester in the absence of PfEMP1. Overall, our results show that an improved understanding of the biological role of small VSAs is essential to completely understand the interplay between rosetting, sequestration and parasite pathology.
Experimental Procedures
Antibodies
For detailed description of different antibodies used see supplemental experimental procedures.
In vitro invasion inhibition assays
Invasion inhibition assays were carried out in triplicate in a 96 well flat-bottom microtitre plate using purified late schizont stage parasites and different concentrations of antisera. Final parasitaemia was determined using SYBR Green I (Molecular Probes) as previously described (Bei et al., 2010b). For comparison of R+ and R- parasites, parasites were cultured in T25 flasks under static and suspension (orbital shaker, ~100 rpm) conditions in the presence of antibodies at 37°C before determining parasitaemia.
Purification of Immunoglobulin G fraction from anti-STEVOR 1 serum
Total IgG to be tested in invasion inhibition was purified from rabbit polyclonal anti-S1 serum using the Saturated Ammonium Sulfate (SAS) precipitation method and the purity was evaluated on non-reducing 10% SDS-PAGE gel.
Constructs for expression of different STEVOR regions on surface of COS7 cells
The semi-conserved (ST1C, ST2C, ST3C), hypervariable (HVR) (ST1H, ST2H, ST3H) and full length (ST1Full, ST2Full, ST3Full) regions of three stevor genes [PF10_0395 (ST1), PFF0850c (ST2) and PFA0105w (ST3)] were were cloned into the pDisplay mammalian expression vector, creating a panel of nine constructs (Figure 2 and Table S1).
COS7 cell culture and transfection
COS7 cell binding assays were carried out as previously described (Chitnis and Miller, 1994) in the presence of absence of different anti-sera to directly determine the impact of these antibodies on binding.
Enrichment of rosetting pigmented trophozoite and schizont iRBCs
P. falciparum cultures were enriched for high rosetting parasites by gelatin sedimentation as previously described (Handunnetti et al., 1992; Rowe et al., 2000). All rosette reversal and blockage assays were performed with late trophozoites and schizont stages parasites determined by Giemsa smear. For reversal of rosetting, rosetting-iRBCs was mixed with anti-STEVOR sera while for blockage of rosetting, purified iRBCs were pre-incubated with anti-STEVOR sera and then allowed to form rosettes with fresh RBCs. Impact of the different treatments on rosette number was assessed.
Immunofluorescent staining of fixed and live iRBCs
Experimental details for immunostaining of acetone-fixed and live iRBCs were as described previously (Fidock and Wellems, 1997; Niang et al., 2009).
pARL-STEVOR1 vector construction and transfection
For the pARL-STEVOR1 vector, a full length stevor gene (PF10_0395) was obtained by modification of the previously described pARL1-STEVOR80-GFP (Przyborski et al., 2005). Transfection strategy and transfectants selection are detailed in supplemental experimental procedures.
Enrichment of culture for high surface-positive STEVOR-iRBCs
Life 3D7ΔMAHRP1-R+ iRBCs were stained with rabbit anti-S1 or anti-S2 sera and sorted by flow cytometry. STEVOR-positive populations (Figure 5E, EB+AF) were sub-cultured with fresh RBCs with weekly enrichment for rosetting and re-evaluated regularly for rosette frequency and surface-positive expression.
Strategy of shRNA-based knockdown of GPC expression in human hematopoietic stem cells
CD34+ HSC precursor cells isolated from cord blood were used for in vitro production of knock down RBCs. The in vitro culture protocol, the production of lentiviral particles containing pLKO plasmids expressing small hairpin RNA (shRNA) against GPC or scrambled shRNA controls, transduction, pyromycin and neomycin selection as well as the flow cytometry measurement of the level of GPC knock down were the same as described previously (Bei et al., 2010a).
Microarray analysis
Total RNA was isolated from highly synchronized P. falciparum trophozoite-stage parasites and hybridized against a reference pool of 3D7 cultures as previously described (Bozdech et al., 2003).
Supplementary Material
Highlights.
