

Abstract
MicroRNA-21 (miR-21) contributes to the pathogenesis of fibrogenic diseases in multiple organs, including the kidneys, potentially by silencing metabolic pathways that are critical for cellular ATP generation, ROS production, and inflammatory signaling. Here, we developed highly specific oligonucleotides that distribute to the kidney and inhibit miR-21 function when administered subcutaneously and evaluated the therapeutic potential of these anti–miR-21 oligonucleotides in chronic kidney disease. In a murine model of Alport nephropathy, miR-21 silencing did not produce any adverse effects and resulted in substantially milder kidney disease, with minimal albuminuria and dysfunction, compared with vehicle-treated mice. miR-21 silencing dramatically improved survival of Alport mice and reduced histological end points, including glomerulosclerosis, interstitial fibrosis, tubular injury, and inflammation. Anti–miR-21 enhanced PPARα/retinoid X receptor (PPARα/RXR) activity and downstream signaling pathways in glomerular, tubular, and interstitial cells. Moreover, miR-21 silencing enhanced mitochondrial function, which reduced mitochondrial ROS production and thus preserved tubular functions. Inhibition of miR-21 was protective against TGF-β–induced fibrogenesis and inflammation in glomerular and interstitial cells, likely as the result of enhanced PPARα/RXR activity and improved mitochondrial function. Together, these results demonstrate that inhibition of miR-21 represents a potential therapeutic strategy for chronic kidney diseases including Alport nephropathy.
Introduction
MicroRNAs (miRNAs) represent distinct small noncoding RNAs that function as important posttranscriptional regulators of gene expression (1). A single miRNA can silence the expression of a number of functionally related genes, and therefore, miRNA can function in an analogous but reciprocal way to transcription factors (2, 3). The importance of miRNAs in the kidney has been underscored by severe developmental defects resulting from blocking miRNA processing in the kidney (4). Several independent studies in human and animal models of kidney disease suggest a number of miRNAs are regulated in response to acute injury and remain dysregulated in chronic disease settings, contributing to the pathogenesis of kidney disease (5). One such miRNA is miR-21, which is thought to be involved in regulating tissue repair responses after injury. Investigations have shown that miR-21 is widely expressed in multiple cell types in the kidney and is upregulated in multiple acute and chronic kidney diseases (CKDs) (1, 4, 6, 7). In short-term rodent models of kidney disease, miR-21 is clearly involved in pathogenic responses, as both genetic deletion and pharmacologic inhibition attenuate disease progression (4, 7). miR-21 also plays an important pathogenic role in disease progression in kidney cancers (8).
Both bioinformatic and experimental approaches can be used to understand which of the multitude of potential targets for any miRNA are relevant for the tissue and disease context being evaluated. Target genes identified as relevant in cancer may or may not be relevant for kidney disease. Bioinformatic analysis of gene signatures obtained from kidney disease models suggests that miR-21 has the potential to regulate metabolic pathways in mitochondria and peroxisomes, leading to impaired fatty acid (FA) metabolism, impaired detoxification, and increased production of ROS in response to kidney injury (4). In particular, pathways regulated by the transcription factor PPARα were identified as important targets for silencing by miR-21 activity in interstitial kidney fibrosis.
Advances in the development of oligonucleotide chemistry have allowed for development of engineered oligonucleotides directed against specific miRNAs that can be administered s.c., taken up freely into cells, bind specifically to individual miRNAs by sequence complementarity, and block the specific miRNA function (9). Therefore, unlike transcription factors, individual miRNAs are readily targetable with oligonucleotides in both animals and humans (10, 11).
Alport syndrome is a genetic disorder characterized by chronic disease of the kidney known as glomerulonephritis, which progresses relentlessly to end-stage renal disease in young adult life (12). Alport syndrome results from mutations in the genes encoding α3, α4, or α5 chains of collagen IV that disturb the normal formation of the capillary basement membranes in the kidney, eye, and inner ear (13). As a result, the integrity of the glomerular filtration barrier is disrupted, resulting in initial glomerular hemodynamic changes and, thereafter, progressive glomerular and tubulointerstitial fibrosis accompanied by severe inflammation (14). Severity of the gene mutation is associated with earlier onset of disease and faster progression (15). Ocular and auditory problems often accompany renal dysfunction because of the defective basement membranes in these organs. Patients with Alport nephropathy lack effective therapies beyond nonspecific blockade of the renin-angiotensin system (16). Therefore, new therapies to counteract the progression of Alport nephropathy are urgently sought. Emerging evidence suggests that CKDs including Alport nephropathy progress to organ failure via similar mechanisms (17). Patients with Alport nephropathy therefore develop features of CKD, a syndrome affecting more than 10% of humans in developed nations. CKD has many causes, including hypertension, diabetes mellitus, vascular disease, infections, and xenobiotic toxicity, but similar to Alport nephropathy, CKD is characterized by progressive loss of kidney function over months and years and is associated with glomerular and interstitial fibrosis, inflammation, capillary destruction, and tubular injury. The advantage of studying Alport nephropathy in animals and potentially in patients is that they lack the comorbid conditions often associated with other, older CKD populations and always progress to end-stage kidney disease.
