

Abstract
Neuromyelitis optica (NMO) is causally linked to autoantibodies (ABs) against aquaporin 4 (AQP4). Here, we focused on the pathogenic effects exclusively mediated by human ABs to AQP4 in vivo. We performed cell-free intrathecal (i.th.) passive transfer experiments in Lewis rats using purified patient NMO immunoglobulin G (IgG) and various recombinant human anti-AQP4 IgG-ABs via implanted i.th. catheters. Repetitive application of patient NMO IgG fractions and of recombinant human anti-AQP4 ABs induced signs of spinal cord disease. Magnetic resonance imaging (MRI) revealed longitudinal spinal cord lesions at the site of application of anti-AQP4 IgG. Somatosensory evoked potential amplitudes were reduced in symptomatic animals corroborating the observed functional impairment. Spinal cord histology showed specific IgG deposition in the grey and white matter in the affected areas. We did not find inflammatory cell infiltration nor activation of complement in spinal cord areas of immunoglobulin deposition. Moreover, destructive lesions showing axon or myelin damage and loss of astrocytes and oligodendrocytes were all absent. Immunoreactivity to AQP4 and to the excitatory amino acid transporter 2 (EAAT2) was markedly reduced whereas immunoreactivity to the astrocytic marker glial fibrillary acid protein (GFAP) was preserved. The expression of the NMDA-receptor NR1 subunit was down-regulated in areas of IgG deposition possibly induced by sustained glutamatergic overexcitation. Disease signs and histopathology were reversible within weeks after stopping injections. We conclude that in vivo application of ABs directed at AQP 4 can induce a reversible spinal cord disease in recipient rats by inducing distinct histopathological abnormalities. These findings may be the experimental correlate of “penumbra-like” lesions recently reported in NMO patients adjacent to effector-mediated tissue damage.
Keywords: Neuromyelitis optica, Aquaporin 4, EAAT2, NMDA receptor, Astrocyte, Intrathecal passive transfer
Introduction
The discovery of aquaporin 4 (AQP4) as the target antigen for IgG autoantibodies (ABs) in neuromyelitis optica (NMO) was a significant advancement in our understanding of central nervous system (CNS) autoimmune disorders (Lennon et al., 2004, 2005). Beside tissue destruction with complement deposition and macrophage infiltrates, NMO lesions show extensive loss of AQP4 pointing to an AB-mediated, target-specific humoral immune reaction with a host of proinflammatory components (Misu et al., 2007; Roemer et al., 2007).
Previous in vivo studies have provided evidence for a pathogenic role of ABs against AQP4 in a co-existing inflammatory milieu (Bennett et al., 2009; Bradl et al., 2009). Moreover, injection of anti-AQP4 ABs into the brain, incubation of spinal cord slice cultures, or primary astrocytes induced AQP4 depletion and death of astrocytes when activated human complement was present (Ratelade et al., 2012; Saadoun et al., 2010; Wrzos et al., 2014). These observations point to cooperative immunopathological events involving anti-AQP4 ABs, complement, and cell-dependent factors as the cause of tissue destruction in NMO.
In AB-mediated neurological diseases of the peripheral and central nervous system, ABs typically have intrinsic effects upon binding to their target antigen e.g. in myasthenia gravis (Toyka et al., 1977), in Lambert–Eaton myasthenic syndrome (Buchwald et al., 2005; Fukunaga et al., 1983), autoimmune encephalitis (Hughes et al., 2010), and stiff-person syndrome (Geis et al., 2010). There is an ongoing debate whether ABs against AQP4 in principal might also have pathogenic effects that are independent from complement activation and secondary cell-mediated inflammatory activity. At the cellular level, it has been shown that the glial excitatory amino acid transporter EAAT2 was co-internalized along with AQP4 in transfected cells upon exposure to anti-AQP4-ABs. This led to the hypothesis that glutamate excitotoxicity might contribute to CNS pathology in anti-AQP4 AB-associated autoimmunity (Hinson et al., 2008; Marignier et al., 2010; Wrzos et al., 2014). Other investigators could not find evidence supporting these mechanisms in primary cell culture experiments and after single intracerebral injection of ABs to AQP4 (Ratelade et al., 2011; Rossi et al., 2012). An autopsy series of NMO patients showed loss of AQP4 on reactive astrocytes in the superficial cortical layers accompanied by neuronal loss and microglia activation in adjacent layers. The authors suggested a clinically relevant neurodegenerative process that was concurrent with inflammatory demyelination (Blanc et al., 2008; Saji et al., 2013). Very recently, “penumbra-like” lesions with loss of AQP4 but preserved astrocytes have been reported in spinal cord sections in autopsy cases of NMO patients adjacent to destructive lesions with complement activation (Misu et al., 2013) and after single intracerebral injection of anti-AQP4 AB in rats (Asavapanumas et al., 2014).
We here investigated the intrinsic effects of anti-AQP4 IgG ABs in vivo in a chronic animal model with repetitive intrathecal (i.th.) application of NMO patient-IgG or recombinant antibodies (rABs) against AQP4 (Geis et al., 2010, 2011). This approach allows testing AB-mediated pathology in the spinal cord in the living rat over several weeks, independent of additional effector mechanisms.
Materials and methods
Patients, therapeutic plasma exchange, and preparation of IgG fractions
All four NMO patients from whom plasma exchange material was available fulfilled the revised diagnostic criteria for NMO (Wingerchuk et al., 2006) and had positive serum titers of ABs against AQP4. Human NMO immunoglobulin G (NMO1-4) was purified from plasma filtrates as described previously (Buchwald et al., 2002) and from disease-control patients with chronic demyelinating inflammatory polyneuropathy who were negative for anti-AQP4 reactivity (control IgG). Purified IgG from one patient (NMO3) was previously utilized and described in another study (Bradl et al., 2009). All IgG fractions were dialyzed against distilled water, freeze dried and stored at − 20 °C. Lyophilized IgG was dissolved in normal saline just before use (100 mg/ml concentration for NMO1, 2, and 4, and control IgG 1 and 2; 12 mg/ml for NMO3). Titers of anti-AQP4 ABs were then measured at a 1 mg/ml concentration by a commercial indirect immunofluorescence test with AQP4 transfected HEK 293 cells (Euroimmun Lübeck, Germany). All purified NMO IgG fractions displayed a strongly positive anti-AQP4 binding pattern (titers ≥1:100).
Recombinant patient antibodies
Purified human recombinant IgG ABs were generated as previously described (Bennett et al., 2009). Recombinant antibodies rABAQP4 and rABcontr2 were generated from plasmablast clones recovered from a NMO patient's cerebrospinal fluid (CSF). rABcontr2 is specific for human AQP4 but does not bind rat AQP4 (Bennett et al., 2009). rABcontr1 is an isotype-matched control human recombinant AB specific for measles virus nucleocapsid protein (Bennett et al., 2009).