The Plasmodium falciparum variant STEVOR is an erythrocyte binding protein that interacts with Glycophorin C on the erythrocyte surface
Antibodies targeting STEVOR semi-conserved regions can interfere with the initial attachment of merozoite to erythrocyte during the invasion process
In P. falciparum, rosetting can be mediated by STEVOR independently of PfEMP1
STEVOR-mediated rosetting confers a growth advantage and provides protection against invasion-blocking antibodies
Acknowledgments
This work was supported by a BioMedical Research Council (BMRC) Singapore grant. We thank Michael J Blackman, Catherine Braun-Breton and Lubin Jiang for the kind gifts of MSP1.19 and SBP1 antibodies and soluble Glycophorin C respectively. We thank Lubin Jiang, Louis Miller, Christine Lomas-Francis and Connie Westhoff for providing the Gerbich negative RBCs. We thank Sue Kyes, Hans-Peter Beck and Jude Przyborski for providing the A4 and 3D7ΔMAHRP1 parasites and pARL-STEVOR80 vector respectively. We thank Goutam Chakraborty for help with the IgG purification and Mark Featherstone for critical reading of the manuscript.
Footnotes
Author contributions
MN, MD and PRP designed and conceived the project and analyzed the data. AKB, SD, UK and generated the GPC knock down cells. KGM, SP, KG, AA, KPY and NSB, contributed to the rosetting and RBC binding assays, chimeric constructs generation, live video microscopy, pARL-STEVOR generation, invasion assays and anti-ATS generation respectively. MN and PRP directed the study, wrote and edited the manuscript.
Conflict of interest
All authors declare that they have no competing interest.
References
- Albrecht L, Moll K, Blomqvist K, Normark J, Chen Q, Wahlgren M. var gene transcription and PfEMP1 expression in the rosetting and cytoadhesive Plasmodium falciparum clone FCR3S1.2. Malar J. 2011;10:17. doi: 10.1186/1475-2875-10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus BJ, Thanikkul K, Silamut K, White NJ, Udomsangpetch R. Short report: Rosette formation in Plasmodium ovale infection. Am J Trop Med Hyg. 1996;55:560–561. doi: 10.4269/ajtmh.1996.55.560. [DOI] [PubMed] [Google Scholar]
- Anstey NM, Handojo T, Pain MC, Kenangalem E, Tjitra E, Price RN, Maguire GP. Lung injury in vivax malaria: pathophysiological evidence for pulmonary vascular sequestration and posttreatment alveolar-capillary inflammation. J Infect Dis. 2007;195:589–596. doi: 10.1086/510756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann A, Esser C, Petter M, Predehl S, von Kalckreuth V, Schmiedel S, Bruchhaus I, Tannich E. Absence of erythrocyte sequestration and lack of multicopy gene family expression in Plasmodium falciparum from a splenectomized malaria patient. PLoS One. 2009;4:e7459. doi: 10.1371/journal.pone.0007459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch DI, Ma XC, Singh HB, Bi X, Pasloske BL, Howard RJ. Identification of a region of PfEMP1 that mediates adherence of Plasmodium falciparum infected erythrocytes to CD36: conserved function with variant sequence. Blood. 1997;90:3766–3775. [PubMed] [Google Scholar]
- Bei AK, Brugnara C, Duraisingh MT. In vitro genetic analysis of an erythrocyte determinant of malaria infection. J Infect Dis. 2010a;202:1722–1727. doi: 10.1086/657157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei AK, Desimone TM, Badiane AS, Ahouidi AD, Dieye T, Ndiaye D, Sarr O, Ndir O, Mboup S, Duraisingh MT. A flow cytometry-based assay for measuring invasion of red blood cells by Plasmodium falciparum. Am J Hematol. 2010b;85:234–237. doi: 10.1002/ajh.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe JE, Surentheran T, Preiser PR. STEVOR--a multifunctional protein? Mol Biochem Parasitol. 