Here, we report the efficacy and mechanism of action of anti–miR-21 oligonucleotides in a mouse model of Alport nephropathy caused by a null mutation of the α3 chain of collagen type IV (Col4a3–/–). This model develops spontaneous CKD by 5 weeks after birth and progresses rapidly to organ failure by 11 weeks.
Results
MicroRNA-21 is upregulated in Col4a3–/– mice.
miR-21 is a highly conserved, small noncoding RNA (Figure 1A) that shows remarkable sequence conservation across species (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI75852DS1). miR-21 is expressed at a higher level in the tubulointerstitium than in the glomeruli in healthy WT kidneys, but is upregulated to similar degrees in both compartments in the Col4a3–/– mouse kidney, such that copy number in diseased glomeruli is similar to that in diseased tubulointerstitium (Figure 1B and Supplemental Experimental Procedures). Podocytes, which likely promote glomerular disease, express high levels of miR-21 (Table 1 and ref. 4), and tubular epithelium as well as interstitial cells upregulate miR-21 in disease (4). In the intact Col4a3–/– kidney, expression is elevated as early as 3 weeks of age and increases further as disease progresses (Figure 1C). Given that upregulation precedes changes in tissue morphology (Figure 1D), it is possible that miR-21 increases as an early response to cellular stresses caused by the abnormal glomerular basement membrane (GBM).
Figure 1. miR-21 upregulation in Alport nephropathy precedes histological changes in the kidney.

(A) Schema showing the sequence and configuration of human (hsa) pre–miR-21, the processed mature miR-21. The domain that binds to 3′ UTR regions of translated mRNA by sequence complementarity is highlighted. (B) Quantitative reverse-transcriptase PCR (qRT-PCR) for miR-21 copy number in tubules and glomeruli of Col4a3–/– kidneys purified by laser-capture microdissection. (C) qPCR levels for miR-21 in whole kidney from Col4a3–/– mice compared with heterozygotes. (D) PAS-stained images of kidney cortex from Col4a3–/– mice or heterozygotes showing essentially normal histology at 3 weeks and rare segmental sclerosis of glomeruli (arrow) at 5 weeks. The tubules appear normal. Scale bars: 50 μm. n = 3–6/group. *P < 0.05, 1-way ANOVA or Mann-Whitney U test)
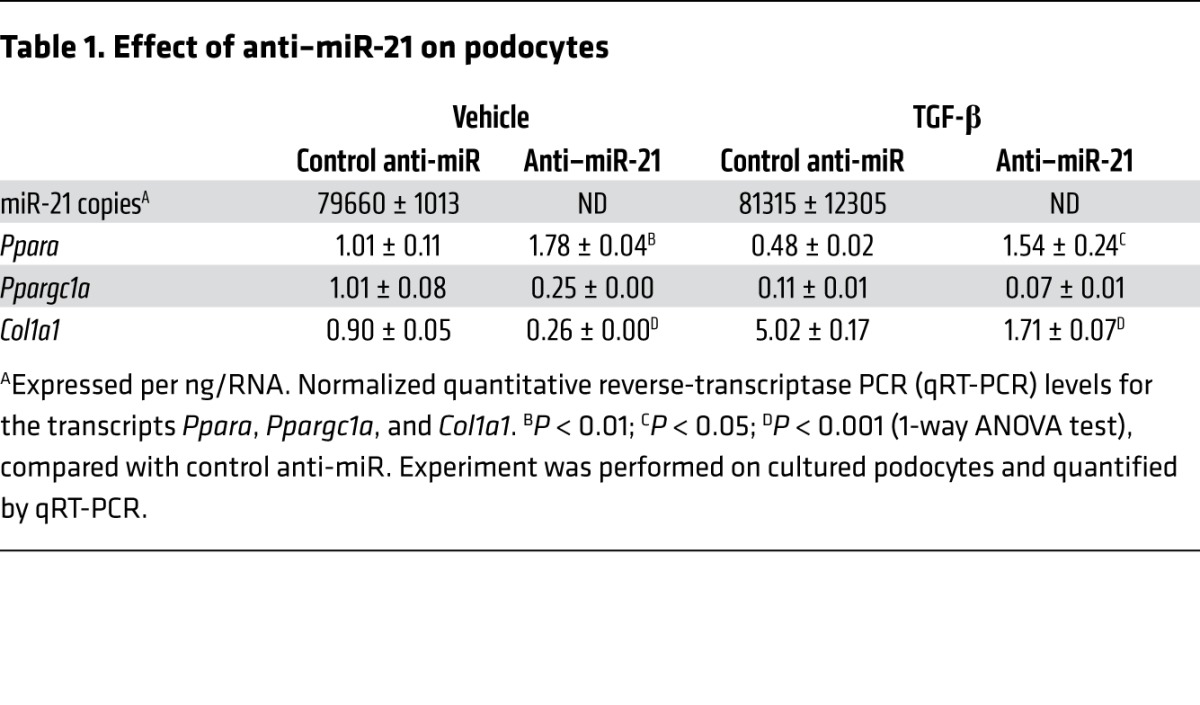
Table 1.