Animals, surgery, and intrathecal injections
A total of 112 female Lewis rats were included in the experiments reported here. Adult rats (8–10 weeks old) were purchased from Harlan-Winkelmann (Borchen, Germany). All experiments were approved by the Bavarian and Thuringian State authorities. Intrathecal (i.th.) catheters (0.61-mm outer diameter, intrathecal length 6.5 cm) were placed as described (Geis et al., 2010; Yaksh and Rudy, 1976). Rats developing signs of paralysis after surgery with catheter placement were sacrificed immediately. All others were allowed to recover for 8–10 days, and rats were randomized to receive either purified patient IgG or recombinant ABs. The principal experimental groups contained a minimum of 6 animals for each treatment mode. All i.th. injections were painless as judged by daily observation of the rats and were done in the awake rat while gently immobilizing them for 30 seconds.
The IgG fractions were injected at the following concentrations: NMO IgG 1, 2, 4 or control IgG 1 or 2 at 100 mg/ml, NMO IgG 3 at 12 mg/ml; for estimating a dose–response relation, NMO IgG 4 was injected at 30 mg/ml and 70 mg/ml in two groups of 3 rats each; rABAQP4 was injected at a concentration of 1 mg/ml (or of 0.5 mg/ml in 7 rats for estimating the dose–response relation) and rABcontr1, rABcontr2 at 1 mg/ml. Because of their monoclonality for the respective antigens, rAB IgG fractions were used in a 100-fold lower concentration than polyclonal NMO-IgG or control IgG fractions. The high concentrations of patient IgG and of control IgG did not produce any unspecific effects as observed in this and in previous experiments (Geis et al., 2010, 2012). Injections of 10 μl purified NMO IgG or rABs or control IgG were performed with a subsequent flush of 10 μl saline in three series of daily injections for 5 days each with a 2 days break before the subsequent series, resulting in 15 total IgG injections over a 3 week period (Fig. 1). This approach was chosen because it had been found useful in earlier studies (Geis et al., 2010, 2011). Moreover, the action of ABs can be studied independent of additional co-factors including complement, and the time course of developing disease signs can be followed. In a further group of animals we studied the recovery potential using the IgG fraction named NMO IgG 3 after a full course of injections observing the rats for another 30 days without any further IgG application.
Fig. 1.
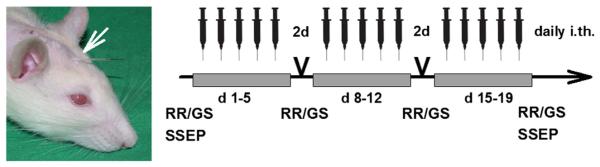
I.th. injections of IgG preparations via implanted catheters (white arrow) were performed in three series of five daily applications with a two-day interval between the three series. Disease severity was scored daily and RotaRod (RR) and grip strength (GS) testing was performed weekly. Magnetic resonance imaging (MRI) and somatosensory evoked potentials (SSEP) testing were done at the indicated time points.
Behavioral analyses
Behavioral analyses were performed by investigators who were masked as to the treatment assignments. Animals were observed daily for at least one hour while moving in their cages and freely over a large table. Body weight was monitored daily. Motor disability was rated on a spinal cord disease score modified from an EAE score (Linker et al., 2002) ranging from 0 (no symptoms) to 10 (death due to complete immobilization; Supplementary Table). Before surgery, rats were trained on an accelerating RotaRod (TSE Systems, Bad Homburg, Germany) and quantitative testing was performed 8–10 days after surgery just before the 1st injection to be taken as baseline, and then after the 5th, 10th and 15th i.th. injections (Fig. 1). Grip strength of the forelimbs was tested with a digital grip force meter (DFIS series, Chatillon, Greensboro, NC, USA) before and after surgery and after the 5th, 10th and 15th i.th. injection.
Magnetic resonance imaging and contrast agent
The experimental contrast agent gadofluorine M (a gift from Bayer Schering Pharma GmbH, Berlin, Germany), which is known to be up to 10 times more sensitive than standard gadolinium in detecting leakage of the blood–brain barrier, was applied intravenously in the tail vein of experimental rats 24 hours before MRI imaging (0.1 mmol/kg) (Bendszus et al., 2008). All measurements of rat spinal cord were performed in the anesthetized rat on a 3T MRI Unit (Siemens TIM Trio, Erlangen, Germany) using a commercial 7 cm coil (Siemens). Rats were placed in a custom-made plastic chamber in a stretched position. The MRI protocol included a scout sequence in three planes, then sagittal and coronal scans were done to identify gadofluorine M enhancing lesions. In total, 22 rats were investigated in this way (NMO IgG1 n = 5, NMO IgG2 n = 3, NMO IgG3 n = 3; control IgG n = 4; rABAQP4n = 4, rABcontr1n = 3).
Functional analyses of afferent spinal cord nerve tracts by recording somatosensory evoked potentials
Somatosensory evoked potentials (SSEP) were recorded in NMO1 and NMO2 IgG injected groups of rats and in the respective control groups in vivo under ketamine–xylazine anaesthesia (100 mg/kg and 5 mg/kg, respectively) before any injections and after terminating i.th. injections. In these groups, two skull recording electrodes and one reference electrode were implanted bilaterally after placing the i.th. catheter. Steel screws were placed according to the following coordinates: 2 mm lateral to midline and 2.5 mm caudal to bregma. Another screw was placed midline 1.5 mm caudal to lambda and served as reference electrode. Holes were drilled (Dremel 300-1/5 driller) under stereomicroscopic control and care was taken not to damage the dura mater. Screws were fixed in the scull using dental cement (Dental Manufaktur GmbH, Berlin, Germany). Recordings were performed using a digital Toennies electromyograph and NeuroScreenplus software (Erich Jaeger GmbH, Hoechberg, Germany). The tibial nerve on either side was serially stimulated 200 times at 1 Hz with a stimulation current 30% above the current needed to obtain the maximal response (supramaximal stimulation) with a stimulus duration of 1 ms. SSEP were recorded from the contralateral somatosensory cortex. Latencies to the first positive deflection (P1) and the amplitudes between P1 and the first negative deflection (N1) were measured.
