

Abstract
Autophagy is a catabolic process mediated by incorporation of cellular material into cytosolic membrane vesicles for lysosomal degradation. It is crucial for maintaining cell viability and homeostasis in response to numerous stressful conditions. In this Review, the role of autophagy in both normal biology and disease is discussed. Emphasis is given to the interplay of autophagy with nutrient signaling through the ULK1 autophagy pre-initiation complex. Furthermore, related cellular processes utilizing components of the canonical autophagy pathway are discussed due to their potential roles in nutrient scavenging. Finally, the role of autophagy in cancer and its potential as a cancer therapeutic target are considered.
The biology of autophagy
Autophagy, or cellular “self-eating,” is a vesicular trafficking pathway that targets intracellular substrates, from entire organelles to protein aggregates and specific proteins, for lysosomal degradation and recycling (1). Identified by genetic and morphologic studies in yeast and mammalian cells, this pathway is now known to be conserved across most of eukaryotic life. At the cellular level, autophagy has a profound impact on the biology of normal and stressed cells. On an organismal level, autophagy is critical for development and adult homeostasis (2). Macroautophagy (hereafter referred to as “autophagy”) is mediated by the action of specialized vesicles called autophagosomes that arise by elongation of membrane precursors called phagophores or isolation membranes (Figure 1 and ref. 3). Because autophagosomes are formed by elongation of precursor vesicles, upon closure they exhibit a characteristic double-membrane structure. As isolation membranes elongate, they engulf intracellular substrates that are subsequently degraded by lysosomal enzymes following autophagosome-to-lysosome fusion (Figure 1). The engulfment and degradation of intracellular aggregates and organelles maintains cellular homeostasis in nutrient-replete conditions. During starvation, autophagy recycles the captured components to sustain macromolecular synthesis, anaplerosis, and energy production for survival (4, 5). Since its first description, research into autophagy has transitioned from morphologic examination of autophagosomes and their contents to identification of the molecular machinery that controls autophagosome formation (3), and later to an emerging view of an expansive role for this pathway in physiology and disease (6). Autophagy defects are linked to numerous diseases including cancer, immune disorders, infectious diseases, heart disease, and neurodegeneration (7, 8).
Figure 1. The autophagy pathway and its role in biology and diseases.

Schematic of the intracellular membrane events involved in the autophagy pathway. Shaded box at the right side lists examples of biological functions that autophagy can accomplish by targeting specific cargos, as well as diseases linked to the deficiency of these autophagy functions. Text at the bottom right describes roles of autophagy-mediated nutrient recycling.
How does this one pathway affect so many different diseases? Some of the answers may lie in the cargos that are delivered for degradation. Although autophagy was initially believed to target bulk cytoplasm non-selectively, it has become clear that autophagy can also target a variety of specific intracellular substrates, from protein aggregates to damaged mitochondria (referred to as mitophagy), peroxisomes (pexophagy) (9), lipid droplets (lipophagy) (10), ferritin (ferritinophagy) (11), and even intracellular microorganisms (xenophagy) (Figure 1). Several of these intracellular substrates are targeted, at least in part, as a result of polyubiquitination, which recruits receptor proteins that bind to both ubiquitin and the autophagosomal protein light chain 3 (LC3), thereby colocalizing sites of autophagosome formation with specific substrates. Notable receptor proteins include p62/SQSTM1 (p62), NBR1, and NDP52 (12, 13). The elimination of targeted substrates by autophagy controls many aspects of normal physiology, from the elimination of parental mitochondria in fertilized oocytes, which is the basis of maternal mitochondrial inheritance, to the complete elimination of mitochondria from developing reticulocytes (14, 15), to the regulation of cellular lipid and iron metabolism (11). A failure to clear potentially dangerous bulk substrates such as damaged mitochondria underlies at least some of the cellular dysfunctions that lead to the development of degenerative conditions such as cardiovascular disease and various forms of neurodegeneration. One clear example is early onset familial Parkinson’s disease, which is associated with mutation of mitochondrial ubiquitin ligase–encoding gene Parkin, and defective clearance of mitochondria through mitophagy (16).
In addition to the autophagy-dependent clearance of targeted substrates, the degradation of engulfed autophagosomal contents can recycle nutrients to support cellular metabolism. The physiologic significance of nutrient recycling by autophagy is emphasized by the phenotype of autophagy-deficient knockout mice, which fail to survive nutrient starvation stress that occurs upon birth, when milk-derived nutrients are not yet available (17), and by the induction of autophagy in multiple tissues, including muscle, liver, pancreas, adipose tissue, and brain, by exercise (18, 19). In cancers, these multiple functions of autophagy, targeted substrate clearance and nutrient recycling, combine to influence disease initiation and progression in a complex, context-dependent (i.e., genotype- and stage-dependent) manner (20).
