

Abstract
Chemokines are important modulators of neuroinflammation and neurodegeneration. In the brains of Alzheimer’s disease (AD) patients and in AD animal models, the chemokine CXCL10 is found in high concentrations, suggesting a pathogenic role for this chemokine and its receptor, CXCR3. Recent studies aimed at addressing the role of CXCR3 in neurological diseases indicate potent, but diverse, functions for CXCR3. Here, we examined the impact of CXCR3 in the amyloid precursor protein (APP)/presenilin 1 (PS1) transgenic mouse model of AD. We found that, compared with control APP/PSI animals, plaque burden and Aβ levels were strongly reduced in CXCR3-deficient APP/PS1 mice. Analysis of microglial phagocytosis in vitro and in vivo demonstrated that CXCR3 deficiency increased the microglial uptake of Aβ. Application of a CXCR3 antagonist increased microglial Aβ phagocytosis, which was associated with reduced TNF-α secretion. Moreover, in CXCR3-deficient APP/PS1 mice, microglia exhibited morphological activation and reduced plaque association, and brain tissue from APP/PS1 animals lacking CXCR3 had reduced concentrations of proinflammatory cytokines compared with controls. Further, loss of CXCR3 attenuated the behavioral deficits observed in APP/PS1 mice. Together, our data indicate that CXCR3 signaling mediates development of AD-like pathology in APP/PS1 mice and suggest that CXCR3 has potential as a therapeutic target for AD.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative brain disorder characterized by the formation of β-amyloid plaques, predominantly in hippocampal and cortical regions (1, 2). Periplaque activation of microglia and astrocytes and induction of proinflammatory molecules suggest a pathogenic role for inflammation in this disease (3, 4). Microglia are resident CNS cells with immune-modulating and phagocytic capabilities (5, 6). Recent studies indicate that the microglial state of activation can determine whether these cells have a protective or detrimental role in AD (7–13). Microglia can generate reactive oxygen species and secrete proinflammatory cytokines and additional neurotoxic factors, which contribute to the pathology of AD (3, 4, 14). In addition, microglia also release Aβ-degrading enzymes and express scavenger receptors, which can mediate Aβ phagocytosis (15–17). There is compelling evidence that microglial cells can modulate the pathological course of AD, although the exact role of microglia in AD remains to be clarified.
Chemokines are cytokines that orchestrate innate and adaptive immune responses and are found to be highly upregulated in several neuroinflammatory disorders (18). CXCL9, CXCL11, and, in particular, CXCL10, are prominent members of the non-ELY CXC chemokines (19). They share the receptor CXCR3 (20, 21), expressed on T cells and NK cells and on resident cells, including neurons, as well (22–29). CXCR3 can be differentially activated by CXCL9, CXCL10, and CXCL11 (21, 30). IFN-γ and TNF-α are major inducers and regulators of both CXCR3 and CXCR3 ligands (31–35).
Previous studies using murine AD models have demonstrated that chemokine receptor systems such as CCR5 (36), CCR2 (37–39), and CX3CR1 (40–43) can modulate the disease course by influencing microglial function, accumulation, and clustering (37, 40–42). In addition, a positive correlation between CXCL10 concentrations in the cerebrospinal fluid and cognitive impairment in AD patients has been demonstrated (44, 45). Moreover, CXCL10 was found to be expressed in astrocytes in AD (27) and detected in close proximity to Aβ plaques in a corresponding AD mouse model (46).
To characterize the role of the CXCR3 chemokine system for AD pathogenesis, we examined the impact of CXCR3 deficiency in amyloid precursor protein (APP)/presenilin 1 (PS1) transgenic mice. This model displays several pathological cellular and behavioral characteristics of AD, including progressive accumulation of cerebral amyloid plaques accompanied by clustering of reactive microglia and astrocytes around amyloid plaques (47–49) and cognitive impairment (13, 50).
Results
Decreased Aβ deposition and Aβ level in CXCR3-deficient APP/PS1 mice.
The APP/PS1 transgenic model exhibits an increase in plaque burden between the ages of 4 and 12 months (51). Therefore, APP/PS1 and APP/PS1/Cxcr3–/– animals were examined in the early stage of Aβ deposition (5 months) and at the stage of compact and diffuse plaque burden (8 months). A widespread distribution of Aβ plaques stained with thioflavin S (ThioS) was found throughout the hippocampus and cerebral cortex of 8-month-old male APP/PS1 mice (Figure 1A). In contrast, APP/PS1/Cxcr3–/– mice revealed a strong reduction in Aβ plaque burden in both regions at 8 months of age. Quantification of the cerebral and hippocampal ThioS+ area (Figure 1B) revealed a strong reduction of Aβ plaques in APP/PS1/Cxcr3–/– animals compared with APP/PS1 mice at both 5 (cerebral cortex, APP/PS1: 0.074% ± 0.009% vs. APP/PS1/Cxcr3–/–: 0.020% ± 0.002%, P < 0.001; hippocampus, APP/PS1: 0.047% ± 0.008% vs. APP/PS1/Cxcr3–/–: 0.004% ± 0.001%, n = 5 mice per group, 16–20 sections, P < 0.001) and 8 months of age (cerebral cortex, APP/PS1: 0.589% ± 0.082% vs. APP/PS1/Cxcr3–/–: 0.047% ± 0.007%, P <0.001; hippocampus, APP/PS1: 0.634% ± 0.145% vs. APP/PS1/Cxcr3–/–: 0.044% ± 0.011%, n = 5 mice per group, 16–20 sections, P < 0.001). Plaque reduction in APP/PS1/Cxcr3–/– mice observed with ThioS was further confirmed by anti-Aβ immunohistochemistry (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI66771DS1). In addition, analysis of the plaque morphology revealed a decreased value for the form factor in CXCR3-deficient APP/PS1 brains (14–19 sections/group, P < 0.05) (Supplemental Figure 1).
Figure 1. CXCR3 deficiency leads to a strong reduction of Aβ deposition in APP/PS1 mice.

(A) Brain sections of 8-month-old APP/PS1 and APP/PS1/Cxcr3–/– male mice were stained with ThioS to detect Aβ deposition. Scale bar: 500 μm. (B) Hippocampal and cortical regions were quantified. (C) ELISA measurement of Aβ1–40 and Aβ1–42 peptides documents a reduction in the insoluble brain fraction of APP/PS1/Cxcr3–/– mice. (D) At 5 months (5 mo), no significant changes in the composition of both soluble peptides were observed in APP/PS1/Cxcr3–/– compared with APP/PS1 mice. At 8 months, both Aβ peptides are significantly reduced in APP/PS1/Cxcr3–/– mice. (E) A scatter plot of soluble Aβ1–40/1–42 composition in 5- and 8-month-old APP/PS1 and APP/PS1/Cxcr3–/– mice is shown. Immunoblot analysis using a holo-APP antibody (CT15) indicates no effect of CXCR3 deficiency on APP processing in APP/PS1 mice at 5 months. (F) Densitometric analysis of holo-APP, α-CTFs, and β-CTFs after normalization to α-tubulin. (G) Unaltered Aβ1–40 and Aβ1–42 secretion of primary cortical APP/PS1/Cxcr3–/– neurons compared with the APP/PS1 genotype. n = 4 cultures of both genotypes. (H) Cxcr3 RNA detection by qPCR confirms the lack of CXCR3 in primary cultured neurons, astrocytes, and microglia. (A–F) Data are shown as mean ± SEM, n = 5–8 mice per group. (H) Data are shown as mean ± SEM, n = 3–5 primary cultures in each group. *P < 0.05; **P < 0.005; ***P < 0.001.
Regarding Aβ peptide concentrations, we found a significantly lower Aβ1–42 concentration in the insoluble SDS fraction of 5-month-old APP/PS1/Cxcr3–/– mice (Aβ1–40: 15.6 ± 0.2 pg/mg, Aβ1–42: 7.8 ± 0.8 ng/mg; n = 5) compared with APP/PS1 mice (Aβ1–40: 26.2 ± 6.3 pg/mg, Aβ1–42: 49.4 ± 8.3 ng/mg; n = 5; P < 0.005 for Aβ1–42; Figure 1C). We found a 5-fold reduction of insoluble Aβ level (Aβ1–40: 32.8 ± 4.7 ng/mg, Aβ1–42: 31.6 ± 1.8 ng/mg; n = 5) in APP/PS1/Cxcr3–/– versus (Aβ1–40: 166.0 ± 41.0; Aβ1–42: 113.5 ± 36.2 ng/mg; n = 7) APP/PS1 mice (P < 0.001; *P < 0.05; Figure 1D), reflecting the strong decrease of ThioS+ Aβ deposition at 8 months. No difference in soluble Aβ isolated from the RIPA fraction was observed at 5 months. However, at 8 months, CXCR3 deficiency showed lowered soluble Aβ peptide concentrations in APP/PS1 mice (APP/PS1: Aβ1–40: 127.8 ± 4.2 pg/mg, Aβ1–42: 59.8 ± 3.5 pg/mg; n = 5 vs. APP/PS1 Cxcr3–/–: Aβ1–40: 85.4 ± 4.2 pg/mg, Aβ1–42: 47.4 ± 2.1 pg/mg; n = 7; P < 0.001 for Aβ1–40; P < 0.05 for Aβ1–42; Figure 1D). In pilot experiments, the characterization of female APP/PS1/Cxcr3+/– mice (n = 4) resulted in the same Aβ peptide concentrations as observed in APP/PS1/Cxcr3+/+ and therefore did not indicate a gene-dosage effect of CXCR3 (Supplemental Figure 2). In summary, CXCR3 deficiency reduces the amyloid plaque deposition and Aβ1–42/Aβ1–40 levels in the APP/PS1 mouse model of AD.
CXCR3 deficiency does not alter APP processing in APP/PS1/Cxcr3–/– mice.