Effect of anti–miR-21 on podocytes
Anti-microRNA oligonucleotides with drug-like properties.
The anti–miR-21 oligonucleotides used in these studies are chemically modified single-stranded RNA molecules with full sequence complementarity to miR-21 (3, 4). Because of the modifications to the phosphate backbone and sugar moieties of the RNA structure, anti–miR-21 oligos are stable in vivo and have high-affinity hybridization-based binding to miR-21, thereby blocking its ability to interact with target genes (ref. 18 and Supplemental Figure 1B). For some studies, a Cy3 moiety was attached to the 5′ end of the oligonucleotide to allow visualization of the compound distribution. For all studies described herein, the oligonucleotides were formulated in PBS using estimated absorption coefficients to calculate oligonucleotide concentrations. Compounds were formulated at concentrations to use 5 ml/kg dosing volumes through s.c. administration. Protocols for study design are shown in Figure 2A.
Figure 2. Anti–miR-21 is widely distributed to kidney cells, increases life span, and protects Col4a3–/– mice from kidney disease progression.

(A) Experimental schema indicating anti–miR-21 delivery from 3.5 weeks after birth and analysis at 63 days (9 weeks) or continued to 110 days (16 weeks). (B) Split panel confocal images of kidney cortex showing distribution of a single s.c. injection of Cy3-conjugated anti–miR-21 oligos (red) or vehicle 48 hours previously in 8-week-old Col4a3–/– mice. Sections were colabeled with antibodies against specific cellular markers of myofibroblasts, fibroblasts/pericytes and mesangial cells, endothelium, or podocytes to highlight uptake in particular cell types (arrowheads). Scale bars: 50 μm. g, glomerulus. (C) Confocal images showing vehicle control. Scale bars: 50 μm. (D) Graph of plasma BUN levels at 9 weeks. A-miR-21, anti–miR-21. (E) Graph of time course of urine albumin concentration normalized to urine creatinine. (F) Curves showing body weight changes with time. (G) Kaplan-Meier survival curve. Arrow indicates last delivery of anti–miR-21. **P < 0.01; ***P < 0.001; ****P < 0.0001, Gehan-Breslow-Wilcoxon test for survival; 1-way ANOVA or Mann-Whitney U test for others. n = 12/group.
Anti–miR-21 oligonucleotides protect against kidney failure and increase survival in Col4a3–/– mice.