The molecular basis of autophagy
An overview of the autophagy machinery
The molecular basis of autophagy was revealed initially by yeast genetics (21–23) and subsequently via studies in higher organisms (3, 24). Up to 35 autophagy-related genes (ATGs) have been identified, and the ATG genes that make up the core machinery of autophagy can be classified into several functional units (mammalian nomenclature is used hereafter): the Unc-51 like autophagy activating kinase 1 (ULK1) protein kinase complex, an initiating point for the autophagic cascade; the VPS34–beclin 1 PI3K complex; two autophagy-specific ubiquitin-like (Ubl) conjugation systems; and phosphatidylinositol-3-phosphate (PI3P) effectors and the transmembrane recycling protein ATG9. Many ATG proteins contribute to the two Ubl conjugation reactions in which LC3 and ATG12 are the Ubl proteins (25). The conjugation of ATG12 to ATG5 is catalyzed by the E1-like enzyme ATG7 and E2-like enzyme ATG10. Strikingly, LC3 is conjugated to phosphatidyl ethanolamine (PE) instead of a protein, a process that is catalyzed by ATG7, E2-like ATG3, and the ATG12-ATG5 conjugate, which is considered to be an E3-like enzyme for LC3-PE conjugation (26, 27). LC3-PE conjugation (or LC3 lipidation) is required for autophagosome formation, cargo recognition, and autophagic membrane tethering (13, 28–30).
Nutrient signals and ULK1 complex–dependent autophagy
Interplay of the ULK1 complex with mTOR complex 1 and AMPK.
The autophagy pathway and nutrient signaling communicate with each other intimately. A central node that coordinates this communication is the nutrient-sensing protein kinase complex mTOR complex 1 (mTORC1), which senses growth factors and amino acid levels. This master regulator of cellular metabolism serves as an autophagy response switch by controlling the phosphorylation status, and thereby the autophagy activity, of the ULK1 complex. The main components of the ULK1 complex include the protein kinase ULK1 and its regulatory proteins ATG13 and FIP200 (31–33). Under nutrient-rich conditions, mTORC1 phosphorylates ATG13 and ULK1 to suppress the autophagy function of the ULK1 complex; when mTORC1 is inactivated by nutrient starvation or other stresses, the ULK1 complex becomes hypo-phosphorylated and active (Figure 2).
Figure 2. The autophagy pathway senses various nutrient signals via the ULK1 complex.
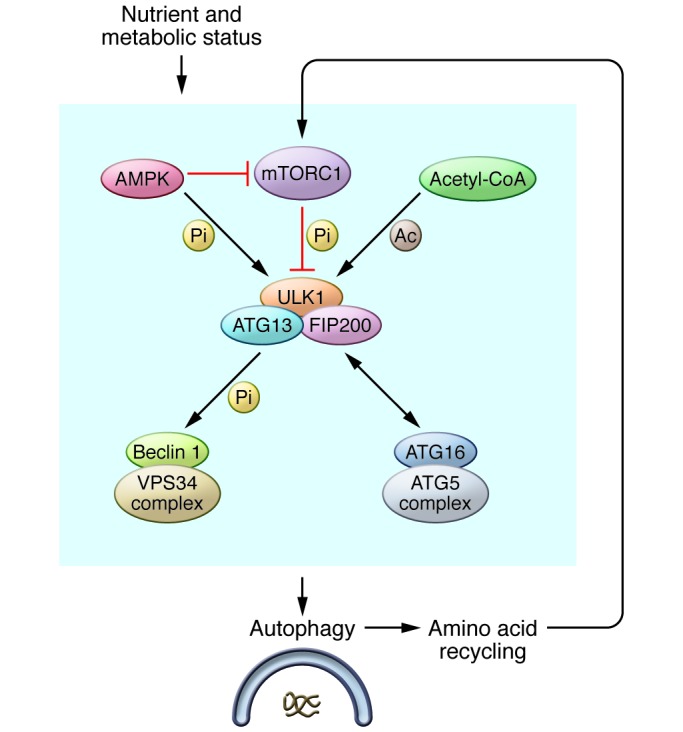
Multiple nutrient-sensing mechanisms, including that modulated by mTORC1, AMPK, and acetyl-CoA, can directly interact with the ULK1 complex and thus regulate autophagy. Autophagy, by recycling amino acids to the cytoplasm, can reactivate mTORC1, and thus feedback suppress the autophagy function of the ULK1 complex. Pi, phosphorylation; Ac, acetylation; ATG16, ATG16L1. The dual-direction arrow indicates protein-protein interaction.