Generally, neurons express functionally relevant CXCR3 (27, 28, 52–54). To examine the impact of CXCR3 on neuronal APP processing as a possible cause of reduced plaque load, we determined the protein concentrations of APP and the APP cleavage products β–C-terminal fragment (β-CTF) and α-CTF in 5-month-old WT, Cxcr3–/–, APP/PS1, and APP/PS1/Cxcr3–/– mice using immunoblot detection with antibody CT15. As expected, control WT and Cxcr3–/– samples showed a weak band of endogenous APP and a weak signal for α- and β-CTF cleavage fragments (Figure 1F). We found no differences in the normalized band intensity of holo-APP between APP/PS1 and APP/PS1/Cxcr3–/– animals (APP/PS1: 2.1 ± 0.3 vs. APP/PS1/Cxcr3–/–: 1.9 ± 0.2 normalized band intensity; Figure 1F). In addition, the concentrations of β-CTF (APP/PS1: 0.3 ± 0.0 vs. APP/PS1/Cxcr3–/–: 0.3 ± 0.0 normalized band intensity) and α-CTF (APP/PS1: 0.3 ± 0.0 vs. APP/PS1/Cxcr3–/–: 0.2 ± 0.0 normalized band intensity; n = 5 each; Figure 1F) were equivalent and showed no effect of CXCR3 deficiency on APP processing. This result was confirmed by detecting β-CTFs using the 82E1 antibody resulting in equal CTF levels (Supplemental Figure 3).
Finally, the secretion of Aβ1–40 and Aβ1–42 from primary neuronal cultures was measured. Here, similar levels of both Aβ peptides (APP/PS1: 1.45 ± 0.17 ng/ml vs. APP/PS1/Cxcr3–/–: 1.50 ± 0.13 ng/ml; Aβ1–40/1–42 ratio; n = 5 each) were detected in supernatants from neuronal cultures, supplying further evidence against a role for CXCR3 in APP processing (Figure 1G).
CXCR3 modulates the in vivo phenotype of microglia and astrocytes in APP/PS1 transgenic mice.
CXCR3 is a well-known modulator of microglial function (23, 25, 26, 55). To elucidate the impact of Cxcr3–/– on the morphological phenotype of microglia in APP/PS1 mice, we characterized periplaque accumulation and activation by morphological criteria. Using combined Aβ-plaque staining with ThioS and IBA1 immunofluorescent labeling of microglia, we specifically detected plaque-associated microglia/macrophages in APP/PS1 and APP/PS1/Cxcr3–/– mice.
In APP/PS1 mice, microglia mostly clustered around Aβ plaques (periplaque position). In CXCR3-deficient APP/PS1 mice, the periplaque accumulation of microglia was strikingly diminished (Figure 2A). We also determined the soma area of microglia as a morphological marker of activation and found a strong reduction of the microglia soma area in APP/PS1/Cxcr3–/– animals compared with APP/PS1 mice in both regions of interest (ROIs) (Figure 2, B and C; cerebral cortex, APP/PS1: 77.2 ± 2.4 μm vs. APP/PS1/Cxcr3–/–: 54.6 ± 1.7 μm, P < 0.001, n ≥ 117; hippocampus, APP/PS1: 71.6 ± 2.1 μm vs. APP/PS1/Cxcr3–/–: 48.7 ± 1.6 μm, n ≥ 154 microglia per group, P < 0.001).
Figure 2. CXCR3 deficiency reduces plaque-associated microglial accumulation, periplaque activation of astrocytes, and levels of inflammatory transcripts in APP/PS1 mice.

(A) Brain sections of APP/PS1 and APP/PS1/Cxcr3–/– mice immunostained with the microglial markers Iba1 and ThioS. Reduced microglial accumulation around Aβ deposition is visible in APP/PS1/Cxcr3–/– compared with the periplaque clusters of IBA1+ microglia in APP/PS1 brains. Microglia soma size was reduced in both frontal cortex (FC) and hippocampus (HC) of APP/PS1/Cxcr3–/– animals. (B and C) Bright-field analysis of Iba1 stain. (D) Immunoblot quantification of GFAP revealed reduced GFAP in APP/PS1/Cxcr3–/– mice compared with APP/PS1 mice. (E) Fewer hypertrophic GFAP+ astrocytes were found around ThioS+ Aβ plaques in APP/PS1/Cxcr3–/– mice compared with APP/PS1 mice. (F) mRNA levels, determined by qPCR, for CXCR3 ligands, inflammatory cytokines/chemokines, and microglial markers were reduced in APP/PS1/Cxcr3–/– mice, whereas the neurotrophic factor Bdnf was higher in APP/PS1/Cxcr3–/– brains. Data are shown as mean ± SEM, n = 4–8 mice per group. *P < 0.05; **P < 0.005; ***P < 0.001. Scale bars: 100 μm; 10 μm (insets).
Furthermore, we colocalized MHCII as an immunological activation marker with the microglial marker Iba1 and regularly found MHCII-positive microglia in the periplaque area, whereas MHCII-positive microglia were only rarely detected in APP/PS1/Cxcr3–/– mice (Supplemental Figure 4).
Astrocytes potentially contribute to the AD-like pathology in APP/PS1 mice. We characterized the astrocytic response in APP/PS1 and APP/PS1/Cxcr3–/– mice by immunoblots and immunohistochemistry against glial fibrillary acidic protein (GFAP), which was strongly reduced in brain tissue from APP/PS1/Cxcr3–/– compared with APP/PS1 mice (Figure 2D).
Histologically there were fewer GFAP-positive cells in APP/PS1/Cxcr3–/– mice compared with APP/PS1 animals. Double staining for GFAP and ThioS demonstrated that most amyloid plaques were surrounded by morphologically activated astrocytes in APP/PS1 mice. In APP/PS1/Cxcr3–/– animals, astrocytes were fewer in number and had less hypertrophic cell bodies (Figure 2E). Hence, CXCR3 deficiency strongly reduces microglial activation and clustering proximal to Aβ plaques. Moreover, morphologically activated astrocytes were strongly reduced in APP/PS1/Cxcr3–/– compared with APP/PS1 brain.
CXCR3 and the corresponding ligands CXCL9 and CXCL10 are induced in APP/PS1 transgenic mice.
To confirm a relevant induction of CXCR3 and its ligands, CXCL9 and CXCL10, in the model examined, we determined corresponding brain RNA concentrations by quantitative PCR (qPCR) analysis from APP/PS1 and APP/PS1/Cxcr3–/– mice. The induction of Cxcr3 transcripts in APP/PS1 mice (7.2 ± 3.0-fold increase, n = 5) relative to WT controls (Figure 2F) was confirmed. Analysis of CXCR3 ligand transcripts revealed elevated concentrations for Cxcl9 (5.8 ± 4.2 fold increase, n = 4) and, in particular, for Cxcl10 (36.7 ± 0.5-fold increase, n = 4) in APP/PS1 mice. As expected, Cxcr3 was not detectable in APP/PS1/Cxcr3–/– mice and, compared with APP/PS1 mice, the levels of the receptor ligands Cxcl9 and Cxcl10 were strikingly diminished (Figure 2F and Supplemental Table 1). Using RNA from primary isolated neurons, astrocytes, and microglia, we confirmed that indeed all cells from Cxcr3–/– mice are CXCR3 deficient (Figure 1H).
CXCR3 modulates the cerebral cytokine milieu of APP/PS1 mice.
The cerebral cytokine levels of APP/PS1/Cxcr3–/– and APP/PS1 mice were examined to further characterize the effect of CXCR3 on a molecular level. The most relevant findings are displayed in Figure 2F. We found remarkably lower transcript levels for Tnfa, Fasl, and Il1b. The RNA concentrations of the microglial activation markers Cd11b and Cd68 were also lower in APP/PS1/Cxcr3–/– versus APP/PS1 mice, further corroborating the finding that microglia are differentially activated in APP/PS1/Cxcr3–/– mice. The determination of cerebral chemokine RNA in APP/PS1 mice revealed the highest level of induction for Cxcl10 and Ccl3 (37-fold vs. 15-fold, respectively, compared with WT controls), whereas other cytokines, such as Cx3cl1, Ccl5, and Ccl19, were only moderately induced. Regarding the chemokine receptors in the APP/PS1 model, we found the highest RNA potency for Cxcr3 (6-fold induction) followed by Cxcr4 (4-fold induction) and Ccr2 (3-fold induction). Comparing the cerebral cytokine milieu of APP/PS1/Cxcr3–/– and APP/PS1 mice, RNA quantities of the inducible cyto- and chemokines in APP/PS1/Cxcr3–/– brain tissue were greatly reduced. However, the cerebral level of the growth factor Bdnf was upregulated in APP/PS1/Cxcr3–/– compared with APP/PS1 mice. Other growth factors, such as Ntf3 and Ntf5, were not significantly regulated (Supplemental Table 1). In summary, the qPCR analysis demonstrates that CXCR3 strongly modulates the cerebral inflammatory milieu in APP/PS1 mice.
Morris water-maze testing displayed improved spatial memory of CXCR3-deficient APP/PS1 animals compared with CXCR3-competent controls.
To investigate whether the attenuated AD-like pathology is associated with improved memory performance, we assessed spatial memory in 8-month-old WT (n = 9), Cxcr3–/– (n = 12), APP/PS1 (n = 16), and APP/PS1/Cxcr3–/– (n = 18) animals using the Morris water-maze test.
All mice learned to find a hidden platform. Spatial learning and memory in comparisons of WT and Cxcr3–/– animals at 8 months of age were unaltered (Figure 3, A–D). Compared with controls, APP/PS1 mice displayed impaired spatial learning with respect to distance travelled to the platform (APP/PS1: 259.8 ± 13.8 cm vs. APP/PS1/Cxcr3–/–: 213.1 ± 9.6 cm; 1-way ANOVA, Tukey’s test, P < 0.05, Figure 3B). CXCR3 deficiency prevented learning and memory dysfunction in APP/PS1 transgenic mice, as shown for the spatial trial on day 9 (Figure 3C; representative runs for all tested genotypes are shown in Figure 3D). Thus, APP/PS1/Cxcr3–/– animals were protected from the memory dysfunction observed in APP/PS1 animals. Visual cued testing confirmed equivalent visual acuity and ability to orient across all groups. In the open-field test, presence of the APP/PS1 transgene yielded greater locomotor activity in CXCR3-deficient and CXCR3-competent animals (Supplemental Figure 5). No differences in distance travelled were found between APP/PS1 and APP/PS1/Cxcr3–/– mice or between WT and Cxcr3–/– mice.
Figure 3. CXCR3 deficiency rescues memory impairment in APP/PS1 mice.

(A) Morris water-maze analysis of 8-month-old mice on 8 consecutive days. (B) Graphical evaluation of the area under the curve from A (mean of n = 9 for WT, n = 12 for CXCR3, n = 16 for APP/PS1, and n = 18 for APP/PS1/Cxcr3–/–). Data are shown as mean ± SEM, 1-way ANOVA followed by Tukey’s post-hoc test. (C) Results of the probe trial at day 9 (mean of n = 9 for WT, n = 12 for CXCR3, n = 16 for APP/PS1, and n = 18 for APP/PS1/Cxcr3–/–). Data are shown as mean ± SEM, Kruskal-Wallis rank sum test, Dunn’s post-hoc test). Data are given as the percentage of time spent in quadrant Q1, where the platform previously was located, compared with the average of the time the animals spent in the other quadrants (Q2–Q4). *P < 0.05; ***P < 0.001. (D) Representative runs from the probe trials.