The 129X1/SvJ strain of mice with homozygous deletion mutation of the Col4a3 gene develops kidney disease, as detected by albuminuria, as early as the second week of life. The first histological signs by light microscopy are seen in some glomeruli at 5 weeks, with occasional basement membrane thickening and increased matrix deposition in the capillary loop regions (Figure 1D). Glomerulosclerosis and tubulointerstitial fibrosis normally progress rapidly thereafter and result in organ failure and death by approximately 10 weeks of age. The distribution of anti–miR-21 oligonucleotides 48 hours following administration of a single s.c. injection of Cy3-conjugated anti–miR-21 was evaluated in 6- and 8-week-old diseased mice (Figure 2, B and C). Anti–miR-21 was highly concentrated in proximal tubules as expected (4), but was also readily identified in other tubules, fibroblasts, endothelial cells, and leukocytes by colocalization (Figure 2, B and C, Supplemental Figure 2, and Supplemental Experimental Procedures). In previous studies in normal kidney or following ureteral obstructive kidney disease, the glomerulus was not a significant target for intracellular uptake, but in mice with early Alport nephropathy, glomerular podocytes, mesangial cells, and endothelium also showed significant uptake of anti–miR-21 (refs. 4, 19, Figure 2B, and Supplemental Figure 2). To study whether blockade of miR-21 would alter the course of disease, Col4a3–/– mice were administered, in a blinded manner, either control (vehicle — PBS) or chemically modified anti–miR-21 oligonucleotides twice weekly by s.c. injection starting at 3.5 weeks of age (Figure 2A). In some studies, kidneys were analyzed at 9 weeks of age to assess renal pathology (Figure 2B) and measure renal function (blood urea nitrogen [BUN]; Figure 2D). In other studies, dosing was continued until 16 weeks. Mutant mice had substantial kidney failure at 9 weeks (Figure 2D), while anti–miR-21–treated mice had preserved kidney function. This observation was supported further by a marked increase in albuminuria in control-treated mice, while albuminuria in anti–miR-21–treated mice was significantly lower (Figure 2E). A reduction in albuminuria was detected by 6 weeks of age. These studies were then repeated in separate cohorts of mice using anti–miR-21 administered at multiple dose levels ranging from 12.5 mg/kg to 50 mg/kg weekly, with 1 group receiving the compound twice a week instead. Again, inhibition of miR-21 decreased BUN concentrations in the blood in a dose-related manner from 68.4 mg/dl in the vehicle-treated mice to 26.8, 32.8, and 25.3 mg/dl for the 12.5, 25, and 50 mg/kg anti–miR-21–treated mice, respectively, at 7 weeks of age (Supplemental Figure 3A). Overall, more than 100 mice have been treated with anti–miR-21 in 7 independent experiments, with highly significant protection from kidney failure.
We additionally evaluated the ability of anti–miR-21 to increase life span in Col4a3–/– mice, which normally have a median survival time of approximately 10 weeks (20). Cohorts of mice were given anti–miR-21 (25 mg/kg twice a week) or vehicle from week 3.5 until week 16 in an operator-blinded manner. Mice receiving anti–miR-21 gained more weight than control-treated mice and maintained stable body weight until week 12, losing weight slowly thereafter (Figure 2F), whereas vehicle-treated mice lost weight earlier and more precipitously. By 11 weeks, all vehicle-treated mice had died as a consequence of renal failure; however, more than half the mice treated with anti–miR-21 survived beyond 16 weeks (Figure 2G). Therefore, anti–miR-21 administration increased median survival by 32 days, from 76 days to 108 days. In the dose-response studies (Supplemental Figure 3), anti–miR-21 enhanced life span in a dose-responsive manner such that 50 mg/kg once weekly increased life span by 46%.
Anti–miR-21 oligonucleotides reduce glomerulosclerosis, crescent formation, and tubulointerstitial kidney disease in Col4a3–/– mice.