There is an elegant feedback loop between mTORC1 activity and autophagy output. Upon nutrient starvation, mTORC1 is suppressed, leading to activation of ULK1-dependent autophagy. Downstream of autophagy, autophagic cargos are degraded in lysosomes to recycle building blocks, including amino acids, which reactivate mTORC1 and thereby attenuate ULK1-dependent autophagy (34, 35). In addition to inhibiting autophagy induction, reactivated mTORC1 also activates lysosome biogenesis by a program called “autophagic lysosome reformation” in order to reset lysosome networks after prolonged autophagy (34).
Another important cellular energy sensor, AMPK, also plays a role in autophagy induction by phosphorylating TSC2 and the mTORC1 component Raptor, leading to inactivation of mTORC1 and subsequent activation of the ULK1 complex. Recently, AMPK has also been shown to directly interact with and phosphorylate ULK1 in a nutrient-dependent manner (Figure 2 and refs. 36–40). One study suggests that AMPK-driven ULK1 phosphorylation is stimulated by glucose starvation, contributing to ULK1 activation (38). However, another study (39) identified AMPK sites on ULK1 that were dephosphorylated upon amino acid starvation, leading to the conclusion that AMPK phospho sites may inhibit autophagy induction. The differences in these observations might reflect the distinct autophagy-related role of AMPK when sensing different triggers; they could also be due to the monitoring of different phosphorylation sites in these two studies. In yet another study, AMPK regulation of ULK1 was linked to mitophagy (40). Further, studies using AMPK-null cells have ruled out an indispensable role of this kinase in autophagy (36, 38). It appears that AMPK functions to fine-tune ULK1 activity and the subsequent autophagy outcome in response to various energy requirements.
Intriguingly, there have also been reports that ULK1 can in turn regulate mTOR and AMPK. ULK1 was reported to phosphorylate Raptor in vitro to negatively regulate mTORC1 activity (41, 42). This could constitute a feed-forward mechanism that ensures rapid shutdown of mTOR signaling during autophagy induction. In terms of AMPK, ULK1 has been reported to be able to phosphorylate all three AMPK subunits (43). Such inhibitory phosphorylation was proposed to form a feedback loop to dampen autophagy induction signals.
Regulation of the ULK1 complex by acetylation.
Acetylation involves the transfer of acetyl group from acetyl-CoA to specific lysine residues of target proteins. Acetyl-CoA is a critical building block for cellular metabolism, and as such, protein acetylation/deacetylation has been implicated as a critical regulatory mechanism for metabolism (44). Given the catabolic nature of autophagy, its regulation by acetylation is an attractive hypothesis. Protein acetylation/deacetylation was initially implicated in autophagy by the studies of the histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA) (45, 46). SAHA induces autophagy in various cell types in a ULK1 complex–dependent manner. However, the role of SAHA in maintaining the acetylation status of ATG proteins was not determined. More recently, a systematic genetic analysis of histone acyltransferase (HAT) complexes in yeast identified Esa1, the catalytic subunit of HAT complex NuA4, as being required for autophagy (47). In a separate study (48), TIP60, the mammalian homolog of Esa1, was reported to acetylate ULK1 to support its autophagy function. Furthermore, TIP60 activation was regulated by glycogen synthase kinase-3 (48), thereby directly linking TIP60 to nutrient sensing and ULK1-mediated autophagy. It should be noted that in addition to specific acetylation of ULK1, a recent study instead indicated that increased cellular acetyl-CoA levels and overall acetylation of cytoplasmic proteins suppress autophagy (49).
Relaying the nutrient signals by the ULK1 complex
Because of its ability to directly sense mTORC1 activity, the ULK1 complex has been suggested to be the most upstream component of the canonical autophagy pathway. However, the exact functional relationship between the ULK1 complex and other upstream ATG proteins is not well defined. In addition to the ULK1 complex, other ATG components functioning upstream of LC3 include the VPS34–beclin 1 complex and the ATG5-ATG12-ATG16 complex (hereafter referred to as the ATG5 complex). It has been suggested that the autophagy cascade proceeds in a linear fashion: the ULK1 complex activates the VPS34 complex, which in turn engages the ATG5 complex (50). More recent studies have indicated a far more complicated relationship among these complexes.