Fibrillar Aβ stimulates microglia to produce TNF-α and CXCL10, while TNF-α but not Aβ induces CXCL10 production by astrocytes.
To determine the cellular source of TNF-α and CXCL10 in response to fibrillar Aβ1–42 (fAβ), primary microglia (PMG) and astrocytes were stimulated with fAβ and TNF-α. LPS-stimulated cultures served as positive controls. The supernatants of cultures stimulated for 4, 12, and 20 hours were analyzed for microglial/astroglial-derived TNF-α and CXCL10 by ELISA (Figure 4A). Stimulation of PMG led to an increase of TNF-α concentrations at 12 hours (105.0 ± 14.0 pg/ml) and 20 hours (264.0 ± 4.0 pg/ml) compared with unstimulated controls (52.8 ± 20.4 pg/ml after 12 hours, 180.0 ± 4.0 pg/ml after 20 hours; P < 0.005, Figure 4B).
Figure 4. Effect of fibrillar fAβ on the induction of microglial and astrocytic TNF-α and CXCL10 secretion.

PMG and astrocytes were stimulated with fAβ, LPS, and TNF-α for 4, 12, and 20 hours. Afterwards, primary culture supernatants were analyzed for secreted TNF-α and CXCL10 levels using ELISA. (A) Schematic diagram. (B) A significant induction of TNF-α is detectable from 12 hours of fAβ stimulation in microglia culture (12 and 20 hours). (C) No TNF-α induction could be found in astrocytes after fAβ treatment over all analyzed time points. (D) fAβ and LPS induced similar levels of CXCL10 secretion at 12 and 20 hours. (E) Primary astrocytes do not respond to fAβ with increased CXCL10 secretion compared with unstimulated controls. Administration of TNF-α strongly stimulates astrocytes to produce CXCL10 after 4 hours. Data are shown as mean ± SEM from 2 to 3 individual experiments. *P < 0.05; **P < 0.01; ***P < 0.001 (1-way ANOVA, Dunn’s post hoc test).
In contrast, primary astrocytes displayed only minimal TNF-α production in response to fAβ stimulation (fAβ, 8 hours: 0.1 ± 0.1 pg/ml vs. control, 8 hours: 0.4 ± 0.2 pg/ml; fAβ,12 hours: 3.5 ± 0.1 pg/ml vs. control, 12 hours: 0.4 ± 0.2 pg/ml; fAβ, 20 hours: 4.2 ± 0.4 pg/ml vs. control, 20 hours: 1.9 ± 0.1 pg/ml; Figure 4C).
Furthermore, microglial synthesis of CXCL10 in response to fAβ was determined. Microglia exposed to fAβ secreted more CXCL10 compared with untreated controls at all time points (P < 0.05, P < 0.005; Figure 4D). Moreover, fAβ was equally potent in inducing CXCL10 when compared with LPS at 12 hours and 24 hours (LPS, 12 hours: 433.5 ± 89.5 pg/ml vs. fAβ, 12 hours: 390.0 ± 84.0 pg/ml; LPS, 24 hours: 409.5 ± 33.5 pg/ml vs. fAβ, 24 hours: 491.0 ± 91.0 pg/ml). Additionally, astrocytic CXCL10 production after exposure to fAβ or TNF-α stimulation was determined. In contrast to the robust production of TNF-α by fAβ-stimulated microglia, astrocytes did not show increased CXCL10 production upon fAβ exposure. However, stimulation of primary astrocytes with TNF-α showed a stronger CXCL10 induction than that by LPS stimulation (Figure 4E, LPS, 12 hours: 2178.0 ± 13.0 pg/ml vs. fAβ, 12 hours: 2258.0 ± 15.0 pg/ml; LPS, 24 hours: 2316.0 ± 61.5 pg/ml vs. fAβ, 24 hours: 2739.0 ± 586.0 pg/ml).
In summary, fAβ-stimulated PMG represent a potent source of TNF-α and CXCL10. Astrocytes are not directly responsive to Aβ, but can contribute to production of CXCL10 when stimulated by TNF-α.
CXCR3 signaling reduces the Aβ phagocytosis of PMG, but increases the secretion of microglial TNF-α.
To examine the impact of CXCR3 on microglial phagocytosis of fAβ, we examined fluorescein amidite–Aβ (FAM-Aβ) uptake in WT and Cxcr3–/– PMG. Using quantitative imaging of FAM-Aβ+ particles in CD68+ microglia cells (Figure 5A), we found greater than 30% higher capacity of Cxcr3–/– microglia (area/cell = 82.1 ± 7.2 μm2, n = 55) to uptake FAM-Aβ than WT microglia (area/cell = 60.7 ± 7.6 μm2, n =43, P < 0.05, Figure 5A). To further elucidate whether CXCR3 signaling conversely reduces phagocytic capacity of WT microglia, microglial cells were pretreated with CXCL9 or CXCL10 and we examined the phagocytotic capacity of FAM-Aβ. Both CXCL9 and CXCL10 pretreatment reduced the phagocytic capacity of WT microglia (1.03 ± 0.04 of unstimulated control vs. 0.62 ± 0.13 of CXCL9 pretreatment and 0.52 ± 0.08 of CXCL10 pretreatment, normalized phagocytosis ± SEM; 3–5 individual experiments, Figure 5B), whereas no effect was observed in pretreated microglia of CXCR3-deficient mice (1.47 ± 0.07 of unstimulated control vs. 1.39 ± 0.09 of CXCL9 pretreatment and 1.50 ± 0.06 of CXCL10 pretreatment, normalized phagocytosis ± SEM; 3–5 individual experiments, Figure 5B). CXCR3 antagonist preincubation of WT microglia led to increased fAβ phagocytosis, to the level of Cxcr3–/– microglia (Figure 5D).
Figure 5. CXCR3 deficiency and CXCR3 antagonism enhance microglial phagocytosis of FAM-Aβ and reduce the production of TNF-α in vitro.

(A) Primary WT and Cxcr3–/– microglia (CD68, red signal) were incubated with or without 0.7 μM FAM-Aβ for 1 hour. Scale bar: 20 μm. (B) A phagocytosis assay revealed a significant increase of FAM-Aβ uptake in Cxcr3–/– compared with WT microglia. mCXCL9 and mCXCL10 (each 250 ng/ml) treatment of WT and Cxcr3–/– microglia diminished phagocytosis in WT but not in Cxcr3–/– cells. (C) Reduced TNF-α concentration in the cell supernatants of Aβ-stimulated Cxcr3–/– microglia cultures. (D) Functional blocking of CXCR3 with an antagonist enhances microglial phagocytosis to levels equal to those found in CXCR3-deficient microglia (CXCR3 antagonist ≥ 100 nM). (E) Analysis of the TNF-α level in CXCR3 antagonist–treated WT microglia exhibits a reduction of TNF-α secretion below WT control level when stimulated with Aβ. Data are shown as mean ± SEM from 3 to 5 individual experiments; *P < 0.05; **P < 0.01; ***P < 0.001 (1-way ANOVA, Dunn’s post hoc test).
To examine the influence of CXCR3 on the production of proinflammatory cytokines during Aβ stimulation further, we analyzed the culture supernatants from WT and Cxcr3–/– microglia after the incubation with fibrillar FAM-Aβ. WT cells secreted TNF-α 10-fold (220.4 ± 10.5 pg/ml) compared with CXCR3-deficient cells (19.1 ± 1.7 pg/ml, P <0.001, Figure 5C). In addition, baseline TNF-α secretion in control Cxcr3–/– (5.9 ± 3.8 pg/ml) was lower compared with that of control WT cells (46.1 ± 14.4 pg/ml, Figure 5C). CXCR3 antagonist–treated WT microglia lowered TNF-α production after fAβ stimulation (18.8 ± 5.5 pg/ml, 100 nM) compared with stimulation without the CXCR3 antagonist (32.7 ± 1.1 pg/ml, 100 nM, Figure 5E). In summary, CXCR3 deficiency yielded increased phagocytotic capacity of microglia and reduced expression of TNF-α. CXCL9/10-CXCR3 signaling can reduce the phagocytic ability in WT, but not in Cxcr3–/–, microglia.
In vivo methoxy-XO4 labeling of Aβ exhibits increased microglial Aβ phagocytosis in CXCR3-deficient APP/PS1 mice.
To verify the impact of CXCR3 on microglial phagocytosis in vivo, APP/PS1 and APP/PS1/Cxcr3–/– mice were treated with methoxy-XO4 (a fluorescent derivative of Congo red, which crosses the blood-brain barrier (BBB) and has high Aβ-binding affinity). Adult microglial cells were isolated and analyzed for methoxy-XO4 fluorescence by flow cytometry as previously described (13). Aβ (IC16) immunofluorescence stainings of methoxy-XO4 brains demonstrated the intense signal of methoxy-XO4–labeled Aβ deposition in both APP/PS1 and APP/PS1/Cxcr3–/– animals (Figure 6A). FACS analysis revealed increased Aβ in microglia from APP/PS1/Cxcr3–/– (1645 ± 179, mean fluorescence intensity [MFI], mean ± SEM, n = 5, P < 0.05) compared with what was found in APP/PS1 mice (1244 ± 54, MFI, mean ± SEM, n = 5; Figure 6B). The percentage of methoxy-XO4+CD11b+CD45+ microglia in APP/PS1/Cxcr3–/– animals slightly increased (32.1% ± 4.2%, mean ± SEM, n = 5) compared with that in APP/PS1 mice (26.9% ± 2.3%, mean ± SEM, n = 5, Figure 6B).
Figure 6. CXCR3 deficiency increases the microglial Aβ uptake in the APP/PS1 model and after intracerebral fAβ injection.