Tissue harvesting and immunohistochemistry
One to 3 days after the last injection animals were deeply anesthetized and blood was collected after vessel transection in an axillary skin pouch. The spinal cord was removed after laminectomy and subdivided into cervico-thoracic and lumbar regions. Spinal cord specimens were then embedded in Tissue Tec OTC embedding compound (Sakura Finetek, Zoeterwoude, Netherlands) and snap-frozen in isobutane cooled in liquid nitrogen and stored at − 80 °C. Serial 10 μm cryosections of the mounted spinal cord were cut at different levels (cervico-thoracic, and lumbar segment) on a cryostat (CM 050S Kryostat, Leica, Wetzlar, Germany). Standard hematoxylin and eosin staining was performed for basic histology. For immunohistochemistry, sections were blocked with 10% BSA, incubated at 4 °C over night with various antibodies, and visualization was done with streptavidin–biotin peroxidase or with immunofluorescence. The following antibodies were used with pre-incubation of 2% milk powder to block non-specific binding: polyclonal rabbit anti-human IgG (Dako, Hamburg, Germany; 1:200), polyclonal rabbit anti-AQP4 (A5971; Sigma Aldrich, St.Louis, USA; 1:500), polyclonal rabbit anti-AQP4 (sc-20812, Santa-Cruz, CA, USA; 1:500), polyclonal rabbit anti-GFAP (EMD Millipore, Corporation, Billerica, MA, USA; 1:5000), monoclonal mouse anti-GFAP (ICN Biomedicals, Irvine, CA, USA; 1:100), polyclonal rabbit anti-Iba1 (Wako, Osaka, Japan; 1:500); polyclonal rabbit anti-EAAT2 (Cell Signaling Technology, Danvers, MA, USA; 1:100), monoclonal mouse anti-ED1 (Linaris, Wertheim, Germany; 1:500), polyclonal rabbit anti-neurofilament H (Affiniti Ltd., Exeter, UK, 1:2000), monoclonal mouse anti-NogoA (kindly provided by Professor M. Schwab, Zurich, Switzerland; 1:5000), monoclonal mouse anti-APP (clone 22C11, EMD-Millipore; 1:2000), rabbit anti-C9neo (kindly provided by B.P. Morgan, Cardiff, Wales, UK; 1:5000), monoclonal mouse NR1 (EMD-Millipore, 1:1000). Visualization with diaminobenzidine was performed after incubation for 30 minutes at room temperature with the biotinylated secondary antibodies and the streptavidin–biotin complex (Vectastain ABC-Peroxidase Kit, Linaris). Washing steps were done using tris-buffered saline. For immunofluorescence staining, we used Cy3 antibody (donkey anti-mouse 1:100 or anti-rabbit 1:100, respectively; Dianova, Hamburg, Germany) and Alexa-Fluor 488 antibody (donkey anti-mouse 1:400 or anti-rabbit 1:400, respectively; Dianova) and DAPI counterstaining. For myelin staining, sections were incubated in Luxol fast blue solution (Sigma Aldrich, Germany; 1 g in 1000 ml ethanol 96% with 5 ml 10% acetic acid) at 60 °C, then washed with ethanol 96% followed by aqua dest., and then differentiated in lithium carbonate (Sigma; 0.5 g in 1000 ml) together with ethanol 70%. After dehydration in ascending alcohol series, sections were mounted with Vitroclud (Langenbrinck, Emmendingen, Germany).
Spinal cord sections were viewed with an Axiophot 2 microscope (Zeiss, Oberkochen, Germany) by an examiner masked as to treatment allocation with the appropriate filter settings and digitized using SPOT imaging software version 4.5 (SPOT imaging solutions, Burroughs, Sterling Heights, USA). Analyses were performed with Image Pro Plus version 4.5 software (Media Cybernetics, Silver Spring, USA).
Densitometric analyses and counting of immunoreactive cells
All immunostains were digitized with the identical microscope and software settings. For quantification, a standardized area of interest (AOI, 62 μm2) was placed in the grey and in the white matter in the ventral and the dorsal horn on either side. Means of grey and white matter values of each animal were then taken for further analyses. Intraspinal regions with high density staining for human IgG, typically near the catheter ending, were defined as a potential lesion areas and AOI were also placed into these regions (see Fig. 3H for scheme of quantitative analyses). Comparisons were then made with the grey and white matter AOI of the contralateral side. In control IgG treated animals, mean values of both sides were taken for analysis. Mean density values on the color scale (red, green, blue scale, 0–255 color grades), areas of positively stained cells, and numbers of positive cells were all determined semi-automatically using custom-made macros with Image Pro Plus software by an investigator masked as to treatments.
Fig. 3.

Longitudinal lesions and human IgG deposition in spinal cord tissue after i.th. passive-transfer A: Representative contrast-enhanced MRI of cervical, thoracic, and lumbar regions (T1 weighted, repetition time 860 ms, echo time 15 ms; slice thickness of 1.0 mm) of a control IgG treated rat (left sagittal sections and right transversal section) with only marginal local enhancement of the experimental contrast agent Gadofluorine M (Gf) adjacent to the tip of the catheter (arrow). Anatomical landmarks are indicated. B: Representative spinal MRI of a rat injected i.th. with NMO1-IgG for 15 days. Twenty-four following Gf administration, T1-weighted contrast enhancement was most pronounced in the region adjacent to the tip of the i.th. catheter (arrow) and extended several segments above it (left panel). Transverse sections (right panel) revealed accumulation of Gf in white and grey matter areas predominantly ipsilateral to the injection side (arrows, slice thickness 1.5 mm). C: Transverse spinal cord sections at low magnification showed intraspinal human IgG deposition in the spinal white and grey matter which was gradually reduced at increasing distance from the site of the catheter ending. D to G: Higher magnification reveals strong IgG deposition in the white (*, D) and grey (#, E) matter, along astrocytic structures, vascular structures and perineural spaces. After i.th. application of control IgG (F,G), immunoreactivity against human IgG was clearly less intense. H: Schematic drawing to illustrate the quantitative evaluation of immunoreactivity. Standardized areas of interest (AOI) were placed in the grey and in the white matter in the ventral and the dorsal horn on either side for densitometric measurements (black squares). At lesion sites, a further AOI was placed in the area of highest human IgG deposition (red square; C: catheter, IgG: area of human IgG deposition) and density measurements were compared with contralateral AOI. Densitometry was done with the red, green, and blue scale (RGB, 0–255 color grades covering the whole color spectrum). Note, that this method reflects large differences in staining intensities by small numeric changes. I: Quantitative evaluation of IgG deposition in spinal cord lesion areas as compared to contralateral white and grey matter for all experimental groups (**p < 0.01, ***p < 0.001). For detailed information on densitometry see Materials and methods.
