

Abstract
Genome integrity is continuously challenged by the DNA damage that arises during normal cell metabolism. Biallelic mutations in the genes encoding the genome surveillance enzyme ribonuclease H2 (RNase H2) cause Aicardi-Goutières syndrome (AGS), a pediatric disorder that shares features with the autoimmune disease systemic lupus erythematosus (SLE). Here we determined that heterozygous parents of AGS patients exhibit an intermediate autoimmune phenotype and demonstrated a genetic association between rare RNASEH2 sequence variants and SLE. Evaluation of patient cells revealed that SLE- and AGS-associated mutations impair RNase H2 function and result in accumulation of ribonucleotides in genomic DNA. The ensuing chronic low level of DNA damage triggered a DNA damage response characterized by constitutive p53 phosphorylation and senescence. Patient fibroblasts exhibited constitutive upregulation of IFN-stimulated genes and an enhanced type I IFN response to the immunostimulatory nucleic acid polyinosinic:polycytidylic acid and UV light irradiation, linking RNase H2 deficiency to potentiation of innate immune signaling. Moreover, UV-induced cyclobutane pyrimidine dimer formation was markedly enhanced in ribonucleotide-containing DNA, providing a mechanism for photosensitivity in RNase H2–associated SLE. Collectively, our findings implicate RNase H2 in the pathogenesis of SLE and suggest a role of DNA damage–associated pathways in the initiation of autoimmunity.
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterized by formation of autoantibodies against nuclear antigens, including nucleic acids. Antiviral type I IFNs induced by innate immune recognition of nucleic acids play a central role in the initiation and perpetuation of SLE (1–3). Type I IFNs have pronounced immunostimulatory effects that promote loss of B cell self-tolerance and autoantibody production. Antinuclear antibodies (ANAs) form complexes with nuclear antigens released from dying cells (4, 5), and deposition of such immune complexes in the capillary bed followed by local complement and leukocyte activation results in destructive tissue inflammation. SLE flares are commonly triggered by viral infection or UV irradiation (6, 7). The latter can provoke not only cutaneous symptoms, but also systemic disease.
Clinical features of SLE alongside type I IFN activation are also observed in the rare inflammatory disorder Aicardi-Goutières syndrome (AGS), an early-onset encephalopathy resembling congenital viral infection (8–11). AGS is caused by biallelic mutations in each of the 3 subunits of ribonuclease H2 (RNase H2): RNASEH2A, RNASEH2B, and RNASEH2C (also known as AGS4, AGS2, and AGS3, respectively) (12). Mutations in other enzymes involved in the intracellular nucleic acid metabolism — the DNA exonuclease 3′ repair exonuclease 1 (TREX1), the triphosphohydrolase SAM domain and HD domain-containing protein 1 (SAMHD1), and the RNA-editing enzyme adenosine deaminase 1 acting on RNA (ADAR) — have also been implicated in a spectrum of autoimmune phenotypes including AGS, familial chilblain lupus, and SLE (13–19). In addition, gain-of-function mutations in the RNA sensor IFN-induced with helicase C domain 1 (IFIH1) can also cause AGS (20). These findings illustrate the importance of coordinated pathways that regulate both degradation and sensing of intracellular nucleic acids in order to prevent an inappropriate innate immune response to self-DNA and -RNA (21).
RNase H2 is a heterotrimeric enzyme that can cleave the RNA strand of RNA/DNA heteroduplexes, such as those found in R-loops, and the 5′ phosphodiester bond of ribonucleotides embedded in a DNA duplex, an activity essential for the removal of ribonucleotides misincorporated in the genome during DNA replication (22–25). Ribonucleotides represent the most common endogenous base lesion in replicating cells, with more than 1 million present in the genome of RNase H2–null mouse cells (23). These are normally removed efficiently in a process termed ribonucleotide excision repair (26), whereas absence of cellular RNase H2 activity causes large-scale genome instability, p53 pathway activation, and early embryonic lethality (23, 24). RNase H2 is therefore a key genome surveillance enzyme required for genome integrity, raising the question as to how DNA repair may be linked to autoimmunity in AGS and SLE.
In the present study, we found that rare variants in RNASEH2 genes were associated with systemic autoimmunity and established that these resulted in impaired ribonucleotide removal. Furthermore, we provided evidence that such embedded ribonucleotides caused increased UV-induced cyclobutane pyrimidine dimer (CPD) formation and enhanced type I IFN signaling, thus linking mutations in a DNA repair enzyme with systemic autoimmunity.
Results
Carriers of AGS mutations frequently develop SLE-associated autoantibodies.
Given the phenotypic overlap of AGS with SLE and the genetic association of TREX1 with both monogenic chilblain lupus and complex SLE (10, 14–16), we asked whether heterozygosity for RNASEH2 mutations increases the risk of systemic autoimmunity. First, we examined whether parents of AGS patients (obligate heterozygous mutation carriers) have subclinical features of SLE. Clinical investigation revealed a high occurrence of autoantibodies, with ANAs detected in AGS parents at a substantially higher frequency than expected (P < 0.001; Figure 1A and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI78001DS1). In addition, other autoantibodies associated with SLE were also found in AGS parents (Supplemental Table 1). Therefore AGS parents appear to have an intermediate autoimmune phenotype, consistent with a recent report that identified an increased risk of autoimmune disease in families of AGS patients (27).
Figure 1. Heterozygosity for RNASEH2 mutations promotes systemic autoimmunity.

(A) Prevalence of ANAs in heterozygous parents of AGS patients. Shown is the percentage of ANA-positive parents of AGS patients (with mutations in TREX1, RNASEH2A–RNASEH2C, or SAMHD1; n = 28; P < 0.001) and in parents carrying RNASEH2 mutations (n = 16; P < 0.01, Fisher’s exact test) compared with data from a control population (n = 1,000) measured in the same laboratory. (B) RNASEH2B, RNASEH2C, and RNASEH2A gene structures. Variants identified in individuals with SLE (n = 600) are shown in bold above; variants in controls (n = 1,056) are shown below (boxes indicate exons). Colors correspond to those used for each subunit in the crystal structure in C: blue, RNASEH2A; green, RNASEH2B; red/pink, RNASEH2C. Except for variants L202S and D205E in RNASEH2A, all variants occurred with allele frequencies of <0.002 (Table 1). Variant A156A in RNASEH2C affected splicing (Table 1). (C) RNase H2 residues (yellow sticks) affected by missense mutations identified in SLE patients, mapped on the structure of the human enzyme (PDB ID, 3PUF; ref. 63).
Hypomorphic variants of RNASEH2 genes are associated with SLE.