To understand the impact of anti–miR-21 on kidney disease progression, histological parameters were evaluated in samples harvested at 9 weeks of age in an observer-blinded manner. Vehicle-treated mice had typical glomerular changes, with extensive glomerulosclerosis (glomerular fibrosis with capillary destruction), the formation of glomerular crescents, and periglomerular fibrosis. Necrosis was not seen (Figure 3, A–C). Anti–miR-21–treated mice had much less glomerulosclerosis, with nearly 60% of glomeruli showing no sclerosis compared with only 12% in control-treated mutant mice. Fewer than 10% of the glomeruli showed extensive sclerosis, as detected by the highest glomerulosclerosis score compared with 30% in control-treated mutants (Figure 3B). This protection of the glomerular tuft was associated with fewer glomerular crescents (Figure 3C). Because glomerulosclerosis is associated with a reduction in podocyte coverage of glomerular capillaries, podocyte number was measured by staining for the transcriptional regulator WT-1 (Figure 3, A and D, and Supplemental Experimental Procedures). Anti–miR-21 administration preserved podocyte numbers in glomeruli. Electron microscopy (EM) of the capillary loops showed severe GBM thickening, splitting, and lamellation in mutant mice, with severe podocyte foot-process effacement. In contrast, many capillary loops in anti–miR-21–treated mice showed normal thickness GBM with occasional subepithelial GBM humps and mild foot-process effacement. In a cohort of mice that survived with treatment to 16 weeks, EM was performed. The GBM showed more humps, but foot processes remained relatively preserved, collectively suggesting that anti–miR-21 benefits podocyte function (Figure 3A and Supplemental Figure 4D). Col4a3–/– mice also have significant tubulointerstitial pathology, characterized by interstitial fibrosis, tubular injury/atrophy, inflammation, and destruction of peritubular capillaries (Figure 3A). The extent of tubule injury was reduced markedly by anti–miR-21 administration (Figure 3, A and E), and the extent of interstitial fibrosis was markedly attenuated (Figure 3, A and F). Therefore anti–miR-21 was highly protective against the development of glomerular, tubular, and interstitial disease.
Figure 3. Anti–miR-21 prevents glomerulosclerosis tubular atrophy and interstitial fibrosis.

(A) Silver methenamine–stained images highlighting glomerulosclerosis (arrowheads); Sirius red–stained images of whole kidney showing interstitial fibrosis; PAS-stained images showing tubule injury; WT-1 immunofluorescence showing podocytes within glomeruli (dotted lines), all at 9 weeks. EM images of glomerular capillary loops that show severe basement membrane thickening, duplication, and podocyte effacement in vehicle-treated Col4a3–/– compared with anti–miR-21–treated Col4a3–/– mice at 9 weeks, which show preserved basement membrane in many glomeruli, with minimal foot-process effacement. At 16 weeks (lowest panel), the GBM in anti–miR-21–treated mice shows small but classical basement membrane humps, whereas foot processes remain partially preserved. U, urinary space; P, podocyte; Pp, podocyte processes; L, capillary lumen; EC, endothelial cell. Scale bars: 100 μm (conventional); 500 nm (EM). (B) Graph of percentage of glomeruli with glomerulosclerosis at 9 weeks, scored from 0 to 4 by extent of sclerosis, where 0 is without sclerosis. (C) Graph showing percentage of glomeruli with glomerular epithelial cell hyperplasia (known as crescent). (D) Podocyte number per glomerular cross section. (E) Tubule injury score. (F) Morphometry of interstitial fibrosis. *P < 0.05; **P < 0.01, 1-way ANOVA or Mann-Whitney U test. n = 12/group.
Anti–miR-21 oligonucleotides reduce tubular injury, inflammation, and myofibroblasts in Col4a3–/– mice.
Histopathological, biochemical, and molecular markers were used to assess effects on tubule injury, fibrogenesis, and inflammation. Anti–miR-21 reduced renal tubule cell apoptosis (Figure 4, A and B) and decreased release of the tubular injury biomarker neutrophil gelatinase–associated lipocalin (NGAL) into both urine and blood (Figure 4, C and D). Inflammation and fibrosis likely occur as a response to capillary and tubular injury. Inhibition of miR-21 significantly reduced both F4/80+ macrophages and CD11b+ leukocytes (Figure 4, A, E, and F), while no effects were seen on the less abundant T lymphocytes (CD3+) and neutrophils (Ly6G+) (Supplemental Figure 4). Consistent with the reduction in fibrosis, kidney interstitial fibroblasts and myofibroblasts detected by PDGFRβ or α-smooth muscle actin (αSMA), respectively, were significantly reduced in number and area (Figure 4, A, G, and H). This was supported by a parallel decrease in the Acta2 gene transcripts, which encode αSMA, and transcripts for encoding fibrillar collagen genes, such as Col1a1 and Col3a1 (Figure 4I and Supplemental Figure 4). Finally, closely associated with the fibrotic process, anti–miR-21 preserved peritubular capillary integrity (Figure 4J). Overall, anti–miR-21 reduced inflammation, tubular and capillary destruction, and fibrogenesis.
Figure 4. Tubular injury and myofibroblast and leukocyte expansion are all attenuated by anti–miR-21.