Investigation of specific IgG tissue binding
In a series of cryosections, we investigated if tissue-bound human IgG could be dissociated to address the question of nonspecific binding. Adjacent sections were either treated with 0.5% polyethylene glycol (PEG) or with 0.5 M acetic acid (pH 2.5) before staining with the specific primary AB (human IgG, AQP4, and EAAT2). To exclude or minimize steric hindrance or blocking of the specific ABs by the high concentrations of tissue-bound human IgG we performed double-immunofluorescence stainings on control spinal cord sections using two different commercial anti-AQP4 ABs (rabbit polyclonal, A5971; Sigma Aldrich, 1:500; rabbit polyclonal, sc-20812, Santa-Cruz, CA, USA, 1:500) both directed at the C-terminal domain of AQP4 which is not recognized by the NMO patient IgG fractions (Crane et al., 2011) in combination with purified NMO-IgG and patient-derived AQP-specific recombinant AB (rABAQP4). These stains showed a reproducible binding pattern to predominantly perivascular astrocytic structures. There was no difference in the staining pattern with rABAQP4, purified NMO IgG, and both commercial anti-AQP4 AB. Double immunofluorescence staining of naive spinal cord tissue with rABAQP4 or purified NMO IgG together with the commercial anti-AQP4 AB gave clear co-labeling (not shown) confirming different binding epitopes of patient and commercial AB and excludes steric hindrance.
Statistical analyses
All data are presented as means and SEMs. Statistical analyses were done using SigmaPlot software version 12.0 (Systat Software Inc., San Jose, CA, USA). Evaluation of normal distribution of data was done by using the Shapiro–Wilk test. Depending on data distribution, the significance of differences between 2 treatment groups was tested by using the Student's t-test or the Mann–Whitney U-test. Multiple comparisons were analyzed with a one-way ANOVA with Bonferroni post-hoc analysis or one-way repeated measures ANOVA with Holm–Sidak pairwise multiple comparison procedures in case of repeated analyses of multiple treatment groups.
Results
Intrathecal passive transfer of NMO-IgG leads to a complement-independent, progressive, and reversible spinal cord disease without affecting the integrity of axons, myelin, and oligodendrocytes.
When NMO-IgG containing high titer anti-AQP4 ABs was repetitively applied i.th. (Fig. 1), rats gradually developed progressive mild to moderate myelopathic signs usually starting with unilateral paresis at the side of the catheter ending and IgG application, later evolving to an asymmetric paraparesis of the hind limbs. Accordingly, rats showed a decline in their forced walking abilities on an accelerating RotaRod (Figs. 2A, B, Supplementary Movie). Grip strength testing of the fore-limbs revealed no significant deficits (not shown), indicating that functional pathology was limited to the thoraco-lumbar level, the region where the antibodies reached highest concentrations. A dose dependent pathology was suggested by experiments in which reduced amounts of i.th. injected NMO-IgG led to diminished disease signs (Fig. 2C). I.th.-injected control IgG led at most to very mild and nonspecific symptoms. When NMO-IgG applications were discontinued after a full series of 15 injections, animals recovered to nearly baseline values in a timeframe of several weeks (Fig. 2D), indicating virtually complete reversibility of NMO-IgG induced disease signs.
Fig. 2.

Induction of spinal cord disease signs upon intrathecal (i.th.) patient IgG antibody (AB) application. A: Progressive increase of disease scores over time in experimental rats upon receiving 15 i.th. injections with NMO-IgG derived from four individual patients (color-coded) in comparison with purified IgG fraction from a control patient. Control IgG injected animals showed only marginally elevated disease scores. Mean disease score values for the injection period are depicted for the group comparisons (**p < 0.01; ***p < 0.001; colored asterisks indicate respective NMO IgG treatment group vs. control IgG; numbers of experimental animals used are indicated). B: Rats injected with NMO-IgG but not with similar amounts of control IgG developed progressive deficits in forced walking abilities on RotaRod testing (abscissa: time on rod, relative to individual baseline values normalized at 1.0; *p < 0.05; **p < 0.01; ***p < 0.001 as compared to baseline values). The individual AB preparations are color-coded. C: Dose-dependency of spinal cord disease signs after i.th. application of NMO-IgG. Separate group of rats injected with 30 mg/ml NMO-IgG 4 showed lower disease scores when compared to animals treated with a concentration of 70 mg/ml. Animals administered 70 mg/ml of NMO-IgG showed roughly comparable severity to that seen in a separate series of rats treated with 100 mg/ml NMO-IgG (in A). D: Disease signs in a separate group of rats gradually declined over several weeks to virtually baseline values when i.th. injections of NMO-IgG ceased and experimental animals were allowed to recover (for interpretation of the references to color in this figure legend, the reader is referred to the web version of the article).
Spinal MRI using the sensitive experimental contrast agent gadofluorine M (Gf) revealed longitudinal contrast enhancing lesions at the side of IgG application in the spinal cord of rats treated with NMO-IgG extending a few segments caudally and rostrally from the site of maximal IgG concentration near the tip of the catheter (Figs. 3A and B). Sequential transverse sections showed Gf enhancement in the white and centromedullary grey matter indicating local disruption of the blood–brain barrier. The focal manifestation of spinal lesions was also seen histologically with marked intraspinal IgG deposition with a high gradient of local IgG concentrations in the region adjacent to the tip of the implanted catheter extending into the adjacent white and grey matter (Figs. 3C–I). The depositions were specific for NMO-IgG as application of control IgG fractions did not produce intraspinal IgG deposition (Figs. 3F,G, and 6, left column). IgG deposits were tightly bound to spinal cord tissue and could not be washed out with polyethylene glycol or low pH acetic acid excluding unspecific binding (Supplementary Fig. 1).
Fig. 6.

Pathology of anti-AQP4 AB induced spinal cord lesions. Representative transverse sections of the lumbar spinal cord of i.th. treated rats are shown at low magnification. At the site of the catheter tip a localized extraspinal swelling of similar size is shown in all experimental groups independent from the type of IgG without causing intramedullary abnormalities. The site of the excised catheter and its lumen can still be identified (large arrows). In control IgG treated rats no abnormalities were seen (vertical column 1). In rats treated with anti-AQP4-IgG there was intense accumulation of human IgG in the white and grey matter of the spinal cord with a local gradient starting highest near the site of the catheter lumen and diffusing radially into the parenchyme (vertical columns 2 and 3). Note, that the immunoreactivity was similar in intensity and extent with i.th. injected recombinant AB (rABAQP4) although the IgG concentration was 100-fold lower as compared to the NMO IgG fractions and to control IgG. In these areas (arrows), expression of AQP4 and EAAT2 was reduced extending in a semicircular pattern perpendicular to the site of the catheter whereas astrocytes showed increased GFAP immunoreactivity. Animals treated with any of the control IgG fractions (left column) showed no changes in AQP4 or EAAT2 staining. In experimental animals allowed to recover for another 30 days (vertical column 4), immunohistological abnormalities were markedly reduced when compared to the spinal cord of animals in the acute phase after i.th. application of NMO IgG. Cervico-thoracic spinal cord sections from NMO-IgG i.th. injected animals (right column) showed only marginal deposition of human IgG and little hypertrophy of GFAP positive astrocytes at the border areas adjacent to the more rostral parts of the catheter where no catheter opening is present (arrow). AQP4 and EAAT2 expression was completely preserved in white and grey matter.