We next sequenced the genes encoding all 3 RNase H2 subunits (RNASEH2A, RNASEH2B, and RNASEH2C) in 600 SLE patients and 1,056 controls and identified rare variants, both nonsynonymous and splice site mutations, associated with SLE (combined P = 0.0011; OR, 2.00; 95% CI, 1.32–3.06; Figure 1B and Table 1). These patients mainly showed cutaneous involvement, photosensitivity, arthritis, lymphopenia, and autoantibody formation; internal organ involvement was less frequent (Supplemental Table 2). Mutations were found in each of the 3 RNase H2 subunits, affecting residues that appear to be evenly distributed throughout the heterotrimeric enzyme complex (Figure 1, B and C). As all 3 subunits are required for enzyme activity (28, 29), we examined the effect of all mutations on recombinant enzyme activity, complex stability, and nuclear localization (Figure 2, A and B, and Supplemental Figure 1). Of the 18 SLE-associated mutations, 17 (94%) impaired RNase H2 function, with mutations either directly impairing enzyme activity, causing reduced complex stability, or altering subcellular localization (Figure 2, A and B, Supplemental Table 3, and Supplemental Figure 1). We therefore concluded that these variants affect cellular enzyme function, which suggests that reduced RNase H2 activity confers an increased risk for autoimmunity. Notably, RNase H2 functional impairment correlated with risk of SLE: as a group, mutations causing mild functional impairment conferred a 1.6-fold relative risk (OR, 1.69; P = 0.03), while mutations that severely affect RNase H2 function led to a 3.8-fold greater risk (OR, 3.94; P = 0.003) (Figure 2C), establishing a direct correlation between enzyme function and disease susceptibility. Furthermore, performing burden testing, weighting mutation count by severity (see Methods), strengthened the significance of the genetic association (P = 0.00026).
Table 1.
Contribution of rare variants of the RNASEH2 genes to SLE risk
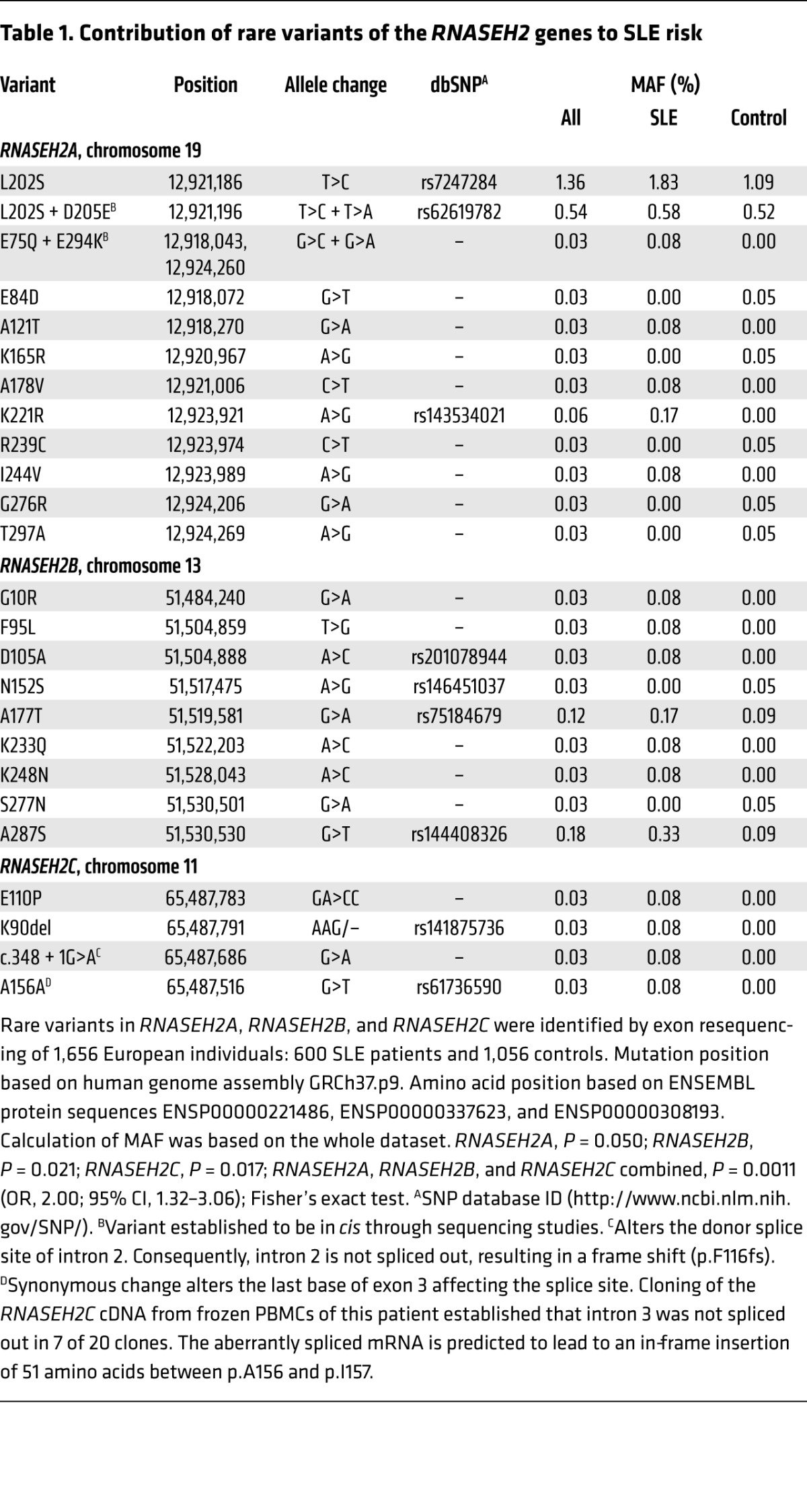
Figure 2. Rare sequence variants impair RNase H2 function in SLE and AGS.

RNASEH2 variants identified by resequencing caused reduced enzyme activity or complex stability. (A) Effects of RNASEH2 variants on recombinant enzyme activity on dsDNA substrate with an embedded ribonucleotide. Cleavage of an RNA/DNA substrate was similarly affected (data not shown). Mean (n = 3) and SD shown. (B) Thermal stability (expressed as thermal shift; ΔTm) of recombinant mutant RNase H2 complexes relative to WT; negative values denote less stable complexes. Mean (n ≥ 9) and SEM shown. (A and B) Unless specifically indicated as P = NS, activity was significantly different compared with WT (P < 0.01, t test). (C) Burden analysis demonstrated that increased risk of SLE correlated with functional severity of the RNASEH2 variants. Variant frequencies for neutral, mild, and severe functional consequences (see Supplemental Table 3) were calculated using number of occurrences as a percentage of total SLE cases (n = 600) or controls (n = 1,056) respectively. Fold change relative to control is also denoted.