(A) Images of apoptotic tubular epithelial cells (arrows), macrophages, and myofibroblasts. (B–D) Quantification of tubular apoptosis and the tubular injury marker NGAL in urine and plasma at 9 weeks. (E–H) Quantification of kidney macrophages (CD11b, F4/80), fibroblasts/PCs (PDGFRβ), and myofibroblasts (αSMA) at 9 weeks. (I) qPCR for fibrogenic transcripts. (J) Quantification of peritubular capillary density. *P < 0.05; **P < 0.01, Mann-Whitney U test. n = 12/group. Scale bars: 50 μm.
Transcriptional analysis indicates anti–miR-21 oligonucleotides restore PPARα signaling pathways and mitochondrial functions.
To obtain an unbiased view of Alport nephropathy, we performed RNA sequencing using the Illumina HiSeq 2000 to compare the global transcriptome of whole kidney from mice with disease at 9 weeks with littermate controls. Similarly, we also obtained RNA-Seq global transcriptome profiles for the 5.5-week-old mice. A total of approximately 32–39 million reads per sample were generated for the 9-week samples, while around 22–30 million reads per sample were generated for the 5.5-week samples. Reads were aligned to the mouse genome, and counts were quantified using RSEM and Cufflinks, respectively, for the 9-week and 5.5-week data. Normalization and differential expression analyses were performed using the R/Bioconductor package limma for RNA-Seq data (Figure 5 and Supplemental Figure 5). Ingenuity Pathway Analysis (IPA) indicated that, of the genes changed by the presence of disease, those genes associated with suppression of mitochondrial function were most enriched (Figure 5A). Genes associated with the pathways of fibroblast activation (as defined through studies of liver disease) were the next most enriched, followed by signaling pathways associated with innate and adaptive immunity. Prominent among upregulated individual genes were those involved in fibroblast activation and matrix deposition, including Pdgfb, Col5a1, Col12a1, Col1a1, and Col3a1 (Supplemental Figure 5A). Prominent among the downregulated genes were the solute carrier protein 22 (Slc22) family of organic anion transporters (OATs); metabolic transcriptional regulators, such as Sim1; peroxisomal and mitochondrial FA metabolism genes, such as Acox2; mitochondrial genes, such as the Pgc1, Cyp450 gene family; and the antioxidant Mpv17l. Overall, these results suggest that, as in other CKDs, such as diabetic kidney disease (21, 22), metabolic and mitochondrial dysfunction are major problems within the mouse Alport kidney.
Figure 5. Global transcriptome sequencing of kidneys from Col4a3–/– mice provides unbiased insight into Alport nephropathy and the actions of anti–miR-21.

(A) Pathways analysis of Col4a3+/+ compared with Col4a3–/– kidneys at 9 weeks showing the most significantly regulated gene ontology pathways, where red denotes upregulated and blue denotes downregulated genes. Height of the bar reflects statistical enrichment (–log10 P value) for the pathways, while x axis reflects the total number of regulated genes in each pathway. (B) Pathways analysis of Col4a3–/– kidneys at 9 weeks treated with vehicle compared with 5.5 weeks of anti–miR-21 treatment, where red denotes upregulated and blue downregulated genes. Note that mitochondrial function genes are derepressed and PPARα signaling is derepressed as identified in 2 pathways (LPS/IL-1 inhibition of RXR function and PPARα/RXRα function) (arrows). (C–F) Western blots at 9 weeks demonstrating the effect of disease and anti–miR-21 on whole kidney levels of PPARα, MCAD, ACAT1, and peroxisomal PMP-70. Normalized densitometry is shown (AU). (D and E) Blots were obtained from the same experiment. Actin blots were derived from parallel samples run on a separate gel, and lines indicate splicing of blots). (G) Schematic showing the effect of disease (no. 1) and anti–miR-21 treatment (no. 2) on PPARα/RXRα-regulated pathways in the nucleus (blue), peroxisome (brown), and mitochondrion (pink) at week 9. PPARα and PGC1α as transcriptionally regulated genes as well as transcription factors are shown regulating downstream genes by green connectors. PPARα target and interactor genes are significantly (*) enhanced (red thermometers) by anti–miR-21 treatment including FABP, ACAA1, CPT, ACADL, whereas PPARα-mediated transcriptional suppression of cJUN is also observed. Analysis performed using Bioconductor LIMMA program. n = 3/group.