(A) Representative micrographs of microglia (Iba1) and Aβ (IC16) stainings together with methoxy-XO4–labeled Aβ in APP/PS1 and APP/PS1/Cxcr3–/– mice. Scale bar: 50 μm. (B) Using flow cytometry, Aβ uptake of CD11b+CD45+ microglia was found enhanced in APP/PS1/Cxcr3–/– animals compared with APP/PS1 animals (MFI). The percentage of methoxy-XO4+ microglia was slightly increased by CXCR3 deficiency. (C) Cxcr3–/– and WT mice were injected with fAβ, and brain sections were stained for IBA1/Aβ. Scale bar: 20 μm. (D) Quantification of IBA1+ microglia cells within the fAβ application center showed a higher number in WT than in Cxcr3–/– mice. (E) Large CD68+ lysosomes (red) containing Aβ immunoreactive content (green) were detected in Cxcr3–/– rather than in WT mice (arrowheads). Scale bars: 20 μm; 10 μm (insets). (F) Fluorescence intensities for CD68 (red stain and line) and Aβ (green stain and line) are shown next to the corresponding RGB image (simplified dimensioning arrow, distance 0-1 = 80 μm). RGB intensity profile localizes peak Aβ (green) intermediate to peak CD68 (red) fluorescence intensity. (G) CD68+ lysosomal content in Cxcr3–/– mice exhibits an increase in Aβ compared with WT mice. (H) A larger lysosomal perimeter was found in Cxcr3–/– microglia compared with WT. Data are shown as mean ± SEM, n = 5 mice per group. *P < 0.05; **P < 0.005; ***P < 0.001.
Intracerebral fAβ injection reveals an enhanced microglial phagocytosis and differences in clustering in Cxcr3–/– mice compared with WT controls.
To evaluate our findings in a second experimental approach not based on the APP/PS1 transgenic model, we injected fibrillar Aβ1–42 into brains of WT and Cxcr3–/– animals (Figure 6, C–H). First, quantification of microglia within an 80-μm radius proximal to the fAβ injection site revealed less microglial accumulation, by 20%, in Cxcr3–/– animals compared with WT mice (WT Aβ: 18.3 ± 0.9 cells vs. Cxcr3–/– Aβ: 14.0 ± 0.7 cells; microglia around fAβ application, n = 17–23 sections out of 4 animals per genotype; Figure 6D). Sham injection showed no difference in respect to the total number of microglia between WT and Cxcr3–/– animals (WT Tris: 11.6 ± 0.8 vs. Cxcr3–/– Tris: 11.0 ± 0.9; microglia around fAβ application site, n = 5 sections of 1 control animal per genotype; Figure 6D). Secondly, intracellular fAβ in CD68+ phagolysosomes from Cxcr3–/– mice was increased compared with that in WT mice (WT Aβ: 50.2 ± 3.1 vs. Cxcr3–/– Aβ: 65.1 ± 3.6, arbitrary units, n ≥ 141 lysosomes per genotype; Figure 6, E–G). Notably, the microglial cells of Cxcr3–/– animals exhibited less ramified morphology, with retracted branches and a more large rounded morphology, with large Iba1-negative vacuoles (Figure 6E). These changes correlated with a larger size of CD68+ lysosomes in these cells (Figure 6H). The lysosomal perimeter of microglia in close proximity to the injected fAβ deposition had a mean size difference of more than 30% in Cxcr3–/– lysosomes compared with WT lysosomes (WT Aβ: 5.6 ± 0.1 μm, n = 717 vs. Cxcr3–/– Aβ: 7.4 ± 0.2 μm, n ≥ 587 lysosomes per genotype; Figure 6H). Using a RGB fluorescence intensity profile, we confirmed the intralysosomal location of fibrillar Aβ (Figure 6F).
Discussion
The present study provides insight into the functional role of the CXCR3 chemokine system on the progression of AD-like pathology in the APP/PS1 AD mouse model. The rationale for our experiments was the recent detection of high levels of CXCL10 in cerebrospinal fluid and brain tissue from AD patients and corresponding animal models, suggesting a role for this chemokine and its receptor, CXCR3, in AD (27, 44, 45). We found strong attenuation of Aβ plaque formation and strikingly diminished Aβ peptide concentrations in brain tissue from CXCR3-deficient APP/PS1 mice, indicating a critical role of CXCR3 in the generation of AD-like pathology in this model. Reduced Aβ pathology in APP/PS1/Cxcr3–/– mice correlated with less severe behavioral impairment, demonstrating that the observed morphological changes may also have a functional impact. A growing number of studies have suggested that Aβ deposition in AD and AD-like models is modulated by the function and activation state of microglia and astrocytes (13, 14, 29, 56, 57). Resident glial cells of the brain functionally express CXCR3, but the impact of glial CXCR3 expression on AD progression is not yet clarified (23–26).
In APP/PS1 mice deficient for CXCR3, we found a strong modulation of microglial and astrocytic activation by both morphological and molecular criteria. This was independent of the local plaque load and was observed around even very large Aβ plaques in aged animals. This indicates primary modulation of the glial response in APP/PS1/Cxcr3–/– mice and not just less-activated glia due to generally reduced plaque load. Consistent with findings for the APP/PS1 model, we detected a distinct microglial phenotype and distribution pattern in fAβ-injected Cxcr3–/– mice. This shows that CXCR3 can modulate the functional state of microglia, in line with recent studies examining the role of CXCR3 in other models of neurodegeneration, showing the absence of CXCR3 is associated with attenuated microglial activation, reduced expression of inflammatory factors, and constrained microglial recruitment (25, 26, 55, 58).
Concerning the mechanisms leading to diminished Aβ load in CXCR3-deficient APP/PS1 mice, based on the in vivo phagocysotis data, we hypothesized that CXCR3-deficient microglia are more capable of phagocytosing Aβ in the APP/PS1 model. Alternatively, an incomplete degradation of Aβ by microglia may foster aggregation and plaque formation, in particular in an acidic environment within the phagolysosome. Thus, deficiency of CXCR3 could also block this effect by reducing microglia mobilization in the first place and consequently reducing the process of plaque formation, contributing to the observed plaque reduction in APP/PS1/Cxcr3–/– mice. Using an in vitro phagocytosis assay, we showed that CXCR3-deficient microglia have higher phagocytic capability for fAβ than WT microglia. Equally, the in vitro activation of CXCR3 by its ligands CXCL9 and CXCL10 reduced the phagocytosis of fAβ by WT microglia but not by CXCR3-deficient microglial cells, indicating a CXCR3-specific effect. In addition, treatment with a CXCR3-specific antagonist elevated phagocytosis in WT microglia. The observation of enhanced microglial fAβ phagocytosis and augmented lysosomal size in Cxcr3–/– animals after intracerebral injection extends our findings on the impact of CXCR3 on microglial phagocytosis in our model: CXCR3 decreases the phagocytic capabilities for fAβ and thereby promotes the deposition of Aβ plaques in APP/PS1 mice.
We also detected strong modulation of the cytokine milieu by CXCR3. Proinflammatory transcripts, such as TNF-α, IL-1β, or FasL, were greatly reduced in CXCR3-deficient APP/PS1 mice. TNF-α and IL-1β are known to be upregulated in senile plaques and dystrophic neurites in AD (59) and in an AD mouse model (49). TNF-α is thought to amplify brain inflammation and cognitive impairment in both AD patients and AD models (60–64). Dysregulated apoptosis has been implicated in several neurodegenerative disorders, including AD. The key apoptosis regulator FasL, but also IL-1β, is potentially involved in both neuronal and immune cell apoptosis in AD and AD models (65–70). It is likely that the reduced levels of TNF-α, IL-1β, and FasL molecules contribute to the alleviated phenotype in APP/PS1/Cxcr3–/– mice. Concerning the induction of chemokines and their receptors, we found the highest levels of induction for CXCR3 and its ligand, CXCL10, further corroborating the pathophysiological role of this chemokine system in the APP/PS1 model. It is well known that the chemokines are promiscuous and have very complex interactions. However, we could not detect any compensatory upregulation of chemokines or chemokine receptors in response to the lack of CXCR3. On the contrary, we found a marked downregulation of key chemokines, such as CX3CL1, CCL3, CCL7, and CXCL12. Hence, CXCR3 is a central molecule in the inflammatory response of the APP/PS1 model, congruent to findings in other neuroinflammatory models (55, 71–73). Cytokine analysis revealed only a single molecule, BDNF, that was upregulated in brain tissue of CXCR3-deficient APP/PS1 mice. Raised levels of BDNF might also contribute to the attenuated disease course observed in CXCR3-deficient APP/PS1 mice. BDNF-producing neural stem cells can also improve the course of APP/PS1 pathology (74). Furthermore, BDNF treatment is reported to reduce neuronal loss in the entorhinal cortex of APP transgenic mice (75).
Our in vitro fAβ-stimulation data of glial cells and the results of numerous experimental studies (76–79) identify microglial cells as the main source for local TNF-α in neuroinflammation, in particular for an AD-like pathology. We found that CXCR3 signaling drastically increases the TNF-α production of microglia after fAβ stimulation.
Because IFN-γ strongly induces CXCR3 ligands (31, 32, 80), we examined Ifng gene expression in our model, but could not find any relevant differences between APP/PS1 and CXCR3-deficient APP/PS1 mice. Cytokines such as TNF-α and IL-1β, found at increased levels in our model, can also induce CXCR3 ligands, in particular CXCL10 (35, 81). To further elucidate the factors that induce CXCL10 in APP/PS1 mice, we stimulated glial cells in vitro with TNF-α and fAβ. Our data revealed that fAβ is a very strong stimulus for microglial secretion of TNF-α and for microglial and astrocytic secretion of CXCL10. Further, TNF-α increased the astrocyte secretion of CXCL10. In summary, fAβ directly induced the production of CXCL10 by microglial cells and astrocytes. Proinflammatory cytokines induced by fAβ, in particular microglial TNF-α, can indirectly contribute to the overall increase by upregulating the astrocytic production of CXCL10.
As neurons are able to functionally express CXCR3 (27, 28, 34), these cells could contribute to the observed plaque reduction in APP/PS1/Cxcr3–/– mice as well. We evaluated the impact of neuronal CXCR3 deficiency on the neuronal expression and processing of APP and found no differences in total APP and cleavage product (CTFs) levels in APP/PS1 and APP/PS1/Cxcr3–/– brain tissue, counterindicative of a role of neuronal CXCR3 in the observed phenotypical changes.