To further validate the genetic association in the sequenced cohort (set 1), we sought to replicate our findings in a second independent cohort of 8,277 European individuals, consisting of 4,212 SLE cases and 4,065 controls (set 2) that had previously been genotyped for a GWAS (see Supplemental Methods). One of the rare variants identified in set 1, rs7247284 (L202S), was also genotyped in set 2. This variant showed 1.7-fold enrichment among SLE cases in set 1, although it did not by itself reach statistical significance. In the larger cohort of set 2, this L202S variant was significantly associated with SLE, after principal component analysis to correct for stratification (Supplemental Methods) and logistic regression resulting in a P value of 0.0046 (additive model, OR, 1.19; recessive model, OR, 2.31). In an additional approach to address that RNASEH2 gene variants in aggregate were associated with SLE in this second dataset, we imputed variants within each of the 3 RNASEH2 genes based on the cleaned GWAS data of set 2 using IMPUTE2, previously prephased with SHAPEIT (30). Association was explored by aggregated tests using RWAS and LRT (version 0.5; ref. 31) including only markers with IMPUTE info value > 0.8, sample call rate > 0.9, and linkage disequilibrium r2 < 0.5 at various minor allele frequency (MAF) thresholds (0.05, 0.01, 0.005, and 0.001). After permutation, aggregate analysis treating the 3 genes as a single unit (based on the fact that the subunits form a single enzyme) resulted in lower P values (e.g., 0.01 MAF threshold, P = 0.0005; Supplemental Table 4). In summary, our findings in 2 independent cohorts support an association of variants within the RNASEH2 genes with a risk for SLE.
RNASEH2 mutations impair ribonucleotide removal in AGS and SLE patient cells.
As removal of misincorporated ribonucleotides from genomic DNA is an essential function of RNase H2 (22–24), we hypothesized that impaired RNase H2 function causes ribonucleotides to accumulate at a low level in the genomic DNA of patient cells. We tested this hypothesis by single-cell gel electrophoresis of patient fibroblasts with mutations in RNASEH2C (patient AGS1, D39Y/D115fs; patient AGS2, P76L homozygous; patient SLE1, c.348 + 1G>A [F116fs] heterozygous) and RNASEH2B (patient SLE2, K233Q heterozygous). Under alkaline conditions (that detect single-strand breaks and alkali-labile sites such as ribonucleotides), RNase H2–deficient mouse embryonic fibroblasts (MEFs) and patient fibroblasts both showed a significant increase in DNA migrating from the nucleus, with significantly larger comet tail moments (see Supplemental Methods), compared with WT cells (Figure 3, A–C). To establish that this DNA alkali sensitivity was due to embedded ribonucleotides, we also performed single-cell gel electrophoresis at neutral pH with and without treatment with RNase H2. Under these conditions, an increase in DNA migrating from the nucleus after RNase H2 treatment should reflect the presence of ribonucleotides incorporated in the DNA. Tail moments were significantly higher for both RNase H2–deficient MEFs and patient fibroblasts compared with WT controls (Figure 3, A, D, and E), in keeping with the presence of higher numbers of misincorporated ribonucleotides in their genomic DNA. Thus, our biochemical and cellular findings were consistent with the notion that partial loss-of-function mutations in genes encoding the RNase H2 enzyme complex cause increased levels of embedded ribonucleotides in genomic DNA in both SLE and AGS patients.
Figure 3. Increased levels of misincorporated ribonucleotides in genomic DNA in cells from patients with AGS and SLE.

(A) Representative images of comets formed by human fibroblasts after single-cell electrophoresis at alkaline and neutral pH without or with RNase H2 pretreatment. Scale bar: 50 μm. (B and C) Quantification of Olive tail moment (see Supplemental Methods) after alkaline comet assay, indicative of single-strand DNA breaks, in (B) fibroblasts from SLE patients (SLE1 and SLE2), AGS patients (AGS1 and AGS2), and WT controls (WT1 and WT2) and in (C) Rnaseh2b-deficient or WT MEFs. (D and E) Quantification of Olive tail moment in comet assays of human fibroblasts and MEFs (as in B and C, respectively) under neutral condition, without and with RNase H2 pretreatment, showed increased Olive tail moments for RNase H2–deficient cells, consistent with elevated levels of embedded ribonucleotides in their genomic DNA. (B–E) Mean and SEM of 30–70 cells from 3 independent experiments per cell line. **P < 0.01, ***P < 0.001 versus WT2 (B and D) or WT (C and E), t test.
SLE and AGS patient fibroblasts have increased p53 signaling and reduced cellular proliferation.
In RNase H2–null mice, loss of enzymatic activity leads to increased embedded ribonucleotides and activation of a p53-dependent DNA damage response, resulting in arrested cell proliferation and embryonic lethality (23, 24). To determine whether reduced RNase H2 function in patients with AGS and SLE also causes a DNA damage response, we examined proliferation rates, cell cycle progression, and p53 activation. Patient fibroblasts proliferated significantly slower than did WT cells (Figure 4A). Flow cytometry of cells synchronized by serum starvation demonstrated that reduced proliferation resulted from delayed cell cycle progression, rather than increased apoptosis (Figure 4B). This was accompanied by elevated phosphorylation of p53 at Ser15 (Figure 4C). In addition, patient cells showed an increased number of p16-positive cells as well as cellular senescence, as shown by β-galactosidase staining (Figure 4, D and E). Fibroblasts from patients with SLE or AGS were also more likely to harbor γH2AX and p53-binding protein (53BP1) foci (Figure 4F), indicative of double-strand DNA (dsDNA) breaks at a low frequency, whereas UVC-induced dsDNA breaks in fibroblasts of patients and controls were repaired with similar efficiency (data not shown). Together, these findings demonstrated activation of a low-level DNA damage response in the absence of exogenous genotoxic stress in AGS and SLE patient cells.
Figure 4. Reduced proliferation and increased DNA damage response activation in SLE and AGS patient fibroblasts.

(A) Reduced proliferation rate of fibroblasts from patients with SLE (SLE1 and SLE2) and AGS (AGS1 and AGS2) compared with the mean of 5 WT control fibroblast lines (2 children, 3 adults). P < 0.05 (3 and 5 hours), P < 0.001 (7 hours), AGS1 vs. WT, t test. (B) Cell cycle analysis of propidium iodide–stained fibroblasts after synchronization in G1 by 24 hours of serum starvation. Representative flow cytometry images depict cell cycle delay in RNase H2–deficient fibroblasts. (C) Activation of a p53-dependent DNA damage response in patient fibroblasts in the absence of exogenous genotoxic stress, as shown by increased levels of phosphorylated p53 (Ser15). The same immunoblot probed for β-actin shows equal loading. (D) Increased number of p16-positive cells in RNase H2–deficient fibroblasts, measured by flow cytometry. (E) Increased senescence in patient fibroblasts, measured by β-galactosidase staining. Scale bar: 100 μm. (F) Increased dsDNA damage in RNase H2–deficient human fibroblasts. dsDNA breaks were visualized by immunostaining of γH2AX (red) and 53BP1 (green). The number of nuclei with 0–3 foci stained for both (yellow) among 50 randomly selected cells was quantified. Original magnification, ×400. Data are mean and SEM (D and E) or mean and SD (A, B, and F) of 3 independent experiments per patient and 5 independent WT control cell lines (A and B) or of 4 experiments per patient and 5 WT controls (D–F). (B–F) *P < 0.05, **P < 0.01, #P < 0.001 versus WT, t test.
Enhanced induction of type I IFN in RNASEH2 mutant cells.