2004;134:11–15. doi: 10.1016/j.molbiopara.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Blythe JE, Yam XY, Kuss C, Bozdech Z, Holder AA, Marsh K, Langhorne J, Preiser PR. Plasmodium falciparum STEVOR proteins are highly expressed in patient isolates and located in the surface membranes of infected red blood cells and the apical tips of merozoites. Infect Immun. 2008;76:3329–3336. doi: 10.1128/IAI.01460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst P, Bitter W, McCulloch R, Van Leeuwen F, Rudenko G. Antigenic variation in malaria. Cell. 1995;82:1–4. doi: 10.1016/0092-8674(95)90044-6. [DOI] [PubMed] [Google Scholar]
- Carlson J, Helmby H, Hill AV, Brewster D, Greenwood BM, Wahlgren M. Human cerebral malaria: association with erythrocyte rosetting and lack of anti-rosetting antibodies. Lancet. 1990a;336:1457–1460. doi: 10.1016/0140-6736(90)93174-n. [DOI] [PubMed] [Google Scholar]
- Carlson J, Holmquist G, Taylor DW, Perlmann P, Wahlgren M. Antibodies to a histidine-rich protein (PfHRP1) disrupt spontaneously formed Plasmodium falciparum erythrocyte rosettes. Proc Natl Acad Sci U S A. 1990b;87:2511–2515. doi: 10.1073/pnas.87.7.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, Crabtree J, Angiuoli SV, Merino EF, Amedeo P, et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455:757–763. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, Ermolaeva MD, Allen JE, Selengut JD, Koo HL, et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002;419:512–519. doi: 10.1038/nature01099. [DOI] [PubMed] [Google Scholar]
- Chen Q, Schlichtherle M, Wahlgren M. Molecular aspects of severe malaria. Clin Microbiol Rev. 2000;13:439–450. doi: 10.1128/cmr.13.3.439-450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis CE, Miller LH. Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J Exp Med. 1994;180:497–506. doi: 10.1084/jem.180.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotivanich KT, Pukrittayakamee S, Simpson JA, White NJ, Udomsangpetch R. Characteristics of Plasmodium vivax-infected erythrocyte rosettes. Am J Trop Med Hyg. 1998;59:73–76. doi: 10.4269/ajtmh.1998.59.73. [DOI] [PubMed] [Google Scholar]
- Claessens A, Ghumra A, Gupta AP, Mok S, Bozdech Z, Rowe JA. Design of a variant surface antigen-supplemented microarray chip for whole transcriptome analysis of multiple Plasmodium falciparum cytoadherent strains, and identification of strain-transcendent rif and stevor genes. Malar J. 2011;10:180. doi: 10.1186/1475-2875-10-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clough B, Atilola FA, Pasvoi G. The role of rosetting in the multiplication of Plasmodium falciparum: rosette formation neither enhances nor targets parasite invasion into uninfected red cells. Br J Haematol. 1998;100:99–104. doi: 10.1046/j.1365-2141.1998.00534.x. [DOI] [PubMed] [Google Scholar]
- Cockburn IA, Donvito B, Cohen JH, Rowe JA. A simple method for accurate quantification of complement receptor 1 on erythrocytes preserved by fixing or freezing. J Immunol Methods. 2002;271:59–64. doi: 10.1016/s0022-1759(02)00368-x. [DOI] [PubMed] [Google Scholar]
- Craig A, Scherf A. Molecules on the surface of the Plasmodium falciparum infected erythrocyte and their role in malaria pathogenesis and immune evasion. Mol Biochem Parasitol. 2001;115:129–143. doi: 10.1016/s0166-6851(01)00275-4. [DOI] [PubMed] [Google Scholar]
- Cunningham D, Lawton J, Jarra W, Preiser P, Langhorne J. The pir multigene family of Plasmodium: antigenic variation and beyond. Mol Biochem Parasitol. 2010;170:65–73. doi: 10.1016/j.molbiopara.2009.12.010. [DOI] [PubMed] [Google Scholar]