In summary, the presented data demonstrate for the first time, to our knowledge, the functional importance of the CXCR3 chemokine system for the clinical and morphological course of a well-established animal model of AD and justify further exploration of the therapeutic potential of CXCR3 antagonists in AD. However, it is important to note that this will be a long and difficult task with an undetermined outcome. Although CXCR3 antagonists have a plethora of conceivable clinical applications and a number of CXCR3 antagonists are available, none of these substances have yet passed clinical studies with success and approval (82). In addition, we know from recent experimental studies that manipulation of the CXCR3 chemokine system can result in very unexpected and potentially harmful effects (83). Finally, CXCR3 antagonists have to be tested and identified for their ability to cross the BBB, allowing a local inhibition of CXCR3 inside the CNS.
We conclude that direct and indirect induction of CXCL10 by fAβ and subsequent activation of the CXCR3 chemokine system are able to modulate the activation state of glial cells and thereby the course of AD-like pathology in the APP/PS1 model. CXCR3 activation reduces the phagocytotic competence of microglia for fAβ, which ultimately promotes plaque formation and behavioral impairment in this model. CXCR3 has a key role in the progression of the AD-like disease in APP/PS1 mice and should be considered as a therapeutic target in AD.
Methods
Animals.
CXCR3-deficient (Cxcr3–/–) mice (originally provided by Bao Lu and Craig Gerard, Children’s Hospital and Harvard Medical School, Boston, Massachusetts, USA) have been described elsewhere (84). The mice were backcrossed at least 8 generations to the C57BL/6 strain. CXCR3-deficient mice displayed no clinical and histological abnormalities when compared with C57BL/6J (WT) mice. APP/PS1 double-transgenic mice (B6C3-Tg [APPswe,PSEN1dE9] 85Dbo/J, The Jackson Laboratory) expressing a chimeric mouse/human APP (Mo/HuAPP695swe) and a mutant human PS1 (PS1-dE9) protein were used for the crossbreeding experiment (51). The animals were hemizygous or littermate control mice and had been backcrossed for at least 8 generations onto the C57BL/6 strain. To obtain CXCR3-deficient mice with a transgenic expression of APP/PS1 (APP/PS1/Cxcr3–/–), heterozygous APP/PS1 mice were successively crossed with Cxcr3–/– mice and subsequently intercrossed. Only male mice were studied in order to avoid possible influence of sex on amyloid plaque formation and inflammation in AD (85, 86). Animals were kept under pathogen-free conditions.
Morris water-maze test.
Eight-month-old WT (n = 9), Cxcr3–/– (n = 12), APP/PS1 (n = 16), and APP/PS1/Cxcr3–/– (n = 18) mice were tested using the Morris water maze. Spatial memory testing was conducted in a pool consisting of a circular tank (1 m diameter) filled with opacified water at 24°C. The water basin was dimly lit (20–30 lux) and surrounded by a white curtain. The maze was virtually divided into 4 quadrants, 1 containing a hidden platform (15 × 15 cm) present 1.5 cm below the water surface. Mice were trained to find the platform using 3 extra maze cues placed asymmetrically as spatial references. Mice were placed into the water in a quasi-random fashion to prevent strategy learning. Mice were allowed to search for the platform for 40 seconds and were placed onto it manually if they did not reach the platform in the allotted time. Mice were allowed to stay on the platform for 15 seconds before the initiation of the next trial. After 4 trials, mice were dried and placed back into their home cages. Mice were trained for 8 consecutive days with 4 trials per day. In spatial probe trials, which were conducted 24 hours after the last training session (day 9), the platform was removed and mice were allowed to swim for 30 seconds. Data are given as percentage of time spent in quadrant Q1, where the platform was located previously, which was compared with the average time the animals spent in the remaining quadrants. In the afternoon of the same day, visual cued testing was performed with a flagged platform and new positions for start and goal during each trial. All movements were recorded by a computerized tracking system that calculated distances moved and latencies required for reaching the platform (Noldus, Ethovision 3.1).
Open-field exploration.
Eight-month-old WT (n = 9), CXCR3 (n = 12), APP/PS1 (n = 16), and APP/PS1/Cxcr3–/– (n = 18) mice were placed in the center of a dimly lit (20–30 lux) chamber of the open-field apparatus. Animal movements were tracked by an automatic monitoring system (Noldus Ethovision) for 5 minutes. The area was virtually divided into a center (square with 40-cm edge lengths), a corridor (7.5 cm along the walls), and 4 corner squares (10-cm edge lengths), which partly overlapped with the corridor area. The time spent in each area, horizontal and vertical activity, and frequency of urination and defecation were monitored. The experiment was repeated on 3 consecutive days.
Immunohistochemistry and ThioS staining.
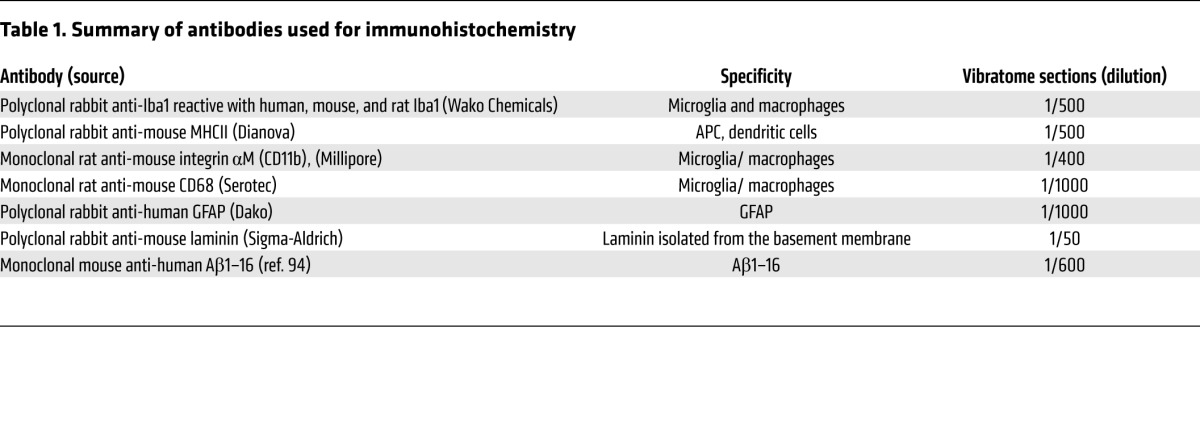
Five- and eight-month-old mice of each examined genotype (WT, Cxcr3–/–, APP/PS1, APP/PS1/Cxcr3–/–) were anesthetized with isoflurane and transcardially perfused with ice-cold PBS. Immediately after euthanasia, the brains were removed and half brains (cut along the sagittal midline) were fixed overnight in PBS-buffered 4% paraformaldehyde at 4°C. The PBS-washed brains were serially sectioned at 40-μm using a vibratome (Leica). Sections were blocked with 5% BSA in PBST and immunolabeled with antibodies against IBA1, MHCII, CD11b, CD68, GFAP, or Aβ using IC16 (Table 1), followed by incubation with Alexa Fluor 488– or Alexa Fluor 594–conjugated secondary antibodies (Life Technologies, Invitrogen). A double-staining protocol was used for combined plaque staining (ThioS and microglial immunostaining). For that purpose, free-floating sections were stained with 0.015% ThioS (50% ethanol) for 10 minutes, then washed in 50% ethanol and water before entering the immunostaining protocol. Finally, sections were mounted on HistoBond (VWR International) slides, dried, and coverslipped with fluorescent mounting medium (Dako) containing 0.1% DAPI (Sigma-Aldrich). The percentage of ThioS+ area or Aβ-immunoreactive plaques was used to estimate amyloid load (3–5 sections of the frontal cortex and hippocampus/animal). To determine the morphological activation of microglia, we applied IBA1 stainings developed for bright-field examination. Vibratome sections were incubated for 1 hour at room temperature (RT) with Iba1. After washing in PBST, a biotinylated secondary antibody (Axxora; 1/500) and HRP-coupled streptavidin (Axxora; 1/500) were applied. The signal was visualized by NovaRED color reagent (Axxora) according to the manufacturer’s instructions. Microglia cell bodies were analyzed using ImageJ software (http://imagej.nih.gov/ij/). Fluorescence microscopy was done with an Olympus BX61 microscope using identical exposure times. Images were processed with Cell^P (Olympus Soft Imaging Solutions). Using AnalySIS 3.2 (Olympus) software, we applied identical fluorescence threshold values to the ThioS signal and created a binary image that was then analyzed using particle features in the ROIs (hippocampus and frontal cortex). To evaluate differences in the morphology of Aβ plaques between APP/PS1 and APP/PS1/Cxcr3–/– mice, we used the shape descriptors plugin (form factor, defined as 4π × area/perimeter2) using ImageJ software. The form factor is usually used as a parameter of macrophage activation by means of morphology (87). The form factor varies between 0 (a 2-dimensional line) and 1 (a circle).
Table 1.
Summary of antibodies used for immunohistochemistry
Tissue processing for protein extraction.
Brain tissue for protein analysis was obtained from 5- and 8–month-old male mice of each examined genotype (WT, Cxcr3–/–, APP/PS1, APP/PS1/Cxcr3–/–). Snap-frozen brain hemispheres were homogenized in ice-cold modified PBS (1 mM EDTA, 1 mM EGTA, 3 μl/ml protease inhibitor mix, pH 7.4) using an UltraTurax T8 Homogenizer (IKA Labortechnik). Homogenates were extracted in RIPA buffer (25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% NP40, 0.5% NaDOC, 0.1% SDS) and centrifuged at 20,000 g for 15 minutes at 4°C; the remaining pellet containing insoluble Aβ was subsequently solubilized in SDS buffer (2% SDS, 25 mM Tris–HCl, pH 7.4). After pulsed sonication for 15 seconds, RIPA and SDS fractions protein concentrations were determined using the BCA Protein Assay Kit (Thermo Scientific).
APP processing and amyloid-β detection.
Proteins were separated using a 4%–12% NuPAGE gel (Life Technologies, Invitrogen) with NuPage MES SDS running buffer (Life Technologies, Invitrogen) at 150 V. PageRuler prestained protein ladder (Thermo Fisher, Fermentas) was used as standard. Proteins were transferred to 0.2 μm nitrocellulose membranes (boiled in water for 5 minutes and blocked for 30 minutes in TBST containing 5% skim milk). Antibody CT-15 raised against the C terminus of APP was used for immunoblotting (88) (Chemicon; 1:2000), and antibody CP06 was used for detecting α-tubulin (Millipore; 1:2000). After incubation with the appropriate HRP-conjugated secondary antibodies, immunoreactivity was detected by HRP chemiluminescent reaction (Millipore). Luminescence intensities were analyzed using Chemidoc XRS Imaging System (Bio-Rad). With the Quantity One (Bio-Rad) program, the densities of bands were determined for each lane and the intensity ratios for the detected proteins were normalized to α-tubulin. Each gel contained a nontransgenic WT and Cxcr3–/– brain extract as controls. GraphPad Prism (GraphPad Software) software was used for statistical analysis.