As induction of DNA damage response signaling can stimulate IFN production (32), this may directly contribute to autoimmunity in SLE and AGS (33). Transcriptional profiling of fibroblasts revealed remarkably similar profiles in patients with SLE and AGS (Supplemental Figures 2–4). Although we did not detect constitutive type I “IFN transcription in unstimulated patient cells (Figure 5A), quantitative RT-PCR analysis demonstrated upregulation of IFN-stimulated genes (Supplemental Figure 2). We therefore hypothesized that the observed DNA damage response in patient fibroblasts, although insufficient in itself to induce IFN secretion, sensitizes the cells to mount type I IFN responses in response to external stimuli. Given that nucleic acids can trigger type I IFN production and promote systemic autoimmunity (34–36), we next challenged patient fibroblasts with polyinosinic:polycytidylic acid [poly(I:C)], a synthetic dsRNA analog; indeed, these displayed stronger and more sustained IFNB expression compared with WT cells (Figure 5A). The type I IFN response was further enhanced when cells were also exposed to UVC irradiation (Figure 5A). These findings suggest that well-known triggers of SLE, nucleic acids (either from viral infection or circulating nucleic acids released from apoptotic cells) and UV light (6, 7), could enhance and prolong IFN production in a susceptible individual, to initiate autoimmunity.
Figure 5. Type I IFN production by patient cells is enhanced in response to nucleic acids and UV light.

(A) Increased type I IFN activation in fibroblasts of patients with SLE and AGS compared with WT controls after treatment with poly(I:C) and/or UVC irradiation (30 J/m2 UVC for 3 seconds). Samples were processed after subsequent culture for 4, 8, and 24 hours. IFNB mRNA was normalized to GAPDH mRNA. Mean and SEM of ≥3 independent experiments per group (SLE and AGS, n = 2; WT, n = 5). *P < 0.05, **P < 0.01, ***P < 0.001 versus poly(I:C) at the same time point, Mann-Whitney U test. (B) Lesional skin from lupus patients carrying the indicated mutations in RNASEH2B and RNASEH2C showed high expression of type I IFN–inducible MxA (red) and CXCL10 (green). Scale bars: 100 μm.
Increased CPD formation in ribonucleotide-containing DNA.
Notably, cutaneous lupus and photosensitivity were the predominant symptoms of SLE patients carrying RNASEH2 mutations (Supplemental Table 2 and Supplemental Figure 5). Furthermore, in keeping with the enhanced type I IFN production we observed in SLE patient fibroblasts with RNASEH2 mutations, the IFN-induced proteins MxA and CXCL10 were detectable in lesional skin (Figure 5B). As exposure to UV irradiation frequently provokes flares of cutaneous lupus erythematosus and SLE, these clinical observations suggested that impaired RNase H2 function might specifically enhance photosensitivity. Consequently, we hypothesized that embedded ribonucleotides enhance the sensitivity of genomic DNA to photodamage. We therefore examined the formation of CPDs, the most abundant UV light–induced DNA lesion. CPDs are formed by the covalent linkage of the ring carbon 5 (C5) and C6 bonds of adjacent pyrimidines (37). Immediately after UVC exposure, CPD formation was significantly higher in fibroblasts from AGS and SLE patients compared with WT fibroblasts (Figure 6A). Increased CPD formation was also observed in patient fibroblasts irradiated with biologically relevant solar-simulated radiation, which, unlike UVC, is not filtered by the atmosphere (Supplemental Figure 6). Differences in CPD staining between mutant and WT cells resolved within hours after irradiation (Figure 6A and Supplemental Figure 6), in keeping with proficient nucleotide excision repair in the patient fibroblasts. Therefore, rather than difficulty in CPD removal by nucleotide excision repair, the patient DNA appeared intrinsically more sensitive to UV exposure, and we postulated that embedded ribonucleotides might enhance CPD formation.
Figure 6. Increased sensitivity to photodamage in ribonucleotide-containing DNA.

(A) Increased CPD formation in RNase H2–deficient fibroblasts (SLE1, SLE2, AGS1, and AGS2) compared with WT in response to 20 J/m2 UVC, measured immediately or 7 and 24 hours after irradiation. Box plots depict interquartile range (box), mean (square), median (line) and SD (whisker) of ≥3 independent experiments per patient and for 5 WT cell lines. *P < 0.05, **P < 0.01, ***P < 0.001 versus WT, t test. (B and C) Dihedral angles between covalent C5-C6 bonds (B) and pyrimidine ring planes (C) of 2 consecutive thymidines of the unmodified (1NAJ) and the ribonucleotide-containing (2L7D) Dickerson dodecamer. Ribonucleotide substitution increased colinearity between C5-C6 covalent bonds and coplanarity between pyrimidine rings, enhancing the probability of a photoreactive dimerization. For each dodecamer, 5 structures deposited in PDB were compared. Mean and SD shown. ***P < 0.001 versus 1NAJ, t test. (D) CPD formation in genomic DNA from fibroblasts of individual RNase H2–deficient versus WT patients and from Rnaseh2b–/– versus WT MEFs (n = 2 per group). Representative dot blot and mean intensities with SEM of 3 independent experiments are shown. *P < 0.05, **P < 0.01, ***P < 0.001 versus WT, t test. (E) CPD formation in double-stranded concatamers of chimeric DNA oligonucleotides containing 3 Dickerson dodecamer sequences in tandem. Replacement of a deoxyriboguanosine at position 4 of the dodecamer by a riboguanosine (R) is indicated.
Thymine dimerization in DNA is an ultrafast photoreaction dependent on DNA conformation prior to light absorption (38, 39). To determine the molecular basis of enhanced CPD formation, we examined the structural consequences of the presence of a ribonucleotide embedded in a DNA duplex. The Dickerson dodecamer, a dsDNA oligonucleotide, is a prototypic B-form DNA (40). Unlike DNA, RNA adopts an A-form helix, which is more resistant to photodamage (41, 42). However, the presence of single riboguanosine positioned 3 bases upstream of 2 adjacent thymines within the Dickerson dodecamer does not convert it from B- to A-form DNA (43). We therefore examined instead whether this ribonucleotide enhances a stereochemical dimerizable conformation resulting from altered thymine stacking. We calculated the dihedral angles between the covalent C5-C6 bonds as well as pyrimidine ring planes of the adjacent thymines based on the NMR structures of the Dickerson dodecamer (1NAJ; ref. 44) and the ribonucleotide-containing dodecamer (2L7D; ref. 43). Comparison of the dihedral angles revealed structural changes indicative of an increase in colinearity between C5-C6 covalent bonds as well as in coplanarity between pyrimidine rings (Figure 6, B and C). Thus, the presence of a ribonucleotide within the dodecamer induces conformational changes of the adjacent thymines that render them more sensitive to photoreactive dimerization. Notably, isolated DNA of RNase H2–deficient human fibroblasts and MEFs displayed a higher rate of CPD formation than WT controls (Figure 6D), which indicated that enhanced CPD formation was an intrinsic feature of the ribonucleotide-containing DNA. Finally, to experimentally confirm that enhanced photodamage in mutant cells may be directly caused by an alteration in DNA structure imposed by ribonucleotides, we examined CPD formation in synthetic DNA oligonucleotides containing the dodecamer sequence with an embedded ribonucleotide. These also had enhanced UVC-induced CPD formation (Figure 6E). We therefore concluded that increased CPD formation in ribonucleotide-containing DNA could contribute to enhanced sensitivity of patients with RNASEH2 mutations to UV irradiation.