- David PH, Handunnetti SM, Leech JH, Gamage P, Mendis KN. Rosetting: a new cytoadherence property of malaria-infected erythrocytes. Am J Trop Med Hyg. 1988;38:289–297. doi: 10.4269/ajtmh.1988.38.289. [DOI] [PubMed] [Google Scholar]
- Deans AM, Lyke KE, Thera MA, Plowe CV, Kone A, Doumbo OK, Kai O, Marsh K, Mackinnon MJ, Raza A, et al. Low multiplication rates of African Plasmodium falciparum isolates and lack of association of multiplication rate and red blood cell selectivity with malaria virulence. The American journal of tropical medicine and hygiene. 2006;74:554–563. [PMC free article] [PubMed] [Google Scholar]
- Deans AM, Rowe JA. Plasmodium falciparum: Rosettes do not protect merozoites from invasion-inhibitory antibodies. Exp Parasitol. 2006;112:269–273. doi: 10.1016/j.exppara.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan SA, Proctor JL, Alling DW, Okubo Y, Wellems TE, Miller LH. Glycophorin B as an EBA-175 independent Plasmodium falciparum receptor of human erythrocytes. Mol Biochem Parasitol. 1994;64:55–63. doi: 10.1016/0166-6851(94)90134-1. [DOI] [PubMed] [Google Scholar]
- Doumbo OK, Thera MA, Kone AK, Raza A, Tempest LJ, Lyke KE, Plowe CV, Rowe JA. High levels of Plasmodium falciparum rosetting in all clinical forms of severe malaria in African children. Am J Trop Med Hyg. 2009;81:987–993. doi: 10.4269/ajtmh.2009.09-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraisingh MT, Maier AG, Triglia T, Cowman AF. Erythrocyte-binding antigen 175 mediates invasion in Plasmodium falciparum utilizing sialic acid-dependent and -independent pathways. Proc Natl Acad Sci U S A. 2003;100:4796–4801. doi: 10.1073/pnas.0730883100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez V, Hommel M, Chen Q, Hagblom P, Wahlgren M. Small, clonally variant antigens expressed on the surface of the Plasmodium falciparum-infected erythrocyte are encoded by the rif gene family and are the target of human immune responses. J Exp Med. 1999;190:1393–1404. doi: 10.1084/jem.190.10.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MU, da Silva Nunes M, Wunderlich G. Antigenic diversity and immune evasion by malaria parasites. Clin Diagn Lab Immunol. 2004;11:987–995. doi: 10.1128/CDLI.11.6.987-995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock DA, Wellems TE. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc Natl Acad Sci U S A. 1997;94:10931–10936. doi: 10.1073/pnas.94.20.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick K, Chen Q. var genes, PfEMP1 and the human host. Mol Biochem Parasitol. 2004;134:3–9. doi: 10.1016/j.molbiopara.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Franke-Fayard B, Janse CJ, Cunha-Rodrigues M, Ramesar J, Buscher P, Que I, Lowik C, Voshol PJ, den Boer MA, van Duinen SG, et al. Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc Natl Acad Sci U S A. 2005;102:11468–11473. doi: 10.1073/pnas.0503386102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Yeo KP, Aw SS, Kuss C, Iyer JK, Genesan S, Rajamanonmani R, Lescar J, Bozdech Z, Preiser PR. Antibodies targeting the PfRH1 binding domain inhibit invasion of Plasmodium falciparum merozoites. PLoS Pathog. 2008;4:e1000104. doi: 10.1371/journal.ppat.1000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JE, Puentes A, Curtidor H, Vera R, Rodriguez L, Valbuena J, Lopez R, Ocampo M, Cortes J, Vanegas M, et al. Peptides from the Plasmodium falciparum STEVOR putative protein bind with high affinity to normal human red blood cells. Peptides. 2005;26:1133–1143. doi: 10.1016/j.peptides.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handunnetti SM, van Schravendijk MR, Hasler T, Barnwell JW, Greenwalt DE, Howard RJ. Involvement of CD36 on erythrocytes as a rosetting receptor for Plasmodium falciparum-infected erythrocytes. Blood. 1992;80:2097–2104. [PubMed] [Google Scholar]