We found no morphological affliction of axons, myelin, or oligodendrocytes even in close proximity to areas of marked human IgG deposition (Figs. 4A to D). Of note, we detected no intraspinal activation of host complement and only very mild intraspinal infiltration with macrophages limited to the area directly adjacent to the tip of the catheter (Figs. 4E and F). To ensure that the anti-AQP4-AB and complement preparations are in principle capable of activating complement, we checked whether our anti-AQP4-AB containing IgG fractions could induce lytic lesions when directly injected intracerebrally together with human complement (Saadoun et al., 2010). We observed complete local depletion of AQP4, destruction of GFAP positive astrocytes, and invasion of T-cells and macrophages (Supplementary Fig. 2). Hence, our IgG preparations are not fundamentally different from those used in previous studies (Bennett et al., 2009; Bradl et al., 2009; Saadoun et al., 2010).
Fig. 4.

Repetitive i.th. application of NMO-IgG lacking human complement does not damage spinal cord integrity and is not activating rat complement nor is there intraspinal inflammatory cell infiltration except marginally near the tip of the catheter. A and B: Anti-NF200 (A) and luxol fast blue (B) staining revealed intact axonal filaments and myelin sheets within and directly adjacent to the lesion sites, respectively (arrows; scale bar: 100 μm). C: Anti-NogoA immunoreactivity showed no differences in the intensity or patterns of oligodendrocyte staining in the lesioned grey and white matter (arrows) when compared to the contralateral side (scale bar: 100 μm). D: Anti-APP staining did not show evidence of acute axonal injury (scale bar: 300 μm). E: Macrophages (ED1) were found in the extraspinal connective tissue around the catheter and extradurally. There was only marginal local intraspinal infiltration restricted to the site of the catheter ending (scale bar: 200 μm). F: anti-rat C9neo staining showed no activated host complement within spinal cord tissue (scale bar: 200 μm).
Anti-AQP4 antibodies are the culprit antibodies inducing myelopathy
To confirm anti-AQP4 AB specificity in this rat model, recombinant antibodies (rABs) derived from human CSF plasma cells (Bennett et al., 2009) were passively transferred to rats using the same i.th. route and injection protocol but at lower concentration. The AQP4-specific rABs (rABAQP4) per se without additional complement application induced disease signs in the rats in a time-dependent manner similar to those with NMO-patient IgG. Control rABs did not induce disease signs. Reducing the dose of rABAQP4 by half led to a significant decrease in disease severity (Fig. 5A). Accordingly, RotaRod testing showed a decline in walking abilities in rats treated with rABAQP4 in both concentrations tested. With control rABs, no significant effects were seen on RotaRod performance (Fig. 5B).
Fig. 5.

Induction of spinal cord disease signs upon i.th. application of recombinant anti-AQP4 ABs. A: I.th. injections of recombinant antibody against AQP4 (rABAQP4) caused signs of myelopathy. Animals treated with half the amount of rABAQP4 (rABAQP4–50%) showed significantly lower disease scores over time corroborating the dose–response relation observed with patient IgG antibodies. Recombinant control ABs (rABcontr1, rABcontr2) had no effect (left: scores over the entire time course, right: mean group score values *p < 0.05; ***p < 0.001; color-coded asterisks indicate comparisons of treatment groups). B: Progressive deficits in RotaRod testing accumulated over time following 5, 10, or 15 i.th. applications of anti-AQP4 rABs (abscissa: time on rod, relative to individual baseline values normalized at 1.0; *p < 0.05; **p < 0.01; as compared to baseline values). The individual AB preparations are color-coded (for interpretation of the references to color in this fiure legend, the reader is referred to the web version of the article).
Anti-AQP4 IgG reduces AQP4 immunoreactivity in situ while GFAP in astrocytes is upregulated
In the areas of anti-AQP4 IgG deposition, the expression of AQP4 was reduced as compared to areas without deposition of human IgG and to rats treated with control IgG (Figs. 6, 7A, B, and E). I.th. injected and already tissue-bound NMO patient IgG or AQP4-specific rAB might theoretically reduce available binding epitopes for the anti-AQP4 ABs used for immunohistochemistry and falsely suggest reduced AQP4 expression in these areas. To rule out this possibility, we used two different commercial anti-AQP4 ABs recognizing the C-terminal region, which is not the target epitope for NMO-IgG or rABAQP4 (Crane et al., 2011) and does not interfere with patient-IgG binding. Staining with both commercial anti-AQP4 ABs also showed a reduced AQP4 immunoreactivity thus suggesting true reduction of AQP4 expression (Fig. 8A). The expression of GFAP positive cells was preserved or even upregulated in lesion sites with marked human IgG deposition (Figs. 6, 7C, D, and E). The observed astrogliosis was accompanied by a hypertrophy of Iba1-positive microglial cells (Fig. 8B). A detailed analysis using double immunofluorescence staining showed a decrease of AQP4 expression in GFAP positive astrocytes in the region adjacent to the catheter (Fig. 7F) that typically was the area of highest IgG deposition (see also Fig. 6). At greater distance, i.e. in areas with low IgG deposition, perivascular GFAP positive astrocytes had preserved co-expression of AQP4 (Fig. 7F). Accordingly, at cervico-thoracic levels, several segments rostral to the catheter ending, we found no or only marginal human IgG deposition with little astrocytic hypertrophy and no reduction of AQP4 in the adjacent white or grey matter (Fig. 6, right column). Thus, the reduction of AQP4 immunoreactivity upon application of NMO-IgG and anti-AQP4 rABs was not associated with a loss of GFAP positive astrocytes in our rat model.
Fig. 7.

AQP4 immunoreactivity was selectively reduced in lesion areas whereas GFAP expression of astrocytes was increased after i.th. injection of NMO-IgG or rABAQP4. A and B: The expression of AQP4 was always reduced ipsilateral to the catheter ending in regions staining positive for human IgG. C: and D: In lesion areas, expression of GFAP was increased and astrocytes appeared hypertrophic (scale bar: 30 μm). E: Densitometric quantification of AQP4 immunoreactivity showed a selectively and significantly reduced expression in spinal cord lesions adjacent to the catheter site (for details see Materials and methods and legend to Fig. 3); GFAP positive cells are more numerous in areas with reduced AQP4 expression (*p < 0.05; **p < 0.01; ***p < 0.001). F: Double immunofluorescence labeling with DAPI counterstain (cell nuclei, blue) revealed a demarcation of reduced AQP4 (red) expression in GFAP positive perivascular astrocytes (green). Higher magnifications from more centromedial (upper right panels) and more lateral (lower right panels) areas of the spinal cord section (marked by rectangles) showed marked loss of AQP immunoreactivity in GFAP positive astrocytes adjacent to the site of the catheter tip. In contrast, at a wider distance, AQP4 expression was still preserved (scale bar: 100 μm) (for interpretation of the references to color in this figure legend, the reader is referred to the web version of the article).