Discussion
Here we report that rare genetic variants in the genes encoding the RNase H2 enzyme increase the risk for systemic autoimmunity. First, we delineated an intermediate phenotype of subclinical autoimmunity among heterozygous parents of AGS patients by demonstrating an increased occurrence of ANAs. The appearance of autoantibodies is one of the first signs of systemic autoimmunity and reflects a series of immunologic events that typically precedes the onset of clinical symptoms by several years. For example, the presence of ANAs or anti-Ro, anti-La, or antiphospholipid antibodies has been shown to increase the risk for clinical disease by at least 40-fold (45). Second, by resequencing the RNASEH2 genes, we identified an excess of rare variants in patients with SLE compared with healthy controls, with biochemically assessed functional severity correlating with SLE risk. We replicated this finding in an independent cohort, in which imputed variants from a large GWAS dataset show significant association in aggregate (P = 0.0005). Future independent, large-scale, sequence-based studies will be valuable in further substantiating this association. Taken together, these findings support a role for RNASEH2 genes as a risk factor for systemic autoimmunity, and moreover highlight the potential contribution of rare variants to the heritability of SLE.
Our findings suggest that RNase H2 dysfunction may contribute to the multistep initiation of SLE autoimmunity at several stages, by enhancing the type I IFN response to both endogenous and external factors (Supplemental Figure 7). First, systemic autoimmunity may be promoted by innate immune recognition of nucleic acid intermediates generated during repair of DNA damage that results from ribonucleotide misincorporation or incomplete resolution of R-loops in cells with impaired RNase H2 activity (25). Indeed, in Trex1–/– cells, type I IFN activation has been attributed to stimulation of pattern recognition receptors by cytosolic DNA (33, 46, 47), which may originate from DNA repair processes (33). However, IFN may also be induced by cytoplasmic nucleic acids arising from replication intermediates of retroviruses/retroelements in TREX1- and/or RNase H2–deficient cells (46, 48). Second, p53 activation in AGS/SLE patient cells may more directly promote innate immune signaling, perhaps through the activation of p53 target genes that stimulate type I IFN or that induce enhanced sensitivity to type I IFN stimuli (32, 49).
AGS and SLE patient fibroblasts do not constitutively express detectable levels of IFN-β, although very weak or intermittent type I IFN production cannot be excluded (50). However, transcriptional regulation of IFN gene expression is complex, with multiple positive and negative regulatory elements (51), and low-level accumulation of intracellular nucleic acids or activation of p53 signaling may therefore not be sufficient to induce type I IFN. Indeed, the IRF3/IRF7/STING/TBK1-dependent activation of antiviral genes in Trex1–/– cells occurs without induction of type I IFN (52), despite IRF3 and IRF7 being established transcriptional activators of IFN-β (53). Consistent with this, transcriptional profiling of patient fibroblasts revealed induction of both p53-dependent genes and antiviral IFN-induced genes in the absence of detectable type I IFN activation. As p53 induction has also been reported in TREX1-null mouse cells (33), this raises the possibility of a common pathogenic mechanism in AGS involving DNA damage signaling. Even without transcriptional IFN induction, priming of IFN signaling through upregulation of pathway components like IRF7 (53) could explain the enhanced sensitivity of cells with impaired RNase H2 function to nucleic acids and UV irradiation (Supplemental Figure 7). Innate immune activation induced by a DNA damage response may be further enhanced by increased CPD formation of ribonucleotide-containing DNA upon UV irradiation. Although we did not observe increased sensitivity to apoptosis in patient cells in response to UV light (Supplemental Figure 8), increased DNA damage in RNase H2–deficient skin cells could render them more susceptible to apoptosis upon repeated exposure to UV light, further promoting the release of immunostimulatory nucleic acids. Such UV light–induced enrichment of lupus nucleic acid–related autoantigens (e.g., Ro/SSA and La/SSB) has been observed in apoptotic surface blebs on keratinocytes (54). As UV light is a major trigger of SLE symptoms (7), our data provide an additional mechanism for the photosensitivity observed in SLE patients and illustrate the interplay of genetic and environmental factors in disease initiation. Given that type I IFN activation underlies initiation of inflammation and autoimmunity in AGS and SLE, targeting the IFN axis by type I IFN blockade may be of potential therapeutic efficacy. Indeed, IFN-α antagonists have been tested in SLE patients in clinical trials with promising results (55), while JAK inhibition was recently reported to ameliorate symptoms in patients with the type I IFN–driven STING-associated vasculopathy (56). TREX1 has been implicated in the restriction of retroelements by metabolizing reverse-transcribed retroelement-derived RNA (46), raising the possibility that retroelement-derived RNA/DNA hybrids may act as substrates of RNase H2. As RNA/DNA hybrids were previously shown to act as a molecular pattern sensed by TLR9 (57), accrual of retroelement-derived RNA/DNA hybrids in RNase H2–deficient cells could contribute to innate immune activation. Further investigations are required to assess the potential role of RNase H2 in retroelement restriction.
In conclusion, our genetic and functional data implicate RNase H2 in SLE, establishing a ribonuclease alongside several DNA nucleases in SLE pathogenesis. Whereas DNase I, DNase II, and DNase 1L3 are involved in clearance of circulating nucleic acids (34–36, 58), RNase H2 and TREX1 (16) are intracellular nucleases involved in DNA replication and repair, and now have established roles in systemic autoimmunity. Furthermore, the DNA damage pathway activation and enhanced UV-induced CPD formation observed in cells of patients with RNASEH2 mutations suggest a role for DNA damage–associated pathways in the pathogenesis of autoimmunity, which warrants further investigation.
Methods
Human samples.
Samples from 600 SLE patients from Germany (n = 471) and the United Kingdom (n = 129) were collected as previously described (16, 59). SLE phenotypes were defined according to the diagnostic criteria by the American College of Rheumatology (60, 61). Control samples (n = 1,056) were obtained from the blood donation programs at University Hospital Dresden and University Hospital Bonn. See Supplemental Methods for details of the replication cohort.
Mutation analysis.
The coding sequences of RNASEH2A, RNASEH2B, and RNASEH2C were amplified by PCR from genomic DNA using gene-specific primers (Supplemental Table 5). Both strands of the purified PCR products were sequenced using fluorescent dye terminator chemistry on ABI 3730 or ABI3010xl instruments. All identified variants were validated by resequencing. Sequence reads were analyzed using Vector NTI, DNASTAR, Sequencher, and Mutation Surveyor.
Autoantibody testing.
Routine serological tests were carried out at the diagnostic laboratory of the Institute of Immunology, Technische Universität Dresden. ANAs were determined using Hep-2 cells; extractable nuclear antigens were analyzed by immunodiffusion. Data on ANAs from a reference population were obtained from 1,000 blood donors (samples collected at the Institute of Immunology, Technische Universität Dresden).