- Hasler T, Handunnetti SM, Aguiar JC, van Schravendijk MR, Greenwood BM, Lallinger G, Cegielski P, Howard RJ. In vitro rosetting, cytoadherence, and microagglutination properties of Plasmodium falciparum-infected erythrocytes from Gambian and Tanzanian patients. Blood. 1990;76:1845–1852. [PubMed] [Google Scholar]
- Helmby H, Cavelier L, Pettersson U, Wahlgren M. Rosetting Plasmodium falciparum-infected erythrocytes express unique strain-specific antigens on their surface. Infect Immun. 1993;61:284–288. doi: 10.1128/iai.61.1.284-288.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell DP, Levin EA, Springer AL, Kraemer SM, Phippard DJ, Schief WR, Smith JD. Mapping a common interaction site used by Plasmodium falciparum Duffy binding-like domains to bind diverse host receptors. Mol Microbiol. 2008;67:78–87. doi: 10.1111/j.1365-2958.2007.06019.x. [DOI] [PubMed] [Google Scholar]
- Janssen CS, Barrett MP, Turner CM, Phillips RS. A large gene family for putative variant antigens shared by human and rodent malaria parasites. Proc Biol Sci. 2002;269:431–436. doi: 10.1098/rspb.2001.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen CS, Phillips RS, Turner CM, Barrett MP. Plasmodium interspersed repeats: the major multigene superfamily of malaria parasites. Nucleic Acids Res. 2004;32:5712–5720. doi: 10.1093/nar/gkh907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemmely NY, Niang M, Preiser PR. Small variant surface antigens and Plasmodium evasion of immunity. Future microbiology. 2010;5:663–682. doi: 10.2217/fmb.10.21. [DOI] [PubMed] [Google Scholar]
- Joannin N, Kallberg Y, Wahlgren M, Persson B. RSpred, a set of Hidden Markov Models to detect and classify the RIFIN and STEVOR proteins of Plasmodium falciparum. BMC Genomics. 2011;12:119. doi: 10.1186/1471-2164-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul DK, Roth EF, Jr, Nagel RL, Howard RJ, Handunnetti SM. Rosetting of Plasmodium falciparum-infected red blood cells with uninfected red blood cells enhances microvascular obstruction under flow conditions. Blood. 1991;78:812–819. [PubMed] [Google Scholar]
- Khattab A, Bonow I, Schreiber N, Petter M, Schmetz C, Klinkert MQ. Plasmodium falciparum variant STEVOR antigens are expressed in merozoites and possibly associated with erythrocyte invasion. Malar J. 2008;7:137. doi: 10.1186/1475-2875-7-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattab A, Meri S. Exposure of the Plasmodium falciparum clonally variant STEVOR proteins on the merozoite surface. Malar J. 2011;10:58. doi: 10.1186/1475-2875-10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyes S, Horrocks P, Newbold C. Antigenic variation at the infected red cell surface in malaria. Annu Rev Microbiol. 2001;55:673–707. doi: 10.1146/annurev.micro.55.1.673. [DOI] [PubMed] [Google Scholar]
- Kyes SA, Rowe JA, Kriek N, Newbold CI. Rifins: a second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proc Natl Acad Sci U S A. 1999;96:9333–9338. doi: 10.1073/pnas.96.16.9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Scanf C, Vigan-Womas I, Contamin H, Guillotte M, Bischoff E, Mercereau-Puijalon O. Rosetting is associated with increased Plasmodium falciparum in vivo multiplication rate in the Saimiri sciureus monkey. Microbes and infection / Institut Pasteur. 2008;10:447–451. doi: 10.1016/j.micinf.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Mackinnon MJ, Walker PR, Rowe JA. Plasmodium chabaudi: rosetting in a rodent malaria model. Exp Parasitol. 2002;101:121–128. doi: 10.1016/s0014-4894(02)00103-0. [DOI] [PubMed] [Google Scholar]