Fig. 8.

Microglia activation in lesion areas with reduced AQP4 expression. A: Reduction of AQP4 expression in areas of human anti-AQP4 IgG deposition is shown by immunohistochemistry using two different commercial polyclonal anti-AQP4 ABs (upper panel: sc-20812, Santa Cruz, USA. Lower panel: S-A5971, Sigma-Aldrich, USA). Dashed lines were drawn to better illustrate the regions of pronounced human IgG deposition (NMO1, red). Both commercial ABs showed a similar reduction of AQP4 immunoreactivity in lesion areas (scale bar: 200 μm). B: Iba-1 expression representing microglial cells was increased largely with hypertrophy and proliferation in lesion areas with identified deposition of human IgG as shown in other sections when compared to control IgG treated animals. Higher magnification (lower panel) showed hypertrophic and ramified microglia in the white matter of the spinal cord (scale bar: upper panel in b: 100 μm, lower panel: 50 μm).
Further, we evaluated whether rats would recover from disease activity after cessation of NMO-IgG injections for 30 days. Indeed, intraspinal human IgG deposition was markedly reduced and abnormalities observed in AQP4 and GFAP immunoreactivity were less obvious (Fig. 6, NMO recovery) indicating that the intrinsic pathogenic effects of AQP4 AB are potentially reversible once AB are eliminated.
Repetitive intrathecal anti-AQP4 IgG application reduces EAAT2 expression
We next investigated the expression of the glutamate transporter EAAT2, which has been proposed to contribute to NMO pathophysiology (Hinson et al., 2008). In the areas with high human IgG deposition and associated high GFAP expression, we found a markedly reduced perivascular and perineuronal astrocytic EAAT2 immunoreactivity. The EAAT2 expression was substantially more reduced near the catheter tip than at more distant areas or at cervico-thoracic levels (Figs. 6 and 9). As for AQP4, in rats that had recovered for 30 days without further injections of NMO-IgG, EAAT2 immunoreactivity was restored confirming a reversible, AB-depending pathomechanism (Fig. 6, NMO recovery).
Fig. 9.

Loss of EAAT2 expression in perivascular and perisynaptic GFAP immunoreactive astrocytes in NMO-IgG and in rABAQP4 i.th. treated animals. A to E: The expression of EAAT2 was inversely correlated with human IgG deposition in the ipsilateral grey matter (in A) as compared to the contralateral side (in B). Conversely, GFAP immunoreactivity was positively correlated with human IgG deposition ipsilaterally (in C) as compared to the contralateral side (in D; scale bar: 30 μm). These observations were quantified by densitometric analyses (E; ***p < 0.001; for details see Materials and methods and legend to Fig. 3). F: Double immunofluorescence showed reduction of EAAT2 immunoreactivity (red) in perivascular and perineuronal astrocyte processes (GFAP, green) in the grey matter of the spinal cord in animals treated with ABs against anti-AQP4 (lower row) (scale bar 25 μm).
Repetitive intrathecal anti-AQP4 IgG application impairs evoked potentials associated and induced reduction of NR1 expression
In two separate experimental series we studied conduction of afferent spinal cord nerve tracts by means of somatosensory evoked potentials (SSEP) in vivo in the symptomatic rats. We found a marked reduction in potential amplitudes indicating axonal conduction failure or transmission deficits predominantly ipsilateral of IgG application (Fig. 10A). Potential latencies were not prolonged which is in line with the observation that myelin was not damaged in the afflicted rats. As an attempt to better understand why these functional abnormalities took place despite morphologically intact structural components, we investigated the expression of NMDA glutamate receptor subunit NR1. NMDA receptors are most vulnerable to extracellular glutamate due to their calcium conductance and increased calcium influx upon receptor activation (Paoletti and Neyton, 2007; Traynelis et al., 2010). In the areas of pathogenic human IgG deposition overlapping with reduced EAAT2 immunoreactivity, we found a highly significant downregulation of NR1 immunoreactive spots and of the overall NR1 signal density possibly resulting from long-lasting perisynaptic glutamatergic overactivation (Fig. 10B).
Fig. 10.

Repetitive i.th. application of anti-AQP4 AB leads to functional consequences in nerve fiber transmission in evoked potential recording and to reduction of NMDA receptor NR1 expression at lesion sites. A: In electrophysiological SSEP recordings, peak N1 amplitudes were reduced after i.th. application of NMO IgG derived from two individual NMO patients (color-coded) but not after i.th. applied control IgG. In contrast, SSEP latencies to the first positive (P1) and negative deflections (N1) were unchanged compared to baseline values *p < 0.05, group comparison). Representative recordings are shown from one of total 28 rats with measurements taken ipsilateral to the side of IgG application, before and after completion of i.th. NMO-IgG applications on day 25. B: The expression of NMDA receptor NR1 subunit was reduced ipsilateral to the catheter ending in comparison to the contralateral side. The amount of NR1 immunoreactive spots and the signal density of NR1-positive structures were diminished after i.th. application of NMO-IgG (middle column) and similarly so with rABAQP4 (right column; scale bar: 50 μm) (for interpretation of the references to color in this figure legend, the reader is referred to the web version of the article).
Discussion
Previous seminal studies have led the groundwork for our current understanding of NMO as an autoimmune disease of the CNS separate from multiple sclerosis (Bennett et al., 2009; Bradl et al., 2009; Lennon et al., 2004, 2005; Roemer et al., 2007; Saadoun et al., 2010; Zhang et al., 2011). Experiments in cell culture and in living animals have demonstrated the important role of ABs to AQP4 in the complex scenario of NMO immunopathology. Some recent publications reported experimental evidence for direct, non-complement dependent actions of AB against AQP4 in vitro (Hinson et al., 2008, 2012; Marignier et al., 2010). However, the intrinsic pathogenicity of AQP4-ABs per se, i.e. without the multifactorial activation of proinflammatory events, has not been elucidated in living animals.
Direct pathogenic effects of IgG-ABs acting on their specific target antigens without co-activation of complement- or cell-mediated cytotoxicity have recently also been shown in other CNS disease models, i.e. in the stiff person-syndrome with ABs to amphiphysin (Geis et al., 2010, 2012) and in NMDA-receptor associated limbic encephalitis (Gleichman et al., 2012; Hughes et al., 2010). IgG antibodies derived from patients with these CNS disorders resulted in impaired function of the respective target proteins, either in presynaptic GABAergic dysfunction (Geis et al., 2010, 2012) or in receptor internalization and prolonged open times of the NMDA-receptor cation channel, respectively (Gleichman et al., 2012; Hughes et al., 2010).