Cells, transfection, and confocal microscopy.
Human fibroblasts were derived from skin biopsies and maintained in DMEM with 10% FCS, 2 mM l-glutamine, and antibiotics. In all experiments, passage-matched fibroblasts (passages 4–11) were used. Rnaseh2b-deficient MEFs were generated as previously described (24). N-terminally GFP-tagged WT RNase H2 subunits were cloned into pEGFP-C1. GFP was replaced by YFP, CFP, or mCherry using AgeI and BsrGI. pRSET-mCherry was provided by R.Y. Tsien (UCSD, La Jolla, California, USA). Mutations were introduced by site-directed mutagenesis (QuikChange Lightning; Agilent) or amplified from patient cDNA. HeLa cells were grown on coverslips in DMEM with 10% FCS, 2 mM l-glutamine, and antibiotics and transfected with 200 ng plasmid DNA of each fluorescently tagged RNase H2 subunit using Fugene HD (Roche). Cells were fixed in 4% paraformaldehyde and mounted in Vectashield Mounting Medium with DAPI (Vector Labs). Confocal microscopy was performed using a Leica TCS SP5 II with a ×40/1.25 oil objective. For subcellular localization, 300 cells total were analyzed per mutation in 3 independent experiments.
Immunohistochemistry.
Paraffin-embedded skin biopsies were cut into 2- to 5-μm sections, rehydrated, and boiled in sodium citrate buffer (pH 6.0). Sections were stained with mouse anti-MxA (provided by O. Haller, Freiburg University, Breisgau, Germany; 1:400 dilution) or rabbit anti-CXCL10 (Bioss; 1:100 dilution), followed by staining with EnVision G|2 System/AP Rabbit/Mouse (Dako) or Alexa Fluor 488–conjugated goat anti-mouse IgG (Invitrogen; 1:200 dilution), respectively. Sections were counterstained with Mayer’s hematoxylin (Merck). For staining of dsDNA breaks, fibroblasts grown on coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS, and probed with mouse anti-γH2AX (Ser139; Novus Biologicals, clone 2F3; 1:500 dilution) and rabbit anti-53BP1 (Novus Biologicals; 1:2,000 dilution). Slides were then probed with Alexa Fluor 546–conjugated goat anti-mouse IgG or Alexa Fluor 488–conjugated goat anti-rabbit IgG (Molecular Probes), respectively, mounted in Vectashield Mounting Medium with DAPI (Vector Labs), and analyzed with a Zeiss Axioimager A1 microscope. For quantification, the number of double-stained dots was counted in 50 randomly selected cells per slide. For detection of CPDs, cells grown on coverslips were fixed with methanol (–20°C) and acetone. DNA was denatured in 0.1 N NaOH. Cells were then dehydrated with increasing concentrations of ethanol (70%–100%) and incubated with mouse anti-CPD (Kamiya Biomedical Co., clone KTM53; 1:1,000 dilution) followed by staining with Alexa Fluor 488–conjugated goat anti-mouse IgG (Molecular Probes; 1:200 dilution). The mean fluorescence intensity from 50 cells per slide was determined using Image J.
RT-PCR.
Total RNA from fibroblasts was extracted with the RNeasy Mini Kit (Qiagen) followed by DNase I digestion. IFNB expression was determined by quantitative real-time RT-PCR using Taqman Universal PCR Master Mix (Applied Biosystems) on an ABI7300 and normalized to GAPDH expression, or using iQ SYBR Green Supermix (Bio-Rad) on an Mx3005P qPCR system (Agilent) and normalized to HPRT1 (Supplemental Table 6).
Western blotting.
Fibroblasts were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM sodium orthovanadate, 20 mM sodium fluoride) supplemented with 1× Complete Protease Inhibitor Cocktail and 1× PhosSTOP phosphatase inhibitors (Roche). 5 μg total protein was subjected to SDS-PAGE followed by Western blotting using antibodies against phosphorylated p53 (Ser15) (Santa Cruz; 1:200 dilution) and β-actin (Sigma-Aldrich, clone AC-74; 1:10,000 dilution). Immunoreactive signals were detected by chemiluminescence (Lumi-Light PLUS; Roche).
RNase H2 activity and thermal stability assays.
Recombinant RNase H2 complexes were purified after expression in E. coli and used in a FRET-based fluorescent substrate release assay to determine enzyme activity, as previously described (29). Thermal stability assays were performed using the fluorescence-based thermal shift (ThermoFluor) method, as previously described (29).
Cell cycle analysis and β-galactosidase staining.
Fibroblasts were synchronized by serum starvation for 24 hours. At the indicated time points after cultivation in complete growth medium, cells were harvested, fixed, and stained with propidium iodide. Flow cytometry was performed on a FACSCalibur instrument using CellQuest software (both BD). Fibroblasts were stained using the Senescence β-Galactosidase Staining Kit (Cell Signaling) and analyzed by light microscopy. For flow cytometry analysis of p16, fibroblasts were fixed in –20°C methanol, permeabilized with 0.01% Tween-PBS, and stained with Alexa Fluor 488–conjugated mouse anti-p16 antibody (Santa Cruz, catalog no. sc-1661).
UV irradiation.
Fibroblasts were grown in 6-well plates or on coverslips and exposed to UVC light (254 nm; low-pressure mercury lamp) at a radiation dosage of 20 or 30 J/m2, or to solar-simulated radiation (Arimed B lamp) at a radiation dose of 100 J/m2. UV lamps were calibrated using a thermopile TS 50-1 (Physikalisch-technische Werkstätten). For the experiment described in Figure 5A, cells were incubated with 100 μg/ml poly(I:C) (Invivogen) for 3 hours. Cells were washed with PBS, and fresh medium was added following UVC irradiation. At the indicated times after irradiation, cells were processed for RNA isolation and RT-PCR or fixed for CPD staining. For flow cytometry analysis of CPDs, cells were fixed in 4% formaldehyde, permeabilized with 0.5% Triton X-100 (in PBS) and stained with mouse anti-CPD (Kamiya Biomedical Co., clone KTM53) and goat F(ab′)2 anti-mouse Alexa Fluor 488 (Molecular Probes).
South Western blotting.