- Maier AG, Duraisingh MT, Reeder JC, Patel SS, Kazura JW, Zimmerman PA, Cowman AF. Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nat Med. 2003;9:87–92. doi: 10.1038/nm807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRobert L, Preiser P, Sharp S, Jarra W, Kaviratne M, Taylor MC, Renia L, Sutherland CJ. Distinct trafficking and localization of STEVOR proteins in three stages of the Plasmodium falciparum life cycle. Infect Immun. 2004;72:6597–6602. doi: 10.1128/IAI.72.11.6597-6602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercereau-Puijalon O, Guillotte M, Vigan-Womas I. Rosetting in Plasmodium falciparum: a cytoadherence phenotype with multiple actors. Transfus Clin Biol. 2008;15:62–71. doi: 10.1016/j.tracli.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Newbold C, Craig A, Kyes S, Rowe A, Fernandez-Reyes D, Fagan T. Cytoadherence, pathogenesis and the infected red cell surface in Plasmodium falciparum. Int J Parasitol. 1999;29:927–937. doi: 10.1016/s0020-7519(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Niang M, Yan Yam X, Preiser PR. The Plasmodium falciparum STEVOR multigene family mediates antigenic variation of the infected erythrocyte. PLoS Pathog. 2009;5:e1000307. doi: 10.1371/journal.ppat.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain A, Bohme U, Berry AE, Mungall K, Finn RD, Jackson AP, Mourier T, Mistry J, Pasini EM, Aslett MA, et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature. 2008;455:799–803. doi: 10.1038/nature07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ME, Holt EH. Erythrocyte receptor recognition varies in Plasmodium falciparum isolates. Mol Biochem Parasitol. 1988;27:23–34. doi: 10.1016/0166-6851(88)90021-7. [DOI] [PubMed] [Google Scholar]
- Petter M, Haeggstrom M, Khattab A, Fernandez V, Klinkert MQ, Wahlgren M. Variant proteins of the Plasmodium falciparum RIFIN family show distinct subcellular localization and developmental expression patterns. Mol Biochem Parasitol. 2007;156:51–61. doi: 10.1016/j.molbiopara.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Przyborski JM, Miller SK, Pfahler JM, Henrich PP, Rohrbach P, Crabb BS, Lanzer M. Trafficking of STEVOR to the Maurer’s clefts in Plasmodium falciparum-infected erythrocytes. Embo J. 2005;24:2306–2317. doi: 10.1038/sj.emboj.7600720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe A, Berendt AR, Marsh K, Newbold CI. Plasmodium falciparum: a family of sulphated glycoconjugates disrupts erythrocyte rosettes. Exp Parasitol. 1994;79:506–516. doi: 10.1006/expr.1994.1111. [DOI] [PubMed] [Google Scholar]
- Rowe A, Obeiro J, Newbold CI, Marsh K. Plasmodium falciparum rosetting is associated with malaria severity in Kenya. Infect Immun. 1995;63:2323–2326. doi: 10.1128/iai.63.6.2323-2326.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe JA, Moulds JM, Newbold CI, Miller LH. P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. 1997;388:292–295. doi: 10.1038/40888. [DOI] [PubMed] [Google Scholar]
- Rowe JA, Obiero J, Marsh K, Raza A. Short report: Positive correlation between rosetting and parasitemia in Plasmodium falciparum clinical isolates. Am J Trop Med Hyg. 2002;66:458–460. doi: 10.4269/ajtmh.2002.66.458. [DOI] [PubMed] [Google Scholar]
- Rowe JA, Rogerson SJ, Raza A, Moulds JM, Kazatchkine MD, Marsh K, Newbold CI, Atkinson JP, Miller LH. Mapping of the region of complement receptor (CR) 1 required for Plasmodium falciparum rosetting and demonstration of the importance of CR1 in rosetting in field isolates. J Immunol. 2000;165:6341–6346. doi: 10.4049/jimmunol.165.11.6341. [DOI] [PubMed] [Google Scholar]