Aβ ELISA.
Levels of Aβ peptides were quantified using human Aβ1–40 and Aβ1–42 ELISA kits (Millipore) according to the manufacturer’s protocol. RIPA and SDS fractions were analyzed for Aβ1–40 and Aβ1–42 peptide concentrations. Samples were analyzed in duplicate. The results were tabulated as mean ± SEM and statistically compared using an unpaired Student’s t test.
Cytokine and chemokine mRNA determination by qPCR.
Total RNA was isolated and purified from aliquots of homogenized brain samples using Trizol reagent (Sigma-Aldrich). RNA quantity was determined using NanoDrop 1000 (Peqlab). Up to 3 μg of total RNA was reverse-transcribed into cDNA using SuperScript III Reverse Transcriptase (Life Technologies, Invitrogen). Real-time qPCR assays were performed on a StepOnePlus Real-Time PCR System (Applied Biosystems) using Power SYBR Green (Applied Biosystems). The composition of reaction mixture was as follows: 1 μl of cDNA corresponding to 40 ng of total RNA, 100 nM of each primer, and 2× Power SYBR Green PCR Master Mix (Applied Biosystems) in a total volume of 25 μl. The primer sequences are listed in Supplemental Table 1.
Samples were analyzed simultaneously for Gapdh mRNA as internal control. Each sample was assayed in duplicate, normalized to Gapdh, and expressed relative to that of nontransgenic C57BL/6J mice. Data were determined as fold change of mRNA ± SEM.
Primary mouse neuronal culture.
Cortical neurons were prepared from postnatal APP/PS1 and APP/PS1/Cxcr3–/– C57BL/6 mice. The whole procedure was performed using individual offspring from hemizygous APP/PS1 × WT and APP/PS1/Cxcr3–/– × Cxcr3–/– breedings to ensure individual genotypes. Meninges and blood vessels of brains were removed mechanically. The brains were homogenized in HBSS buffer (1% HEPES) by chopping and pipetting through 10-ml and 5-ml pipettes. The homogenate was filtered through a cell strainer (70 μm). 3 × 105 cells/well were seeded on poly-l-lysine–coated (100 μg/ml) 6-well plates and grown at 37°C with 5% CO2 in modified 1× neurobasal medium (Gibco, Life Technologies) containing 1% fetal calf serum (PAN Biotech), 3% GlutaMAX (Gibco, Life Technologies), 2% B-27 supplement (×50; Gibco, Life Technologies), and 100 U/ml penicillin/streptomycin (PAA). After 2 hours of incubation, nonadherent cells were washed out; adherent cells were overlayed with 2.5 ml of modified 1× neurobasal medium. Half of the medium was changed once after 4 days; cultures were incubated for 3 additional days without medium change. The neurons were found in large aggregates interconnected with bundles of processes. The morphological changes of neurons were observed under an inverted phase-contrast microscope. Immunofluorescence staining for NeuN (Chemicon) was performed to identify the neurons. The level of Cxcr3 gene expression of the neuronal cultures from WT and Cxcr3–/– mice was determined by qPCR. Cxcr3 RNA was detectable in neurons from APP/PS1 and WT control mice, but not in Cxcr3–/– nor in APP/PS1/Cxcr3–/– mice (Figure 1H). At day 7, the supernatants were taken from each neuronal culture representing individual brains from APP/PS1 (n = 5) and APP/PS1/Cxcr3–/– (n = 4) mice and were analyzed using human Aβ1–40 and Aβ1–42 ELISA kits (Millipore). Secreted Aβ1–40 and Aβ1–42 were normalized to total neuronal culture protein concentration evaluated by BCA assay of harvested neurons.
Retrieval of primary mouse microglia and astrocytes.
Cortical murine microglia were prepared from brains of pups at postnatal day 1 as previously described (89). Briefly, meninges of isolated brains were removed mechanically, and brains were dissociated by trituration to culture cells in high-glucose (4.5 mg/ml) DMEM (Gibco, Life Technologies) supplemented with 10% fetal calf serum (PAN Biotech) and 1% penicillin/streptomycin (PAA) for up to 14 days. Microglia cells were harvested by shake-off. The media containing detached microglia were collected, and the isolated microglia were reseeded and allowed to settle for at least 1 hour.
Primary astrocyte cultures were obtained from microglial/astrocytic coculture maintained for a minimum of 7 days to generate a confluent glial culture. Prior to trypsinization, contaminating microglial cells were separated by repeated mechanical agitation and removed by subsequent washing in HBSS (PAA). Astrocytic monolayers were then dislodged from flasks by trypsinization (0.25% trypsin in HBSS with 1 mM EDTA). Cells were seeded in 9-cm diameter plates (1 × 105 cells/well) and grown for 7 days until confluent prior to stimulation. Culture purity was determined by double-labeling immunohistochemistry for GFAP and CD11b to identify astrocytes (>95%) and microglia (<5%). The levels of microglial and astrocytic Cxcr3 gene expression were determined by qPCR and allowed the clear differentiation of cells derived from WT and Cxcr3–/– mice. Cxcr3 RNA was detectable in astrocytes and microglia from APP/PS1 and WT control mice, but not in Cxcr3–/– nor in APP/PS1/Cxcr3–/– mice. We could not observe any significant difference in Cxcr3 RNA levels in cells from APP/PS1 animals compared with WT controls (Figure 1H).
Microglia and astrocytes were stimulated with either unlabeled aggregated 0.7 μM Aβ1–42 (referred to as fAβ) (Peptide Speciality Laboratories), LPS (100 ng/ml, Sigma-Aldrich, from E. coli 0127:B8), or mouse recombinant TNF-α (10 ng/ml, Roche Diagnostics) in low-serum DMEM (1% FBS). Three separate cultures were stimulated, so that data represent the mean of 3 independent experiments. Additional astrocytes were incubated in medium to serve as unstimulated controls. Supernatants were collected after stimulation for 4, 12, or 20 hours and stored at -80°C until analysis, while cells were washed in PBS and scraped for collection with 1× RIPA buffer containing protease inhibitor cocktail (Sigma-Aldrich).
Microglial in vitro phagocytosis of FAM-labeled fAβ.
PMG were seeded on cover glasses and incubated with 0.7 μM of fibrillar FAM-labeled Aβ1–42 (FAM-Aβ, AnaSpec). Fibrillar FAM-Aβ was generated using NaOH-treated peptide lyophilizate after incubation in acetate buffer to initiate aggregation (90). The conditions during aggregation (18 hours at 37°C, 50 mM Tris-HCl, pH 7) result in a mixture of n-meric Aβ1–42 ranging from monomeric to high–molecular-weight–aggregated (fibrillar) Aβ1–42. Aggregation was controlled by 4%–12% NuPAGE, and aggregates were detected using antibody 6E10 (Signet). Microglia cells were washed with PBST fixed with 4% PFA and stained with rat polyclonal anti-CD68 (1:500; Serotec) after a 1-hour FAM-Aβ incubation period. Secondary detection antibody was conjugated to Alexa Fluor 594 (Life Technologies, Invitrogen). Furthermore, a microglial fAβ phagocytosis reader assay was applied as described previously (91). Briefly, 5 × 104 cells/well were seeded and incubated with 0.7 μM fibrillar FAM-Aβ for 1 hour. Afterwards, the medium was removed and extracellular FAM-Aβ fluorescence signal was quenched with trypan blue. Fluorescence intensity was measured at 485 nm excitation/535 nm emission using a fluorescence plate reader (Infinite 200 M, Tecan). To compensate for different cell counts, results were normalized to the Hoechst Dye 33342 (Sigma-Aldrich) nuclear stain signals. To evaluate the influence of CXCR3 ligands on microglial phagocytosis, recombinant mouse CXCL9 or CXCL10 (250 ng/ml, R&D systems) was applied 30 minutes following FAM-Aβ incubation.
In additional experiments, a CXCR3 antagonist (30–300 nM, agonist 120) (92), was added 30 minutes prior to the phagocytosis experiment.
Detection of cytokine proteins in culture supernatant.
Commercially available mouse Quantikine Immunoassays (R&D Systems) were used to quantify the production of the chemokines CXCL9 and CXCL10 in culture supernatants. For the detection of TNF-α, the ELISA Ready-Set-Go!Kit (eBioscience) was applied. Each sample was measured in duplicate.
Microglial in vivo phagocytosis of methoxy-XO4–labeled Aβ.
In vivo Aβ phagocytosis was determined using mice intraperitoneally injected with 10 mg/kg methoxy-XO4 (50% DMSO/50% NaCl [0.9%], pH 12) 3 hours before sacrifice (provided by Alfons Verbruggen, Katholieke Universiteit Leuven, Leuven, Belgium). Mice were perfused with ice-cold PBS, and the brains were removed, minced using scalpels, and incubated in HBSS and 10% FCS containing 0.144 mg/ml collagenase type IV (Sigma-Aldrich) for 1 hour at 37°C. Homogenization was achieved by pipetting through a 19-gauge needle. The homogenate was filtered through a cell strainer (70 μm) and centrifuged at 155 g at RT for 10 minutes without using the brake. The pellet was resuspended in 37% Percoll in PBS, underlayered with 70% Percoll in PBS, and overlayered with ice-cold PBS. The gradient was centrifuged at 800 g at 4°C for 25 minutes without using the brake. Microglial cells were recovered from the 37%/70% Percoll interphase, diluted with 3 vol PBS, and centrifuged at 880 g at 4°C for 25 minutes without a break. The pellet containing the microglial cells was resuspended in 200 μl PBS. For flow cytometry analysis, 50 μl of cells was diluted with 0.5 ml HBSS and centrifuged at 250 g for 5 minutes at 4°C. Binding of antibodies to Fc receptors was prevented by adding 1 μg of Fc block (BD Bioscience). Cells were taken up in 50 μl of primary antibody mix (CD11b-APC [1:100, BioLegend] and CD45-FITC [1:100, eBioscience]) and incubated for 30 minutes on ice. Cells were centrifuged at 250 g for 5 minutes at 4°C and resuspended in 200 μl HBSS. Corresponding isotype control antibodies were used for control and compensation. Cells were measured on a FACSCanto II Flow Cytometer (BD Bioscience), and data were analyzed using the flow cytometry software FlowJo (TreeStar). For analysis, the CD11b+ CD45+ population was gated. WT mice injected with methoxy-XO4 were used to determine the methoxy-XO4 threshold for nonphagocytosing cells, and unstained WT cells were used to determine background fluorescence.