After UV irradiation, genomic DNA was isolated as follows. Cell pellets were resuspended in 200 μl lysis buffer (100 mM Tris, 5 mM EDTA, 0.2% SDS, 0.2 M NaCl, pH 7.0) containing 1 mg/ml proteinase K and centrifuged at 13,000 g in an Eppendorf minifuge. The supernatant was mixed with an equal volume of isopropanol, incubated at –80°C for 1 hour, and centrifuged at 13,000 g. Pellets were washed with 70% and 100% ethanol, air dried, and dissolved in TE (pH 7). 400 ng genomic DNA in a volume of 200 μl 150 mM NaCl was then exposed to 30 J/m2 UVC in a 6-well plate. After mixing with 200 μl NE buffer (10 mM EDTA, 0.4 M NaOH), samples were denatured at 95°C and placed on ice. Next, DNA samples were blotted to a nitrocellulose membrane pre-soaked in 2× SSC using a dot blotter and vacuum pump. The membrane was washed in neutralization buffer (1 M Tris-HCl, 2 M NaCl, pH 7) and baked at 80 °C for 2 hours, followed by blocking in 4% milk in PBS and probing with mouse anti-CPD (Kamiya Biomedical Co., clone KTM53; 1:1,000 dilution). After 3 washes in PBS with 1% Tween, the membrane was incubated with anti-mouse IgG (Dako; 1:1,000 dilution) for 1 hour. Immunoreactive signals were detected by chemiluminescence. Signal density was determined using Image J. DNA oligonucleotides (BioSpring) containing the Dickerson dodecamer sequence 3 times in a row with or without riboguanosine (underlined) at position 4 (oligo 1, CGCGAATTCGCG-CGCGAATTCGCG-CGCGAATTCGCG; oligo 2, CGCGAATTCGCG-CGCGAATTCGCG-CGCGAATTCGCG; oligo 3, CGCGAATTCGCG-CGCGAATTCGCG-CGCGAATTCGCG) were concatamerized by heating to 96°C followed by slow cooling to room temperature. 2 nmol of each of the oligonucleotides was then exposed to UVC for 20 minutes and subjected to South Western dot blotting using anti-CPD antibody.
Modeling of Dickerson dodecamers.
Dihedral angles were measured from atomic coordinates within each strand of the 5 representative models of the modified (1NAJ; ref. 44) and unmodified (2L7D; ref. 43) NMR Dickerson structures deposited in the Protein Data Bank (PDB; www.PDB.org). The dihedral angle between the covalent bonds linking C5 and C6 of the 2 consecutive thymidines (θ1; Figure 6B) that constitute the 4 members of the cyclobutane ring reflects the degree of colinearity. The angle is 0° when the 2 bonds are perfectly colinear and 90° when perpendicularly arranged. The dihedral angle between the 2 vectors perpendicular to the planes defined by the heterocyclic rings of the adjacent thymidines (θ2; Figure 6C) indicates the degree of coplanarity. This angle is 0° when the 2 heterocyclic rings are perfectly coplanar and 90° when perpendicularly arranged.
Comet assay and RNA sequencing.
See Supplemental Methods and Supplemental Figures 2–4 for details. RNA sequencing data were deposited in GEO (accession no. GSE63755).
Statistics.
Data are presented as mean ± SD or mean ± SEM as indicated. Statistical significance was determined by 2-tailed Student’s t test, Fisher’s exact test, or Mann-Whitney U test as indicated. A P value less than 0.05 was considered statistically significant. The number of mutations between cases and controls was compared with a 2 × 2 contingency table using 2-sided Fisher’s exact test. The burden analysis was carried out after grouping the mutations into 3 categories (neutral, mild, and severe functional impact) using logistic regression; disease ORs reported are for comparisons of mutation carriers and noncarriers within the same category. Finally, logistic regression was also carried out on a continuous mutation burden, using a weighted approach (62), giving mutations with mild functional defects a conservative score of 0.5, those with severe functional defects a score of 1, and neutral ones a score of 0. Severity of functional deficiency was determined using data from enzyme activity, complex stability, and nuclear localization studies (Supplemental Table 3). Statistical analysis was carried out using SPSS and R (www.r-project.org). See Supplemental Methods for association analysis of the replication cohort using imputation and aggregated analysis.
Study approval.
DNA samples from SLE patients and controls for genetic analysis were collected with written informed consent after approval by the institutional ethics review boards of the participating centers. Human primary fibroblasts were derived from skin biopsies with written informed consent from patients or their parents. The study was approved by the ethics committee of the Medical Faculty, Technische Universität Dresden.
Supplementary Material
Acknowledgments
We thank the patients, their families, the German AGS patient organization, and the Lupus Erythematodes Selbsthilfegemeinschaft e.V. for participation in this study. We thank Kerstin Engel, Susan Hunger, Nick Zimmermann, Monika Schwarz, Rachel Rigby, and Ailsa Revuelta for technical assistance; Peter Knuschke for calibration of the UV light source; Andrea Petzold and Christian Kupka (Technische Universität Dresden) for providing skin samples from healthy individuals; Bridget Griffiths (Freeman Hospital, Newcastle upon Tyne, United Kingdom) for contributing patients and Bernd Pötzsch (University of Bonn) for help with collecting DNA samples from anonymous blood donors; Alexander Rapp and Cristina Cardoso (Technical University Darmstadt) for advice on comet assay and helpful discussion; Angela-Rösen-Wolff, Herbert Schulz, Karen Mackenzie, Björn Hiller, David Hunt, and Veronique Vitart for helpful discussions; Silke Tulok (Technische Universität Dresden) for assistance with confocal microscopy; Otto Haller for anti-MxA antibody; Caroline Hayward (MRC Human Genetics Unit Edinburgh) for providing sera from healthy individuals; and Roger Y. Tsien for pRSET-mCherry. ThermoFluor assays were carried out at the Edinburgh Centre for Translational and Chemical Biology (ECTCB), which is funded by the Wellcome Trust and the Biotechnology and Biological Sciences Research Council. M.M. Nöthen is a member of the DFG-funded excellence cluster ImmunoSensation. This study was supported by grants from the DFG (KFO 249/GU1212/1-1 and GU1212/1-2 to C. Günther; KFO 249/LE1074/4-1 and LE1074/4-2 to M.A. Lee-Kirsch; KFO 249/AR2133/6-1 to A. Roers); by Friede Springer Stiftung (to M.A. Lee-Kirsch); by the German Federal Ministry of Education and Research (BMBF 01 GM 0310/0634 to I. Melchers); by an MRC Senior Clinical Fellowship and Lister Institute Prize Fellowship (to A.P. Jackson); by the ISCIII (grant no. 02558), in part through FEDER funds of the European Union (to M.E. Alarcon-Riquelme); and by the Fundación Ramón Areces and the European Science Foundation support for the BIOLUPUS project (coordinated by M.E. Alarcon-Riquelme).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(1):413–424. doi:10.1172/JCI78001.
See the related Commentary beginning on page 102.