To understand the action of anti–miR-21 on the mouse Alport kidney, we analyzed the transcriptomes of kidneys from 5.5-week-old Col4a3–/– mice treated with vehicle or anti–miR-21 for 2 weeks, using MetaCore Pathway Analysis. At this early time point, miR-21 was elevated, albuminuria was modestly elevated, and a minority of glomeruli showed basement membrane thickening, but tubules looked normal histologically (Figure 1). Pathway analysis showed that genes involved in the PPARα/retinoid X receptor–regulated (PPARα/RXR-regulated) pathways of FA metabolism were inversely modulated with disease and anti-miR treatment (Supplemental Figure 5, C and D). The effect of anti–miR-21 was also assessed using IPA after 5.5 weeks of anti–miR-21 treatment, when mice were 9 weeks old (Figure 5B). More genes were regulated by the cumulative treatment with anti–miR-21, but with much of the analysis reflecting the histological observations described above. Strikingly though, genes in the mitochondrial function pathways were the most significantly enriched, but with directionality opposite from that of the comparison of Col4a3–/– with Col4a3+/+ kidneys (Figure 5B and Supplemental Figure 5). In addition, genes involved in the retinoic acid receptor (RAR) and RXR transcription families of metabolic detoxification pathways, which includes PPARα signaling (referred to as LPS/IL-1 inhibition of RXR function), were highly enriched, with most genes upregulated as a result of treatment (Figure 5B). PPARα signaling was also found to be enhanced in a separate but related ontology pathway (PPARα/RXRα activation) (Figure 5B). These observations indicated that at both early and late time points in disease progression, PPARα signaling was enhanced by anti–miR-21 and that the most enriched genes at late time points were those regulating mitochondrial functions. PPARα, a seed sequence-matched target gene for miR-21, was confirmed to be enhanced in the setting of anti–miR-21 treatment (Figure 5C) as were downstream target genes for PPARα, including the β-oxidation enzymes MCAD (ACADM) and ACAT1 (also a target of miR-21), providing clear evidence for enhanced β-oxidation by the action of anti–miR-21 (Figure 5, D and E, and Supplemental Figure 6A; see complete unedited blots in the supplemental material). PPARα has a wide range of effects on metabolic function beyond FA metabolism (β-oxidation), acting in concert with RXRs to regulate transcriptional activation (23), glucose homeostasis, glycolysis, detoxification, and antiinflammatory pathways as well as mitochondrial and peroxisome biogenesis. The peroxisome marker PMP70 was enhanced by anti–miR-21 treatment, which is indicative of enhanced peroxisome biogenesis (Figure 5F). Collectively, these results point to PPARα and its downstream targets as important in anti–miR-21 effects on the kidney. The interactions among PPARα signaling, lipid metabolism, β-oxidation and mitochondrial functions, inflammatory signaling, and oxidative phosphorylation (not shown) as functionally related targets for anti–miR-21 were independently confirmed by MetaCore pathway analysis (Figure 5F). Upstream Regulatory Analysis (IPA) also showed that a number of genes modulated by treatment with anti–miR-21 would be consistent with activation of PPARα (z score, 2.167; P value, 2.7 × 10–12). In addition to influencing lipid metabolism, the analyses suggested PPARα was playing roles in mitochondrial biogenesis and may contribute to inhibition of inflammation and fibrosis. PPARα has been reported to inhibit NF-κB signaling via activation of IκB-α, inhibit JNK signaling via inhibition of c-JUN (Figure 5G), and inhibit TGF-β signaling via direct inhibition of SMAD2/3 complexes. Consistent with such reports, the SMAD2/3 gene target and TGF-β activator integrin β6, was suppressed by anti–miR-21 treatment in epithelial cells, independently of any regulation of inhibitory SMADs by anti–miR-21 (Supplemental Figure 6, B and C). The pathways analysis also showed that anti–miR-21 treatment resulted in enrichment of fibroblast activation, with genes generally decreased. This group of genes consists of fibrogenic as well as inflammatory genes, including genes encoding signaling components downstream of NF-κB (Figure 5B). Such transcriptional downregulation triggered by anti–miR-21 is likely secondary to derepression of the aforementioned PPARα and mitochondrial pathways (Figure 5B and Supplemental Figure 5) rather than direct regulation of TGF-β pathway inhibitors such as SMAD7 (Supplemental Figure 6A). The unbiased transcriptional analysis, therefore, provided strong evidence that anti–miR-21 operates by derepressing pathways already known to be silenced by the effects of miR-21 (4).