VPS34 is a lipid kinase, and its enzymatic product PI3P is required, directly or indirectly, for the recruitment of multiple autophagy components to the pre-autophagosomal membrane structure (51, 52). Similar to the ULK1 complex, the VPS34 complex is also a major point of regulation for autophagy induction, as reviewed in depth by others (53, 54). Here we focus on its functional interplay with the ULK1 complex.
It has been reported that in response to nutrient starvation, AMBRA1, a binding partner of the VPS34 complex, is phosphorylated by ULK1 (55). This phosphorylation releases the AMBRA1-VPS34 complex from dynein and the microtubule network, freeing the complex to translocate to autophagy initiation sites on the endoplasmic reticulum. This appears to be a reasonable explanation for the ULK1-VPS34 connection and the autophagic function of ULK1 kinase activity. However, AMBRA1 is unlikely to be the universal mediator for these two autophagy complexes, as it does not have a clear functional counterpart in yeast.
More recently, ULK1 has been reported to directly phosphorylate beclin 1 (56), an essential component of the VPS34 complex. The VPS34 complex is required for both autophagy and endocytic processes. ULK1-driven phosphorylation of beclin 1 specifically activates a subset of the VPS34 complex, which is the ATG14-containing population dedicated to autophagy, thus unveiling how the ULK1 complex stimulates autophagy via the VPS34 complex (Figure 2).
The ATG5 complex was considered to function downstream of the VPS34 complex and not directly communicate with the ULK1 complex. Surprisingly, two recent studies revealed a direct interaction between FIP200 and ATG16L1 (57, 58), essential components of the ULK1 and ATG5 complexes, respectively (Figure 2). Importantly, the FIP200-ATG16L interaction is essential for mediating amino acid starvation–induced autophagy, which requires the ULK1 complex; on the other hand, this interaction is not required for glucose deprivation–induced autophagy, a ULK1 complex–independent process (57). Therefore, this specific physical interaction between the ULK1 complex and the ATG5 complex may distinguish ULK1-dependent and -independent autophagy processes.
ULK1 complex–independent autophagy
LC3 lipidation still occurs in cells lacking expression of ATG13, FIP200, or ULK1/2 (31, 39, 59), suggesting that the ULK1 complex is not essential for activation of the LC3 lipidation machinery and that there are ULK-independent ways to set off the autophagy cascade. Recent work has shown that glucose deprivation–induced LC3 lipidation was dependent on ATG5 yet did not require ULK1 or ULK2, likely due to the accumulation of ammonia associated with amino acid catabolism within the cell (59, 60). It should be noted that glucose deprivation has also been reported to stimulate autophagy in a ULK1-dependent manner (38). Unlike the ammonium scenario, this is a typical bioenergetic response involving mTOR suppression plus AMPK activation, which subsequently activates the ULK1 complex. Seemingly contradictory to each other, glucose starvation might induce autophagy via both mechanisms, depending on exact biological contexts such as growth conditions and cell types.
Non-autophagy processes utilizing autophagy machinery
While ULK1 complex–dependent and –independent mechanisms of autophagy induction are apparent, it is also becoming clear that autophagy machinery directs the lipidation of LC3 onto non-autophagosomal membranes in a ULK1-independent manner. Although referred to elsewhere as “non-canonical autophagy” (61), here we will refer to this activity as “non-canonical LC3 lipidation,” as the majority of membranes onto which LC3 is lipidated are endocytic (thus non-autophagic) in nature, harboring engulfed extracellular- rather than intracellular-derived substrates. The first discovery of ATG protein–dependent, non-autophagosomal LC3 lipidation was that phagosomes containing engulfed microorganisms in macrophages are lipidated by LC3 in a manner requiring the core autophagy proteins ATG5 and ATG7 but independent of the appearance of double-membrane autophagosome structures or regulation by mTORC1 (62). Subsequently, this non-autophagosomal activity of the autophagy lipidation machinery (VPS34 complex and ATG Ubl conjugation systems) was reported to be involved in macroendocytic processes, including apoptotic or necrotic cell phagocytosis (63, 64), Fc receptor and Dectin-1 receptor–mediated phagocytosis (61, 65, 66), macropinocytosis (64), the live cell engulfment program entosis (64), as well as osteoclast resorption of bone (67) and the retinal pigment epithelium–mediated engulfment of photoreceptor outer segments in vivo (ref. 68 and Figure 3). As these processes occur under nutrient-replete conditions, it is perhaps not surprising that LC3 lipidation in these contexts is activated independently of mTORC1 signaling (62) and accordingly requires neither ULK1 (63) nor the ULK1 complex protein FIP200 (64, 68). Importantly, while phagosomes and endosomes can be targeted by autophagosomes, particularly when damaged (69, 70), and endocytic vesicles may contribute to the membrane pool from which autophagosomes derive (71), these findings identify instead the lipidation of LC3 directly onto the limiting membrane of endocytic vacuoles in a manner distinct from autophagy.