We utilized an established rat model that allows the study of CNS-targeted ABs over longer time in situ in the spinal cord (Geis et al., 2010, 2011, 2012). Using the i.th. passive transfer model, we found that the chronic administration of NMO-IgG or anti-AQP4-specific rABs induced a reversible spinal cord disorder in recipient rats. We did not obtain immunohistological evidence of complement activation nor did we find inflammatory cell infiltration in areas of high NMO-IgG deposition precluding a relevant role for complement and other effector activation in our rat passive transfer model. Within a few days of i.th. AB administration we observed signs of a progressive spinal cord disease. Histologically, this was characterized by loss of AQP4, EAAT2, and NR1 immunoreactivity in areas of NMO-IgG deposition and astrogliosis but no astrocyte loss or demyelination.
The time course of our rat passive transfer model with slowly developing and potentially reversible disease signs is basically different from the characteristic disease course in the previously described autoimmune NMO animal models (Bennett et al., 2009; Bradl et al., 2009; Saadoun et al., 2010). This is not unexpected since we intended to study the intrinsic effects of IgG-ABs in isolation. Collectively, the observations in our rat model represent one aspect of the multifaceted nature of the immune reaction in NMO models. Very recently, “penumbra”-like lesions of AQP4 loss in preserved astrocytes have also been demonstrated after single intracerebral injection of anti-AQP4 AB in rats (Asavapanumas et al., 2014).
In this context it now becomes likely that CNS pathology in NMO may be more complex than previously appreciated. Recently, in an autopsy study, Saji et al. 2013 reported the loss of AQP4 in cortical grey matter accompanied by the presence of hypertrophic, GFAP-positive astrocytes, in the absence of complement deposition or demyelination. Another autopsy study found similar histopathology (type 4 lesion) in the vicinity of so-called typical NMO lesions in the spinal cord (Misu et al., 2013). Moreover, non-destructive injury due to intrinsic effects of AB to AQP4 may be the histopathological correlate of prominent MRI signal abnormality that is noted in some NMO patients with minimal neurologic signs (Asgari et al., 2011) and it may be one reason for substantial and fast recovery in some patients with relapses and successful treatment with plasma exchange.
After up to 3 weeks of applying purified NMO-patient IgG or anti-AQP4 rABs i.th. we found significantly reduced AQP4 and EAAT2 expression in spinal cord lesion areas but no loss of GFAP-positive astrocytes. It is unclear by what mechanism NMO-IgG binding leads to the reduction of EAAT2 expression. The reduction in EAAT2 expression could be (1) the result of a disruption of the AQP4-EAAT2 association (Hinson et al., 2008) or (2) a consequence of complex neuroinflammatory signalling by microglia or activated astrocytes (Graeber, 2010; Prinz and Priller, 2014; Walsh et al., 2014). Assuming a reduced clearance of perisynaptic glutamate due to glial transporter dysfunction, excessive extracellular glutamate may result in chronic overactivation and subsequent downregulation of surface expression of NMDA receptors (Paoletti and Neyton, 2007; Traynelis et al., 2010). These receptors are particularly vulnerable to glutamatergic oversupply due to increased calcium influx upon prolonged receptor activation (Nakanishi, 1992). While we propose that reduced NR1 receptor expression contributes to the behavioral and neurophysiological abnormalities observed in this model, future experiments are needed addressing the transporter currents in living astrocytes directly which may help to determine whether the effects of anti-AQP4 ABs on EAAT2 expression exert a functionally relevant action on glutamate transporter function.
Conclusions
I.th. administration of ABs against AQP4 can induce a potentially reversible spinal cord disorder in vivo that is concentration dependent and independent of complement activation. Spinal cord pathology is characterized by novel histopathologic changes. These findings may be the experimental correlate of distinct, presumably AB-mediated histopathological changes described in recent autopsy studies (Misu et al., 2013; Saji et al., 2013) that are clearly different from the lesions described in animal models with full complement activation and tissue destruction and from lesions in patients with advanced NMO. These results are in support of the experimental treatment strategies aiming at effective therapeutic strategies for NMO by reducing antibody-mediated pathology (Saadoun et al., 2012; Tradtrantip et al., 2012a, 2012b, 2013).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This work was supported by a grant from the Interdisciplinary Clinical Research Center (IZKF) of the University of Würzburg School of Medicine (to CG N-151), a neuroimmunology research grant by Merck-Serono, Germany (K.V.T. and C.G.), by the Integrated Research and Treatment Center for Sepsis Control and Care (CSCC, Jena to C.G.) and by intramural University Research Funds of the University of Würzburg School of Medicine. J.L.B. was supported by grants from the Guthy-Jackson Research Foundation and the National Institutes of Health (EY022936). K.V.T. is the recipient of a senior professorship awarded by the University of Würzburg Medical School. The authors declare no competing financial interests. We thank L. Biko, H. Bruenner, B. Dekant, S. Hellmig, V. Michels, C. Staudenmaier, and C. Varallyay for expert technical assistance. This project was inspired by Professor John Prineas, Sydney. We are grateful to Professor Hans Lassmann, Vienna, for helpful discussions and critical comments.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.expneurol.2014.12.015.
Conflict of interest
The authors declare no competing financial interests.