- Ruangjirachuporn W, Afzelius BA, Helmby H, Hill AV, Greenwood BM, Carlson J, Berzins K, Perlmann P, Wahlgren M. Ultrastructural analysis of fresh Plasmodium falciparum-infected erythrocytes and their cytoadherence to human leukocytes. The American journal of tropical medicine and hygiene. 1992;46:511–519. doi: 10.4269/ajtmh.1992.46.511. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Egee S, Bouyer G, Perrot S, Safeukui I, Bischoff E, Buffet P, Deitsch KW, Mercereau-Puijalon O, David PH, et al. Plasmodium falciparum STEVOR proteins impact erythrocyte mechanical properties. Blood. 2011 doi: 10.1182/blood-2011-08-370734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S, Egee S, Bouyer G, Perrot S, Safeukui I, Bischoff E, Buffet P, Deitsch KW, Mercereau-Puijalon O, David PH, et al. Plasmodium falciparum STEVOR proteins impact erythrocyte mechanical properties. Blood. 2012;119:e1–8. doi: 10.1182/blood-2011-08-370734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul A. The role of variant surface antigens on malaria-infected red blood cells. Parasitol Today. 1999;15:455–457. doi: 10.1016/s0169-4758(99)01534-3. [DOI] [PubMed] [Google Scholar]
- Scherf A, Lopez-Rubio JJ, Riviere L. Antigenic variation in Plasmodium falciparum. Annu Rev Microbiol. 2008;62:445–470. doi: 10.1146/annurev.micro.61.080706.093134. [DOI] [PubMed] [Google Scholar]
- Sherman IW, Eda S, Winograd E. Cytoadherence and sequestration in Plasmodium falciparum: defining the ties that bind. Microbes Infect. 2003;5:897–909. doi: 10.1016/s1286-4579(03)00162-x. [DOI] [PubMed] [Google Scholar]
- Sim RB. Large-scale isolation of complement receptor type 1 (CR1) from human erythrocytes. Proteolytic fragmentation studies. Biochem J. 1985;232:883–889. doi: 10.1042/bj2320883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadafora C, Awandare GA, Kopydlowski KM, Czege J, Moch JK, Finberg RW, Tsokos GC, Stoute JA. Complement receptor 1 is a sialic acid-independent erythrocyte receptor of Plasmodium falciparum. PLoS Pathog. 2010;6:e1000968. doi: 10.1371/journal.ppat.1000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spycher C, Rug M, Pachlatko E, Hanssen E, Ferguson D, Cowman AF, Tilley L, Beck HP. The Maurer’s cleft protein MAHRP1 is essential for trafficking of PfEMP1 to the surface of Plasmodium falciparum-infected erythrocytes. Mol Microbiol. 2008;68:1300–1314. doi: 10.1111/j.1365-2958.2008.06235.x. [DOI] [PubMed] [Google Scholar]
- Tiburcio M, Niang M, Deplaine G, Perrot S, Bischoff E, Ndour PA, Silvestrini F, Khattab A, Milon G, David PH, et al. A switch in infected erythrocyte deformability at the maturation and blood circulation of Plasmodium falciparum transmission stages. Blood. 2012;119:e172–180. doi: 10.1182/blood-2012-03-414557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udomsangpetch R, Brown AE, Smith CD, Webster HK. Rosette formation by Plasmodium coatneyi-infected red blood cells. Am J Trop Med Hyg. 1991;44:399–401. doi: 10.4269/ajtmh.1991.44.399. [DOI] [PubMed] [Google Scholar]
- Udomsangpetch R, Wahlin B, Carlson J, Berzins K, Torii M, Aikawa M, Perlmann P, Wahlgren M. Plasmodium falciparum-infected erythrocytes form spontaneous erythrocyte rosettes. J Exp Med. 1989;169:1835–1840. doi: 10.1084/jem.169.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt AM, Barragan A, Chen Q, Kironde F, Spillmann D, Wahlgren M. Heparan sulfate on endothelial cells mediates the binding of Plasmodium falciparum-infected erythrocytes via the DBL1alpha domain of PfEMP1. Blood. 2003;101:2405–2411. doi: 10.1182/blood-2002-07-2016. [DOI] [PubMed] [Google Scholar]
- Wahlgren M, Carlson J, Udomsangpetch R, Perlmann P. Why do Plasmodium falciparumm-infected erythrocytes form spontaneous erythrocyte rosettes? Parasitol Today. 1989;5:183–185. doi: 10.1016/0169-4758(89)90141-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.