Figure 3. LC3 lipidation in non-autophagic processes.

The phagocytosis of apoptotic bodies and macropinocytic uptake of serum albumin are shown as examples of extracellular substrates that may be degraded in an autophagy protein–dependent manner. Shaded box at the right lists multiple non-autophagic processes utilizing LC3 lipidation, as well as their cargos and biological functions. Their potential common roles in nutrient scavenging are emphasized.
These findings demonstrate that autophagy proteins participate in multiple lysosomal degradative processes, including those that target bulk extracellular substrates, in a manner akin to the phagocytic scavenging of nutrients (phagotrophic nutrition) that supports the metabolism of ancestral unicellular eukaryotes. This activity may allow cancer cells to utilize extracellular nutrient scavenging to support metabolism (discussed below) and may also play important roles in normal physiology. Therefore, autophagy proteins may generally participate in lysosomal nutrient recovery under nutrient-replete as well as starvation conditions, and from both intracellular and extracellular sources. In all cases the targeted removal of specific substrates (e.g., apoptotic cells and microorganisms engulfed by phagocytosis, damaged organelles engulfed by autophagy) may contribute to a homeostatic function, while the additional turnover of non-specific bulk substrates (serum albumin engulfed by macropinocytosis, bulk cytoplasm engulfed by autophagy) may more generally support metabolism (Figure 3). Whether these multiple LC3-associated mechanisms act cooperatively or antagonistically remains to be determined.
Autophagy and cancer
The role of autophagy in cancer
Autophagy is implicated in multiple human diseases, particularly cancer. The exact role of autophagy in cancer is complicated and most likely context dependent (72–76). On one hand, autophagy might be a tumor-suppressive mechanism. For example, mice with heterozygotic deletion of beclin 1 are susceptible to tumorigenesis in multiple tissues (77, 78), and liver-specific deletion of ATG7 leads to development of benign liver adenomas (79). Mechanistically, the role of autophagy in clearance of mutagens such as damaged mitochondria (a source of ROS) might prevent insults to genomic DNA and hence suppress tumor initiation (73–75). The accumulation of specific protein substrates that are normally targeted for lysosomal degradation may also underlie tumor suppression by autophagy, as p62, an autophagy receptor protein, is reported to promote tumor formation through several oncogenic signaling pathways including NF-κB and mTORC1, as well as NRF2-dependent antioxidant signaling resulting from p62-dependent sequestration of the KEAP1 ubiquitin ligase (80–85). In addition, although more frequently a pro-survival mechanism (86–88), autophagy may cause cell death under certain conditions (“type II programmed cell death” or “autophagic cell death”; refs. 89–93). This may also contribute to its tumor-suppressive function. On the other hand, as a critical and conserved cellular survival mechanism, it is clear that autophagy promotes malignant progression following the initiation of tumor growth (72–75). During the progression of solid tumors, an increase in tumor size produces stress on cells within central regions, as these areas become hypoxic and nutrient deficient due to inadequate blood supply (94). Even as angiogenesis occurs, regions within solid tumors may still experience metabolic stress as a result of low glucose and glutamine availability (95–97), due to the leakiness of tumor-associated vessels as well as continued hypovascularization (98). Under such conditions, tumor cells can use autophagy to provide an alternative energy source for survival and proliferation (99, 100). Similarly, various cancer therapies have been shown to block tumor tissue angiogenesis or inhibit the kinase signaling that normally suppresses autophagy, thus leading to activation of cancer cell autophagy as a survival mechanism. Indeed, many observations have been made recently to support this view: in multiple tissue origins and multiple oncogenic conditions, inhibition of autophagy can potentiate tumor cell death and reduce the growth of tumors of both xenograft and endogenous origin (101–111).