Intracerebral injection of fibrillar Aβ.
Six- to eight-month-old Cxcr3–/– (n = 5) and WT (n = 5) mice were anesthetized with ketamine/xylazine (30 mg/kg, 4 mg/kg) and immobilized using a stereotactic device. A 0.5-mm burr hole was drilled in the skull, and 1 μl of fibrillar Aβ1–42 solution injected intracortically into the right hemisphere (anteroposterior –2.5, lateral 2.0 at 1.0 mm cortex relative to the bregma) at a rate of 1 μl/min using a 5-μl Hamilton syringe as described previously (93). Control animals received a buffer solution into the right hemisphere, accordingly. Mice were sacrificed 48 hours after injection and prepared for histology as previously described. 30-μm–thick horizontal brain sections, including the needle track, were immunostained with the primary antibodies Iba1 and IC16 or CD68 as listed in Table 1. The antibody binding was detected with Alexa Fluor dye–conjugated secondary antibodies. Sections were analyzed using a BX61 microscope equipped with a confocal disk-scanning unit (Olympus). Image stacks were deconvoluted using Cell^P (Olympus). IBA1-positive microglia within a 80-μm radius of multiple 10-μm z-stacks of the needle track were counted and examined for the presence of intralysosomal fAβ1–42. Analysis of microglia fAβ content within CD68+ microglial lysosomes was performed using ImageJ software. Microglial bodies were identified in z-projections from confocal images. For the quantification of intralysosomal fAβ, ROI was drawn around the CD68+ area and the MFI values were obtained from the resulting area after subtraction of CD68+ microglia regions. Aβ+ deposit not located inside microglia was considered to be in the extracellular space. For the determination of mean lysosomal perimeter of microglia in proximity to the fAβ injection site, CD68+ lysosomes in WT and Cxcr3–/– were analyzed. The RGB fluorescence intensity profile plot was obtained using ImageJ.
Statistics.
Data from real-time PCR, ELISA, Western blot, and quantitative histological studies were analyzed where appropriate by a 2-tailed Student’s t test, with P < 0.05 considered to be statistically significant. The behavioral experiments as summarized in Figure 3 and the in vitro studies as summarized in Figures 4 and 5 were statistically analyzed using 1-way ANOVA followed by Tukey’s post-hoc test (Morris water maze) the Kruskal-Wallis rank sum test, or Dunn’s post-hoc test (probe trial, in vitro studies). The cytokine study as listed in Supplemental Table 1 was further analyzed by a false discovery rate analysis (Q value set to 0.05). All statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software).
Study approval.
All procedures were carried out according to the guidelines of the Animal Care and Use Committee of the University of Bonn. All animal studies were approved by the ethical committee of the University of Bonn Medical Center.
Supplementary Material
Acknowledgments
Richard M. Ransohoff, Iain L. Campbell, and Daniel R. Getts are gratefully acknowledged for fruitful discussions. We thank Cathy Widmann for critical reading of the manuscript and editing. This research has received funding from the German Research Council/DFG Clinical Research Group 177 (to M.T. Heneka and M. Müller), the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement HEALTH-F2-2011-278850 (INMiND to M.T. Heneka), and the German Research Council/DFG Cluster of Excellence “Immunosensation.”
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(1):365–378. doi:10.1172/JCI66771.
References
- 1.Katzman R, Saitoh T. Advances in Alzheimer’s disease. FASEB J. 1991;5(3):278–286. [PubMed] [Google Scholar]
- 2.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430(7000):631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heneka MT, O’Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer’s disease. J Neural Transm. 2010;117(8):919–947. doi: 10.1007/s00702-010-0438-z. [DOI] [PubMed] [Google Scholar]
- 4.Parpura V, et al. Glial cells in (patho)physiology. J Neurochem. 2012;121(1):4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends Neurosci. 1988;11(6):273–277. doi: 10.1016/0166-2236(88)90110-5. [DOI] [PubMed] [Google Scholar]
- 6.Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992;48(2):405–415. doi: 10.1016/0306-4522(92)90500-2. [DOI] [PubMed] [Google Scholar]
- 7.Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25(36):8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shie F-S, Breyer RM, Montine TJ. Microglia lacking E Prostanoid Receptor subtype 2 have enhanced Aβ phagocytosis yet lack Aβ-activated neurotoxicity. Am J Pathol. 2005;166(4):1163–1172. doi: 10.1016/S0002-9440(10)62336-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto M, et al. Overexpression of monocyte chemotactic protein-1/CCL2 in β-amyloid precursor protein transgenic mice show accelerated diffuse β-amyloid deposition. Am J Pathol. 2005;166(5):1475–1485. doi: 10.1016/S0002-9440(10)62364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Town T, et al. Blocking TGF-[beta]-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14(6):681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krause DL, Müller N. Neuroinflammation, microglia and implications for anti-inflammatory treatment in Alzheimer’s disease. Int J Alzheimer Dis. 2010;2010:732806. doi: 10.4061/2010/732806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reed-Geaghan EG, Reed QW, Cramer PE, Landreth GE. Deletion of CD14 attenuates Alzheimer’s disease pathology by influencing the brain’s inflammatory milieu. J Neurosci. 2010;30(46):15369–15373. doi: 10.1523/JNEUROSCI.2637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heneka MT, et al. NLRP3 is activated in Alzheimer/’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease — a double-edged sword. Neuron. 2002;35(3):419–432. doi: 10.1016/S0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 15.Paresce D, Ghosh R, Maxfield F. Microglial cells internalize aggregates of the Alzheimer’s disease Amyloid β-protein via a scavenger receptor. Neuron. 1996;17(3):553–565. doi: 10.1016/S0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 16.Weldon DT, et al. Fibrillar β-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci. 1998;18(6):2161–2173. doi: 10.1523/JNEUROSCI.18-06-02161.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J Neurosci. 2004;24(44):9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354(6):610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 19.Müller M, Carter S, Hofer MJ, Campbell IL. Review: The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity — a tale of conflict and conundrum. Neuropathol Appl Neurobiol. 2010;36(5):368–387. doi: 10.1111/j.1365-2990.2010.01089.x. [DOI] [PubMed] [Google Scholar]
- 20.Loetscher M, et al. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184(3):963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weng Y, et al. Binding and functional properties of recombinant and endogenous CXCR3 chemokine receptors. J Biol Chem. 1998;273(29):18288–18291. doi: 10.1074/jbc.273.29.18288. [DOI] [PubMed] [Google Scholar]
- 22.Sallusto F, Lanzavecchia A. Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol Rev. 2000;177:134–140. doi: 10.1034/j.1600-065X.2000.17717.x. [DOI] [PubMed] [Google Scholar]
- 23.Biber K, et al. Functional expression of CXCR3 in cultured mouse and human astrocytes and microglia. Neuroscience. 2002;112(3):487–497. doi: 10.1016/S0306-4522(02)00114-8. [DOI] [PubMed] [Google Scholar]
- 24.Flynn G. Regulation of chemokine receptor expression in human microglia and astrocytes. J Neuroimmunol. 2003;136(1–2):84–93. doi: 10.1016/s0165-5728(03)00009-2. [DOI] [PubMed] [Google Scholar]
- 25.Rappert A, et al. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci. 2004;24(39):8500–8509. doi: 10.1523/JNEUROSCI.2451-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Jong EK, et al. Expression of CXCL4 in microglia in vitro and in vivo and its possible signaling through CXCR3. J Neurochem. 2008;105(5):1726–1736. doi: 10.1111/j.1471-4159.2008.05267.x. [DOI] [PubMed] [Google Scholar]
- 27.Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer’s disease. J Neuroimmunol. 2000;108(1–2):227–235. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- 28.Nelson TE, Gruol DL. The chemokine CXCL10 modulates excitatory activity and intracellular calcium signaling in cultured hippocampal neurons. J Neuroimmunol. 2004;156(1–2):74–87. doi: 10.1016/j.jneuroim.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Zhang B, Patel J, Croyle M, Diamond MS, Klein RS. TNF-alpha-dependent regulation of CXCR3 expression modulates neuronal survival during West Nile virus encephalitis. J Neuroimmunol. 2010;224(1–2):28–38. doi: 10.1016/j.jneuroim.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28(11):3696–3705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 31.Luster AD, Unkeless JC, Ravetch JV. γ-Interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. 1985;315(6021):672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- 32.Cole KE, et al. Interferon-inducible T cell α chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187(12):2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carter SL, Müller M, Manders PM, Campbell IL. Induction of the genes for Cxcl9 and Cxcl10 is dependent on IFN-γ but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microglia in vitro. Glia. 2007;55(16):1728–1739. doi: 10.1002/glia.20587. [DOI] [PubMed] [Google Scholar]
- 34.Zhang B, Patel J, Croyle M, Diamond MS, Klein RS. TNF-alpha-dependent regulation of CXCR3 expression modulates neuronal survival during West Nile virus encephalitis. J Neuroimmunol. 2010;224(1–2):28–38. doi: 10.1016/j.jneuroim.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi K, Ni L, Jonakait GM. Fas ligation and tumor necrosis factor alpha activation of murine astrocytes promote heat shock factor-1 activation and heat shock protein expression leading to chemokine induction and cell survival. J Neurochem. 2011;116(3):438–448. doi: 10.1111/j.1471-4159.2010.07124.x. [DOI] [PubMed] [Google Scholar]
- 36.Lee YK, et al. CCR5 deficiency induces astrocyte activation, Abeta deposit and impaired memory function. Neurobiol Learn Mem. 2009;92(3):356–363. doi: 10.1016/j.nlm.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 37.El Khoury J, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13(4):432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 38.Kiyota T, et al. CCL2 accelerates microglia-mediated Aβ oligomer formation and progression of neurocognitive dysfunction. PLoS One. 2009;4(7):e6197. doi: 10.1371/journal.pone.0006197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Semple BD, Frugier T, Morganti-Kossmann MC. CCL2 modulates cytokine production in cultured mouse astrocytes. J Neuroinflammation. 2010;7:67. doi: 10.1186/1742-2094-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuhrmann M, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;13(4):411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee S, et al. CX3CR1 deficiency alters microglial activation and reduces β-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. 2010;177(5):2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J Neurosci. 2010;30(50):17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nash KR, et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol Aging. 2013;34(6):1540–1548. doi: 10.1016/j.neurobiolaging.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galimberti D, Schoonenboom N, Scarpini E, Scheltens P. Chemokines in serum and cerebrospinal fluid of Alzheimer’s disease patients. Ann Neurol. 2003;53(4):547–548. doi: 10.1002/ana.10531. [DOI] [PubMed] [Google Scholar]