References
- Asavapanumas N, Ratelade J, Verkman AS. Unique neuromyelitis optica pathology produced in naive rats by intracerebral administration of NMO-IgG. Acta Neuropathol. 2014;127:539–551. doi: 10.1007/s00401-013-1204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari N, Lillevang ST, Skejoe HP, Falah M, Stenager E, Kyvik KO. A population-based study of neuromyelitis optica in Caucasians. Neurology. 2011;76:1589–1595. doi: 10.1212/WNL.0b013e3182190f74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendszus M, Ladewig G, Jestaedt L, Misselwitz B, Solymosi L, Toyka K, Stoll G. Gadofluorine Menhancement allows more sensitive detection of inflammatory CNS lesions than T2-w imaging: a quantitative MRI study. Brain. 2008;131:2341–2352. doi: 10.1093/brain/awn156. [DOI] [PubMed] [Google Scholar]
- Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, Gilden D, Nessler S, Stadelmann C, Hemmer B. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009;66:617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc F, Zephir H, Lebrun C, Labauge P, Castelnovo G, Fleury M, Sellal F, Tranchant C, Dujardin K, Vermersch P, de Seze J. Cognitive functions in neuromyelitis optica. Arch. Neurol. 2008;65:84–88. doi: 10.1001/archneurol.2007.16. [DOI] [PubMed] [Google Scholar]
- Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, Adzemovic M, Bauer J, Berger T, Fujihara K, Itoyama Y, Lassmann H. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann. Neurol. 2009;66:630–643. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- Buchwald B, Ahangari R, Weishaupt A, Toyka KV. Intravenous immunoglobulins neutralize blocking antibodies in Guillain–Barre syndrome. Ann. Neurol. 2002;51:673–680. doi: 10.1002/ana.10205. [DOI] [PubMed] [Google Scholar]
- Buchwald B, Ahangari R, Weishaupt A, Toyka KV. Presynaptic effects of immunoglobulin G from patients with Lambert–Eaton myasthenic syndrome: their neutralization by intravenous immunoglobulins. Muscle Nerve. 2005;31:487–494. doi: 10.1002/mus.20269. [DOI] [PubMed] [Google Scholar]
- Crane JM, Rossi A, Gupta T, Bennett JL, Verkman AS. Orthogonal array formation by human aquaporin-4: examination of neuromyelitis optica-associated aquaporin-4 polymorphisms. J. Neuroimmunol. 2011;236:93–98. doi: 10.1016/j.jneuroim.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga H, Engel AG, Lang B, Newsom-Davis J, Vincent A. Passive transfer of Lambert–Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc. Natl. Acad. Sci. U. S. A. 1983;80:7636–7640. doi: 10.1073/pnas.80.24.7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geis C, Weishaupt A, Hallermann S, Grunewald B, Wessig C, Wultsch T, Reif A, Byts N, Beck M, Jablonka S, Boettger MK, Uceyler N, Fouquet W, Gerlach M, Meinck HM, Siren AL, Sigrist SJ, Toyka KV, Heckmann M, Sommer C. Stiff person syndrome-associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain. 2010;133:3166–3180. doi: 10.1093/brain/awq253. [DOI] [PubMed] [Google Scholar]
- Geis C, Weishaupt A, Grunewald B, Wultsch T, Reif A, Gerlach M, Dirkx R, Solimena M, Perani D, Heckmann M, Toyka KV, Folli F, Sommer C. Human stiff-person syndrome IgG induces anxious behavior in rats. PLoS One. 2011;6:e16775. doi: 10.1371/journal.pone.0016775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geis C, Grunewald B, Weishaupt A, Wultsch T, Toyka KV, Reif A, Sommer C. Human IgG directed against amphiphysin induces anxiety behavior in a rat model after intrathecal passive transfer. J. Neural Transm. 2012;119:981–985. doi: 10.1007/s00702-012-0773-3. [DOI] [PubMed] [Google Scholar]
- Gleichman AJ, Spruce LA, Dalmau J, Seeholzer SH, Lynch DR. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J. Neurosci. 2012;32:11082–11094. doi: 10.1523/JNEUROSCI.0064-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB. Changing face of microglia. Science. 2010;330:783–788. doi: 10.1126/science.1190929. [DOI] [PubMed] [Google Scholar]
- Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL, Howe CL, Pittock SJ, Lennon VA. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J. Exp. Med. 2008;205:2473–2481. doi: 10.1084/jem.20081241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, Fallier-Becker P, Noell S, Lennon VA. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc. Natl. Acad. Sci. U. S. A. 2012;109:1245–1250. doi: 10.1073/pnas.1109980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, Parsons TD, Lynch DR, Dalmau J, Balice-Gordon RJ. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 2010;30:5866–5875. doi: 10.1523/JNEUROSCI.0167-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linker RA, Maurer M, Gaupp S, Martini R, Holtmann B, Giess R, Rieckmann P, Lassmann H, Toyka KV, Sendtner M, Gold R. CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuro-inflammation. Nat. Med. 2002;8:620–624. doi: 10.1038/nm0602-620. [DOI] [PubMed] [Google Scholar]
- Marignier R, Nicolle A, Watrin C, Touret M, Cavagna S, Varrin-Doyer M, Cavillon G, Rogemond V, Confavreux C, Honnorat J, Giraudon P. Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain. 2010;133:2578–2591. doi: 10.1093/brain/awq177. [DOI] [PubMed] [Google Scholar]
- Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, Takahashi T, Nakashima I, Takahashi H, Itoyama Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain. 2007;130:1224–1234. doi: 10.1093/brain/awm047. [DOI] [PubMed] [Google Scholar]
- Misu T, Hoftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S, Nakashima I, Konno H, Bradl M, Garzuly F, Itoyama Y, Aoki M, Lassmann H. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013;125:815–827. doi: 10.1007/s00401-013-1116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- Ratelade J, Bennett JL, Verkman AS. Evidence against cellular internalization in vivo of NMO-IgG, aquaporin-4, and excitatory amino acid transporter 2 in neuromyelitis optica. J. Biol. Chem. 2011;286:45156–45164. doi: 10.1074/jbc.M111.297275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861–872. doi: 10.1007/s00401-012-0986-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W, Mandler RN, Weinshenker BG, Pittock SJ, Wingerchuk DM, Lucchinetti CF. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130:1194–1205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- Rossi A, Ratelade J, Papadopoulos MC, Bennett JL, Verkman AS. Neuromyelitis optica IgG does not alter aquaporin-4 water permeability, plasma membrane M1/M23 isoform content, or supramolecular assembly. Glia. 2012;60:2027–2039. doi: 10.1002/glia.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, MacDonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann. Neurol. 2012;71:323–333. doi: 10.1002/ana.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saji E, Arakawa M, Yanagawa K, Toyoshima Y, Yokoseki A, Okamoto K, Otsuki M, Akazawa K, Kakita A, Takahashi H, Nishizawa M, Kawachi I. Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann. Neurol. 2013;73:65–76. doi: 10.1002/ana.23721. [DOI] [PubMed] [Google Scholar]
- Toyka KV, Drachman DB, Griffin DE, Pestronk A, Winkelstein JA, Fishbeck KH, Kao I. Myasthenia gravis. Study of humoral immune mechanisms by passive transfer to mice. N. Engl. J. Med. 1977;296:125–131. doi: 10.1056/NEJM197701202960301. [DOI] [PubMed] [Google Scholar]
- Tradtrantip L, Zhang H, Anderson MO, Saadoun S, Phuan PW, Papadopoulos MC, Bennett JL, Verkman AS. Small-molecule inhibitors of NMO-IgG binding to aquaporin-4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB J. 2012a;26:2197–2208. doi: 10.1096/fj.11-201608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, Bennett JL, Verkman AS. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann. Neurol. 2012b;71:314–322. doi: 10.1002/ana.22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L, Ratelade J, Zhang H, Verkman AS. Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti-aquaporin-4 immunoglobulin G into therapeutic antibody. Ann. Neurol. 2013;73:77–85. doi: 10.1002/ana.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014;15:84–97. doi: 10.1038/nrn3638. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- Wrzos C, Winkler A, Metz I, Kayser DM, Thal DR, Wegner C, Bruck W, Nessler S, Bennett JL, Stadelmann C. Early loss of oligodendrocytes in human and experimental neuromyelitis optica lesions. Acta Neuropathol. 2014;127:523–538. doi: 10.1007/s00401-013-1220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol. Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bennett JL, Verkman AS. Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann. Neurol. 2011;70:943–954. doi: 10.1002/ana.22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.