Intriguingly, recent studies also indicate that some tumors driven by specific oncogenes such as mutant Ras and B-Raf can develop “autophagy addiction”; a highly active autophagy pathway appears to be essential for tumor cell mitochondrial homeostasis and metabolism, and autophagy inhibition leads to tumor cell death (101, 105, 108). In this specific case, autophagy may facilitate oncogenic Ras/B-Raf–triggered tumor initiation and maintenance, and autophagy inhibition might be effective as a monotherapy. In support of this, although loss of ATG5 accelerates the initiation of KrasG12D-driven lung tumors, it increases tumor-free survival by inhibiting the progression to malignancy (112). Autophagy-deficient, oncogenic Ras-driven lung tumors exhibit increased cell death (112, 113) and the accumulation of defective mitochondria, which inhibits tumor progression and leads to the formation of benign tumors resembling oncocytomas (113). Similarly, oncogenic Ras-driven pancreatic tumors require autophagy in order to progress to malignant pancreatic ductal adenocarcinoma in vivo (114). Notably, these antitumor effects of inhibiting autophagy in multiple tumor types in the context of oncogenic Ras have been reported to be dependent on p53, suggesting that the accumulation of cell stress (e.g., genome damage) in the context of defective autophagy is surveilled by this important tumor suppressor, or alternatively that p53 could contribute to promoting cellular metabolism in the context of defective autophagy (76, 112–114).
Autophagy or autophagy proteins?
While B-Raf/Ras-driven carcinogenesis requires an intact autophagy pathway to maintain healthy mitochondria and supply glutamine through lysosomal recycling (115), Ras-driven cancer cells are also able to utilize a scavenging activity linked to macropinocytic uptake of extracellular fluid that allows for the recovery of amino acids and unsaturated lysophospholipids from extracellular sources (116, 117). Amino acid recovery is linked to the uptake and lysosomal digestion of serum albumin, which apparently acts in concert with autophagy-mediated amino acid scavenging from intracellular protein sources to support macromolecular synthesis and TCA cycle anaplerosis, in order to meet the metabolic demands imposed by Ras activation. Similar to autophagy gene deletion, the inhibition of macropinocytosis suppresses tumor formation that is driven by activated K-Ras (116). In light of these apparently parallel pathways supporting amino acid recovery, it is interesting to note that the autophagy lipidation machinery, including ATG5 and ATG7, which are required for Ras-driven tumorigenesis, has been shown to lipidate LC3 onto the limiting membranes of macropinosomes (64). The extent to which LC3 lipidation onto macropinosomes could contribute to the requirement of ATG5 and ATG7 for Ras-driven tumorigenesis remains to be explored.
The function that lipidated LC3 serves on endocytic vacuoles remains uncertain (118). Several groups have provided evidence that the lipidation of LC3 onto phagosomes or entotic vacuoles in macrophages and epithelial cells promotes lysosome fusion and the degradation of engulfed extracellular cargo (62, 118), which would be predicted to promote amino acid scavenging by macropinocytosis. However, a recent report concluded that LC3 may delay lysosome fusion in dendritic cells (119). The more precise function of lipidated LC3 at these non-autophagosomal endocytic membranes awaits further elucidation of the mechanisms whereby LC3 influences vacuole maturation.
Targeting autophagy for cancer therapy
Inhibition of autophagy has been demonstrated to be beneficial in treating various cancers. Being kinases, the ULKs are attractive candidates as drug targets. One of the main uses of a ULK-targeted drug in cancer treatment might be its combination with other therapeutics. For example, the lack of clinical impact of mTOR inhibitors in the treatment of many cancers may be due in part to the fact that they upregulate tumor-protective autophagy by activating the ULK1 complex (120). Furthermore, a plethora of currently used anticancer agents can induce autophagy through mTORC1 suppression, thereby activating the ULK1 complex. Hence, combining these treatments with ULK1 inhibitors might be more effective therapeutically. Although targeting other components of the autophagy pathway (such as VPS34, lysosomal activity, and autophagy-specific Ubl reactions) will serve the same autophagy inhibition purpose, there are potential problems associated with these alternative targets. VPS34 and lysosomes are also required for other cellular functions, such as all endocytic processes; thus, inhibition of these entities is more likely to cause additional side effects. Autophagic Ubl reactions are presumably specific to autophagy. However, their inhibition will ablate not only mTOR suppression–induced, ULK1 complex–dependent autophagy in cancer cells, but also basal levels of autophagy in normal cells that is important for maintaining cellular homeostasis. Therefore, broad-based, Ubl-targeted inhibition of autophagy may also be more detrimental than specific inhibition of the ULK1 complex, whose activity is specifically upregulated upon mTOR-suppressing therapies. It should be mentioned that the kinase activity of ULK1 may have other functions in addition to mediating mTOR-regulated autophagy, but the ULK1-ATG13 interaction, which stimulates ULK1 kinase activity and is required for the autophagy function of the complex, is more likely to be specific only for ULK1-dependent autophagy.