- 45.Galimberti D, et al. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch Neurol. 2006;63(4):538–543. doi: 10.1001/archneur.63.4.538. [DOI] [PubMed] [Google Scholar]
- 46.Duan R-S, et al. Decreased fractalkine and increased IP-10 expression in aged brain of APP(swe) transgenic mice. Neurochem Res. 2008;33(6):1085–1089. doi: 10.1007/s11064-007-9554-z. [DOI] [PubMed] [Google Scholar]
- 47.Jankowsky JL, et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum Mol Genet. 2004;13(2):159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 48.Garcia-Alloza M, et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24(3):516–524. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 49.Ruan L, Kang Z, Pei G, Le Y. Amyloid deposition and inflammation in APPswe/PS1dE9 mouse model of Alzheimer’s disease. Curr Alzheimer Res. 2009;6(6):531–540. doi: 10.2174/156720509790147070. [DOI] [PubMed] [Google Scholar]
- 50.Hammerschmidt T, et al. Selective loss of noradrenaline exacerbates early cognitive dysfunction and synaptic deficits in APP/PS1 mice. Biol Psychiatry. 2013;73(5):454–463. doi: 10.1016/j.biopsych.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jankowsky JL, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17(6):157–165. doi: 10.1016/S1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 52.Cho J, Nelson TE, Bajova H, Gruol DL. Chronic CXCL10 alters neuronal properties in rat hippocampal culture. J Neuroimmunol. 2009;207(1–2):92–100. doi: 10.1016/j.jneuroim.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sui Y, et al. CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur J Neurosci. 2006;23(4):957–964. doi: 10.1111/j.1460-9568.2006.04631.x. [DOI] [PubMed] [Google Scholar]
- 54.Coughlan CM, et al. Expression of multiple functional chemokine receptors and monocyte chemoattractant protein-1 in human neurons. Neuroscience. 2000;97(3):591–600. doi: 10.1016/S0306-4522(00)00024-5. [DOI] [PubMed] [Google Scholar]
- 55.Riemer C, et al. Accelerated prion replication in, but prolonged survival times of, prion-infected CXCR3–/– mice. J Virol. 2008;82(24):12464–12471. doi: 10.1128/JVI.01371-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koistinaho M, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med. 2004;10(7):719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- 57.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28(3):138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 58.Van Weering HRJ, et al. CXCL10/CXCR3 signaling in glia cells differentially affects NMDA-induced cell death in CA and DG neurons of the mouse hippocampus. Hippocampus. 2011;21(2):220–232. doi: 10.1002/hipo.20742. [DOI] [PubMed] [Google Scholar]
- 59.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129(2):154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramos EM, et al. Tumor necrosis factor α and interleukin 10 promoter region polymorphisms and risk of late-onset Alzheimer disease. Arch Neurol. 2006;63(8):1165–1169. doi: 10.1001/archneur.63.8.1165. [DOI] [PubMed] [Google Scholar]
- 61.Tobinick E, Gross H, Weinberger A, Cohen H. TNF-α modulation for treatment of Alzheimer’s disease: a 6-month pilot study. Med Gen Med. 2006;8(2):25. [PMC free article] [PubMed] [Google Scholar]
- 62.Pardridge WM. Biologic TNFα-inhibitors that cross the human blood-brain barrier. Bioeng Bugs. 2010;1(4):231–234. doi: 10.4161/bbug.1.4.12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frankola KA, Greig NH, Luo W, Tweedie D. Targeting TNF-α to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol Disord Drug Targets. 2011;10(3):391–403. doi: 10.2174/187152711794653751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McAlpine FE, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34(1):163–177. doi: 10.1016/j.nbd.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hofmann K, Tschopp J. The death domain motif found in Fas (Apo-1) and TNF receptor is present in proteins involved in apoptosis and axonal guidance. FEBS Lett. 1995;371(3):321–323. doi: 10.1016/0014-5793(95)00931-X. [DOI] [PubMed] [Google Scholar]
- 66.Felderhoff-Mueser U, et al. Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic-ischemic injury to the developing rat brain. Brain Pathol. 2000;10(1):17–29. doi: 10.1111/j.1750-3639.2000.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ethell DW, Buhler LA. Fas ligand-mediated apoptosis in degenerative disorders of the brain. J Clin Immunol. 2003;23(6):439–446. doi: 10.1023/B:JOCI.0000010420.96419.a8. [DOI] [PubMed] [Google Scholar]
- 68.Su JH, et al. Fas and Fas ligand are associated with neuritic degeneration in the AD brain and participate in β-amyloid-induced neuronal death. Neurobiol Dis. 2003;12(3):182–193. doi: 10.1016/S0969-9961(02)00019-0. [DOI] [PubMed] [Google Scholar]
- 69.Shaftel SS, et al. Sustained hippocampal IL-1 β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117(6):1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pinteaux E, Trotter P, Simi A. Cell-specific and concentration-dependent actions of interleukin-1 in acute brain inflammation. Cytokine. 2009;45(1):1–7. doi: 10.1016/j.cyto.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 71.Müller M, et al. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol. 2007;179(5):2774–2786. doi: 10.4049/jimmunol.179.5.2774. [DOI] [PubMed] [Google Scholar]
- 72.Miu J, et al. Chemokine gene expression during fatal murine cerebral malaria and protection due to CXCR3 deficiency. J Immunol. 2008;180(2):1217–1230. doi: 10.4049/jimmunol.180.2.1217. [DOI] [PubMed] [Google Scholar]
- 73.Christensen JE, et al. Efficient T-cell surveillance of the CNS requires expression of the CXC chemokine receptor 3. J Neurosci. 2004;24(20):4849–4858. doi: 10.1523/JNEUROSCI.0123-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang W, Wang PJ, Sha HY, Ni J, Li MH, Gu GJ. Neural stem cell transplants improve cognitive function without altering amyloid pathology in an APP/PS1 double transgenic model of Alzheimer’s disease. Mol Neurobiol. 2014;50(2):423–437. doi: 10.1007/s12035-014-8640-x. [DOI] [PubMed] [Google Scholar]
- 75.Nagahara AH, et al. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J Neurosci. 2013;33(39):15596–15602. doi: 10.1523/JNEUROSCI.5195-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klegeris A, Walker DG, McGeer PL. Interaction of Alzheimer β-amyloid peptide with the human monocytic cell line THP-1 results in a protein kinase C-dependent secretion of tumor necrosis factor-alpha. Brain Res. 1997;747(1):114–121. doi: 10.1016/S0006-8993(96)01229-2. [DOI] [PubMed] [Google Scholar]
- 77.Gregersen R, Lambertsen K, Finsen B. Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2000;20(1):53–65. doi: 10.1097/00004647-200001000-00009. [DOI] [PubMed] [Google Scholar]
- 78.Lue LF, et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia. 2001;35(1):72–79. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- 79.Hanisch U-K. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 80.Ferber I, et al. Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. 1996;156(1):5–7. [PubMed] [Google Scholar]
- 81.Majumder S, et al. Regulation of human IP-10 gene expression in astrocytoma cells by inflammatory cytokines. J Neurosci Res. 1998;54(2):169–180. doi: 10.1002/(SICI)1097-4547(19981015)54:2<169::AID-JNR5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 82. Smit MJ, Lira SA, Leurs R. Chemokine Receptors as Drug Targets. Malden, Massachusetts, USA: John Wiley & Sons; 2010. [Google Scholar]
- 83.Krauthausen M, et al. Opposing roles for CXCR3 signaling in central nervous system versus ocular inflammation mediated by the astrocyte-targeted production of IL-12. Am J Pathol. 2011;179(5):2346–2359. doi: 10.1016/j.ajpath.2011.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hancock WW, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192(10):1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Candore G, et al. Age-related inflammatory diseases: role of genetics and gender in the pathophysiology of Alzheimer’s disease. Ann N Y Acad Sci. 2006;1089:472–486. doi: 10.1196/annals.1386.008. [DOI] [PubMed] [Google Scholar]
- 86.Casadesus G, et al. The estrogen myth: potential use of gonadotropin-releasing hormone agonists for the treatment of Alzheimer’s disease. Drugs R D. 2006;7(3):187–193. doi: 10.2165/00126839-200607030-00004. [DOI] [PubMed] [Google Scholar]
- 87.Müller M, Wacker K, Getts D, Ringelstein EB, Kiefer R. Further evidence for a crucial role of resident endoneurial macrophages in peripheral nerve disorders: Lessons from acrylamide-induced neuropathy. Glia. 2008;56(9):1005–1016. doi: 10.1002/glia.20674. [DOI] [PubMed] [Google Scholar]
- 88.Martin LJ, et al. Amyloid precursor protein in aged nonhuman primates. Proc Natl Acad Sci U S A. 1991;88(4):1461–1465. doi: 10.1073/pnas.88.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hanisch U-K, et al. The microglia-activating potential of thrombin. J Biol Chem. 2004;279(50):51880–51887. doi: 10.1074/jbc.M408318200. [DOI] [PubMed] [Google Scholar]
- 90.Teplow DB. Preparation of amyloid beta-protein for structural and functional studies. Meth Enzymol. 2006;413:20–33. doi: 10.1016/S0076-6879(06)13002-5. [DOI] [PubMed] [Google Scholar]
- 91.Fleisher-Berkovich S, et al. Distinct modulation of microglial amyloid β phagocytosis and migration by neuropeptides (i). J Neuroinflammation. 2010;7:61. doi: 10.1186/1742-2094-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hayes ME, et al. Discovery of small molecule benzimidazole antagonists of the chemokine receptor CXCR3. Bioorg Medicinal Chem Lett. 2008;18(5):1573–1576. doi: 10.1016/j.bmcl.2008.01.074. [DOI] [PubMed] [Google Scholar]
- 93.Kummer MP, et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011;71(5):833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 94.Jäger S, et al. α-secretase mediated conversion of the amyloid precursor protein derived membrane stub C99 to C83 limits Aβ generation. J Neurochem. 2009;111(6):1369–1382. doi: 10.1111/j.1471-4159.2009.06420.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.