References
- 1.Atianand MK, Fitzgerald KA. Molecular basis of DNA recognition in the immune system. J Immunol. 2013;190(5):1911–1918. doi: 10.4049/jimmunol.1203162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6(11):823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ronnblom L, Alm GV, Eloranta ML. Type I interferon and lupus. Curr Opin Rheumatol. 2009;21(5):471–477. doi: 10.1097/BOR.0b013e32832e089e. [DOI] [PubMed] [Google Scholar]
- 4.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416(6881):603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 5.Lovgren T, Eloranta ML, Bave U, Alm GV, Ronnblom L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50(6):1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 6.James JA, Harley JB, Scofield RH. Role of viruses in systemic lupus erythematosus and Sjogren syndrome. Curr Opin Rheumatol. 2001;13(5):370–376. doi: 10.1097/00002281-200109000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Bijl M, Kallenberg CG. Ultraviolet light and cutaneous lupus. Lupus. 2006;15(11):724–727. doi: 10.1177/0961203306071705. [DOI] [PubMed] [Google Scholar]
- 8.Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15(1):49–54. doi: 10.1002/ana.410150109. [DOI] [PubMed] [Google Scholar]
- 9.Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-α in infants with progressive familial encephalopathy. J Neurol Sci. 1988;84(2–3):201–208. doi: 10.1016/0022-510x(88)90125-6. [DOI] [PubMed] [Google Scholar]
- 10.Ramantani G, et al. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutieres syndrome. Arthritis Rheum. 2010;62(5):1469–1477. doi: 10.1002/art.27367. [DOI] [PubMed] [Google Scholar]
- 11.Rice GI, et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12(12):1159–1169. doi: 10.1016/S1474-4422(13)70258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crow YJ, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38(8):910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 13.Crow YJ, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38(8):917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 14.Rice G, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80(4):811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee-Kirsch MA, et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med. 2007;85(5):531–537. doi: 10.1007/s00109-007-0199-9. [DOI] [PubMed] [Google Scholar]
- 16.Lee-Kirsch MA, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39(9):1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 17.Rice GI, et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41(7):829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A. 2011;155A(1):235–237. doi: 10.1002/ajmg.a.33778. [DOI] [PubMed] [Google Scholar]
- 19.Rice GI, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44(11):1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rice GI, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46(5):503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee-Kirsch MA, Wolf C, Gunther C. Aicardi-Goutieres syndrome: a model disease for systemic autoimmunity. Clin Exp Immunol. 2014;175(1):17–24. doi: 10.1111/cei.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nick McElhinny SA, et al. Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol. 2010;6(10):774–781. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reijns MA, et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149(5):1008–1022. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med. 2012;209(8):1419–1426. doi: 10.1084/jem.20120876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chon H, et al. RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res. 2013;41(5):3130–3143. doi: 10.1093/nar/gkt027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sparks JL, et al. RNase H2-initiated ribonucleotide excision repair. Mol Cell. 2012;47(6):980–986. doi: 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt JL, et al. Family history of autoimmune disease in patients with Aicardi-Goutieres syndrome. Clin Dev Immunol. 2012;2012:206730. doi: 10.1155/2012/206730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chon H, et al. Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucleic Acids Res. 2009;37(1):96–110. doi: 10.1093/nar/gkn913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reijns MA, et al. The structure of the human RNase H2 complex defines key interaction interfaces relevant to enzyme function and human disease. J Biol Chem. 2011;286(12):10530–10539. doi: 10.1074/jbc.M110.177394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39(7):906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 31.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9(2):179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 32.Brzostek-Racine S, Gordon C, Van SS, Reich NC. The DNA damage response induces IFN. J Immunol. 2011;187(10):5336–5345. doi: 10.4049/jimmunol.1100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131(5):873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 34.Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25(2):177–181. doi: 10.1038/76032. [DOI] [PubMed] [Google Scholar]
- 35.Yasutomo K, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. 2001;28(4):313–314. doi: 10.1038/91070. [DOI] [PubMed] [Google Scholar]
- 36.Nagata S. Autoimmune diseases caused by defects in clearing dead cells and nuclei expelled from erythroid precursors. Immunol Rev. 2007;220:237–250. doi: 10.1111/j.1600-065X.2007.00571.x. [DOI] [PubMed] [Google Scholar]
- 37.Novarina D, Amara F, Lazzaro F, Plevani P, Muzi-Falconi M. Mind the gap: keeping UV lesions in check. DNA Repair (Amst). 2011;10(7):751–759. doi: 10.1016/j.dnarep.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schreier WJ, et al. Thymine dimerization in DNA is an ultrafast photoreaction. Science. 2007;315(5812):625–629. doi: 10.1126/science.1135428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Law YK, Azadi J, Crespo-Hernandez CE, Olmon E, Kohler B. Predicting thymine dimerization yields from molecular dynamics simulations. Biophys J. 2008;94(9):3590–3600. doi: 10.1529/biophysj.107.118612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drew HR, et al. Structure of a B-DNA dodecamer: conformation and dynamics. Proc Natl Acad Sci U S A. 1981;78(4):2179–2183. doi: 10.1073/pnas.78.4.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Middleton CT, de La HK, Su C, Law YK, Crespo-Hernandez CE, Kohler B. DNA excited-state dynamics: from single bases to the double helix. Annu Rev Phys Chem. 2009;60:217–239. doi: 10.1146/annurev.physchem.59.032607.093719. [DOI] [PubMed] [Google Scholar]
- 42.Kundu LM, Linne U, Marahiel M, Carell T. RNA is more UV resistant than DNA: the formation of UV-induced DNA lesions is strongly sequence and conformation dependent. Chemistry. 2004;10(22):5697–5705. doi: 10.1002/chem.200305731. [DOI] [PubMed] [Google Scholar]
- 43.DeRose EF, Perera L, Murray MS, Kunkel TA, London RE. Solution structure of the Dickerson DNA dodecamer containing a single ribonucleotide. Biochemistry. 2012;51(12):2407–2416. doi: 10.1021/bi201710q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Z, Delaglio F, Tjandra N, Zhurkin VB, Bax A. Overall structure and sugar dynamics of a DNA dodecamer from homo- and heteronuclear dipolar couplings and 31P chemical shift anisotropy. J Biomol NMR. 2003;26(4):297–315. doi: 10.1023/A:1024047103398. [DOI] [PubMed] [Google Scholar]
- 45.Arbuckle MR, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 46.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134(4):587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol. 2014;192(12):5993–5997. doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- 48.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11(11):1005–1013. doi: 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Munoz-Fontela C, et al. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med. 2008;205(8):1929–1938. doi: 10.1084/jem.20080383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol. 2001;2(5):378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 51.Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I IFN induction: a current view. Int Immunol. 2005;17(11):1367–1378. doi: 10.1093/intimm/dxh318. [DOI] [PubMed] [Google Scholar]
- 52.Hasan M, et al. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol. 2013;14(1):61–71. doi: 10.1038/ni.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Honda K, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 54.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179(4):1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petri M, et al. Sifalimumab, a human anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013;65(4):1011–1021. doi: 10.1002/art.37824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rigby RE, et al. RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 2014;33(6):542–558. doi: 10.1002/embj.201386117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Al-Mayouf SM, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011;43(12):1186–1188. doi: 10.1038/ng.975. [DOI] [PubMed] [Google Scholar]
- 59.Aitman TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439(7078):851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 60.Tan EM, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 61.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 62.Price AL, et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010;86(6):832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Figiel M, Chon H, Cerritelli SM, Cybulska M, Crouch RJ, Nowotny M. The structural and biochemical characterization of human RNase H2 complex reveals the molecular basis for substrate recognition and Aicardi-Goutieres syndrome defects. J Biol Chem. 2011;286(12):10540–10550. doi: 10.1074/jbc.M110.181974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.