Concluding remarks
Extensive studies over the last two decades have provided great insights into the mechanisms and function of autophagy in normal biology and disease; however, many fundamental questions remain. For example, as autophagy may be involved in both cell survival and cell death (autophagic cell death), is there a specific molecular switch to dictate these two completely opposite outcomes? What are the mechanisms and functions underlying non-autophagic LC3 lipidation? How is autophagy regulated in a context-dependent manner to target distinct cargos and thus achieve specific functions (e.g., erythrocyte differentiation–associated mitophagy)? Since autophagy is involved in multiple diseases including cancer, how can one develop autophagy-targeted small-molecule modulators as therapeutic agents? Which ATGs should be targeted? Also, while we have learned much about how and from where autophagosomes originate and what substrates they target, comparatively little is known about the terminal stages of substrate degradation and nutrient recycling, which are required for the clearance of targeted substrates as well as the nutrient scavenging function that may be a therapeutic target in cancer. Perhaps as we contemplate therapeutic interventions to inhibit autophagy, a deeper understanding of the identity of the critically recovered metabolites and what cancer cells derive from them may reveal therapeutic strategies that target the steady-state outputs of autophagy in addition to autophagy induction.
Acknowledgments
This work is supported in part by a grant from the Cycle for Survival of Memorial Sloan Kettering Cancer Center (to X. Jiang) and NIH grants R01CA166413 (to X. Jiang), R01CA154649 (to M. Overholtzer), NIH R01 CA105463 (to C.B. Thompson), and NIH P01CA104838 (to C.B. Thompson).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(1):47–54. doi:10.1172/JCI73942.
References
- 1.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24(1):24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6(6):439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 4.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–455. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- 7.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 8.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellu AR, Kiel JA. Selective degradation of peroxisomes in yeasts. Microsc Res Tech. 2003;61(2):161–170. doi: 10.1002/jemt.10325. [DOI] [PubMed] [Google Scholar]
- 10.Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol. 2012;2012:282041. doi: 10.1155/2012/282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8(13):1986–1990. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- 13.Pankiv S, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 14.Novak I, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schweers RL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104(49):19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy. 2009;5(5):706–708. doi: 10.4161/auto.5.5.8505. [DOI] [PubMed] [Google Scholar]
- 17.Kuma A, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 18.He C, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481(7382):511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He C, Sumpter R, Sumpter R, Jr, Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy. 2012;8(10):1548–1551. doi: 10.4161/auto.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333(1):169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 22.Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131(3):591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thumm M, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349(2):275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- 24.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15(2):231–236. doi: 10.1016/j.semcdb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Hanada T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282(52):37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 27.Noda NN, Fujioka Y, Hanada T, Ohsumi Y, Inagaki F. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013;14(2):206–211. doi: 10.1038/embor.2012.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bjorkoy G, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komatsu M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 30.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130(1):165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 31.Hara T, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181(3):497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganley IG, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung C, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rong Y, et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci U S A. 2011;108(19):7826–7831. doi: 10.1073/pnas.1013800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mack HI, Zheng B, Asara JM, Thomas SM. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy. 2012;8(8):1197–1214. doi: 10.4161/auto.20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5(11):e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. 2011;108(12):4788–4793. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. 2011;7(7):737–747. doi: 10.4161/auto.7.7.15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jung CH, Seo M, Otto NM, Kim DH. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7(10):1212–1221. doi: 10.4161/auto.7.10.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Löffler AS, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7(7):696–706. doi: 10.4161/auto.7.7.15451. [DOI] [PubMed] [Google Scholar]
- 44.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13(4):270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- 45.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101(52):18030–18035. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A. 2012;109(17):6561–6565. doi: 10.1073/pnas.1204429109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yi C, et al. Function and molecular mechanism of acetylation in autophagy regulation. Science. 2012;336(6080):474–477. doi: 10.1126/science.1216990. [DOI] [PubMed] [Google Scholar]
- 48.Lin SY, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science. 2012;336(6080):477–481. doi: 10.1126/science.1217032. [DOI] [PubMed] [Google Scholar]
- 49.Marino G, et al. Regulation of autophagy by cytosolic acetyl-coenzyme a. Mol Cell. 2014;53(5):710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 50.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6(6):764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsunaga K, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190(4):511–521. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Polson HE, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6(4):506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 53.Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex — at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20(6):355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410(1):1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 55.Di Bartolomeo S, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191(1):155–168. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Russell RC, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gammoh N, Florey O, Overholtzer M, Jiang X. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat Struct Mol Biol. 2013;20(2):144–149. doi: 10.1038/nsmb.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishimura T, et al. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep. 2013;14(3):284–291. doi: 10.1038/embor.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A. 2011;108(27):11121–11126. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal. 2010;3(119):ra31. doi: 10.1126/scisignal.2000911. [DOI] [PubMed] [Google Scholar]
- 61.Henault J, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–997. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanjuan MA, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 63.Martinez J, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108(42):17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011;13(11):1335–1343. doi: 10.1038/ncb2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang J, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106(15):6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ma J, Becker C, Lowell CA, Underhill DM. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J Biol Chem. 2012;287(41):34149–34156. doi: 10.1074/jbc.M112.382812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeSelm CJ, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011;21(5):966–974. doi: 10.1016/j.devcel.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim JY, et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154(2):365–376. doi: 10.1016/j.cell.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maejima I, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013;32(17):2336–2347. doi: 10.1038/emboj.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482(7385):414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12(8):747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22(2):212–217. doi: 10.1016/j.ceb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155(6):1216–1219. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takamura A, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mathew R, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Valencia T, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26(1):121–135. doi: 10.1016/j.ccr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Komatsu M, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 84.Inami Y, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193(2):275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duran A, et al. The signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 86.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16(6):663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 87.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115(10):2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Berry DL, Baehrecke EH. Autophagy functions in programmed cell death. Autophagy. 2008;4(3):359–360. doi: 10.4161/auto.5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8(6):569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 90.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23(16):2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 91.Yu L, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304(5676):1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 92.Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6(12):1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 93.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131(6):1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Naumov GN, Folkman J, Straume O. Tumor dormancy due to failure of angiogenesis: role of the microenvironment. Clin Exp Metastasis. 2009;26(1):51–60. doi: 10.1007/s10585-008-9176-0. [DOI] [PubMed] [Google Scholar]
- 95.Reid MA, Wang WI, Rosales KR, Welliver MX, Pan M, Kong M. The B55α subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol Cell. 2013;50(2):200–211. doi: 10.1016/j.molcel.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 96.Roberts E, Frankel S. Free amino acids in normal and neoplastic tissues of mice as studied by paper chromatography. Cancer Res. 1949;9(11):645–648. [PubMed] [Google Scholar]
- 97.Urasaki Y, Heath L, Xu CW. Coupling of glucose deprivation with impaired histone H2B monoubiquitination in tumors. PLoS One. 2012;7(5):e36775. doi: 10.1371/journal.pone.0036775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Agrawal V, et al. Direct endothelial junction restoration results in significant tumor vascular normalization and metastasis inhibition in mice. Oncotarget. 2014;5(9):2761–2777. doi: 10.18632/oncotarget.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ladoire S, et al. Immunohistochemical detection of cytoplasmic LC3 puncta in human cancer specimens. Autophagy. 2012;8(8):1175–1184. doi: 10.4161/auto.20353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang S, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Calabretta B, Salomoni P. Inhibition of autophagy: a new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk Lymphoma. 2011;52(suppl 1):54–59. doi: 10.3109/10428194.2010.546913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Munshi A. Chloroquine in glioblastoma — new horizons for an old drug. Cancer. 2009;115(11):2380–2383. doi: 10.1002/cncr.24288. [DOI] [PubMed] [Google Scholar]
- 104.Amaravadi RK, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17(4):654–666. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guo JY, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11(4):448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 107.Fan QW, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3(147):ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Strohecker AM, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3(11):1272–1285. doi: 10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huo Y, et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013;3(8):894–907. doi: 10.1158/2159-8290.CD-13-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cufi S, et al. The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci Rep. 2013;3:2469. doi: 10.1038/srep02469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Han W, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One. 2011;6(6):e18691. doi: 10.1371/journal.pone.0018691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rao S, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 113.Karsli-Uzunbas G, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–927. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rosenfeldt MT, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 115.Guo JY, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27(13):1447–1461. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Commisso C, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kamphorst JJ, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A. 2013;110(22):8882–8887. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Florey O, Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol. 2012;22(7):374–380. doi: 10.1016/j.tcb.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Romao S, et al. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. J Cell Biol. 2013;203(5):757–766. doi: 10.1083/jcb.201308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huang S, Yang ZJ, Yu C, Sinicrope FA. Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J Biol Chem. 2011;286(46):40002–40012. doi: 10.1074/jbc.M111.297432. [DOI] [PMC free article] [PubMed] [Google Scholar]