Anti–miR-21 directly enhances PPARα and mitochondrial metabolic functions in proximal tubule cells and podocytes during cell stress.
To explore the close link among mitochondrial function, miR-21, and PPARα, we compared the effect of miR-21 deficiency or anti–miR-21 treatment on unpassaged primary cultures of proximal kidney tubule cells (PTECs), since these cells are rich in mitochondria, peroxisomes, undergo FA metabolism, and are important in detoxification. Using TGF-β receptor activation as a profibrotic stress, miR-21–deficient PTECs showed robust derepression of Ppara within 6 hours, which was not seen in WT PTECs (Figure 6A and Supplemental Experimental Procedures). TGF-β is known to upregulate miR-21 in PTECs approximately 3-fold in response to TGF-β stress (4). Downstream target genes of PPARα in the FA metabolism pathway, such as Mcad and Cpt1, involved in transport of long-chain FAs and FA oxidation, were also decreased by TGF-β and derepressed when miR-21 was absent (Figure 6, A–D). In the same experiment, mitochondrial ATP generation was quantified (Figure 6E). Whereas TGF-β treatment reduced mitochondrial ATP, in the absence of miR-21, mitochondria maintained normal levels of ATP, an effect associated with increased mitochondrial and peroxisome density and elevated levels of the mitochondrial biogenesis transcriptional coactivator PGC1α (which is reported to be coregulated with PPARα) after 24 hours (Figure 6, D, F, and G). Therefore, in the setting of cell stress, miR-21 normally silences PPARα and mitochondrial function. To determine whether anti–miR-21 could recapitulate these genetic studies, PTECs were isolated from 7-week-old Col4a3–/– mice following 3 weeks of treatment with anti–miR-21 or control anti-miR. These PTECs showed higher levels of Ppara, mitochondrial biogenesis, and mitochondrial ATP generation ex vivo when previously treated with anti–miR-21 (Figure 6, H–I). Similarly, human primary PTEC cultures treated in vitro with anti–miR-21 (Figure 6, J–L) increased expression of PPARA and PGC1α, particularly in the setting of cell stress, and maintained normal levels of mitochondrial ATP in response to TGF-β–mediated stress. Podocytes are implicated as important cells that directly act in the glomerulosclerosis process and are glomerular epithelial cells. Cultured podocytes produce miR-21 at high levels (Table 1 and ref. 4). The effect of anti–miR-21 on mouse podocyte responses to TGF-β was also evaluated in vitro (Table 1). Podocytes respond to anti–miR-21 by upregulating Ppara similarly to PTECs. Consistent with anti–miR-21 having direct activity on the PPARα pathway in glomerular cells, purified glomeruli from 9-week-old kidneys contained higher levels of Ppara following systemic treatment with anti–miR-21 (Supplemental Figure 7). Collectively, these studies suggest miR-21 acts through PPARα to regulate mitochondrial function and FA metabolism in the kidney epithelium.
Figure 6. Anti–miR-21 administration recapitulates miR-21 gene deficiency by preventing miR-21–mediated suppression of the PPARα FA metabolism and mitochondrial biogenesis pathways in the kidney proximal epithelium.

(A–C) Graphs of qPCR results showing the effect of miR-21 gene deficiency on PPARα lipid metabolism pathway transcriptional responses (Ppara, Cpt1, and Mcad) in primary cultured mouse PTECs in resting conditions or in response to active TGF-β–induced cell stress. (D) Graph of Ppargc1a levels in steady-state and response to TGF-β stress in miR21-deficient PTECs. (E) Graph of the effect of TGF-β on mitochondrial ATP generation by mouse PTECs in the absence of miR21. (F) Representative images of MitoTracker-labeled PTECs showing increased mitochondrial density in miR21-deficient PTECs, particularly in response to TGF-β stress. (G) Western blot showing levels of the peroxisome protein PMP70 in mouse PTECs at rest and in response to TGF-β stress. (H–I) The effect of anti–miR-21 treatment of mouse PTECs from Col4a3–/– mouse kidneys on Ppara expression and mitochondrial ATP generation (J–L) Graphs showing the effect of anti–miR-21 treatment on human primary PTEC levels of PPARA or PPARGC1A or mitochondrial ATP production under resting or TGF-β stress conditions. *P < 0.05; **P < 0.01, Mann-Whitney U test. n = 3–5/group. Scale bars: 25 μm.
Further support for an important link between miR-21 and PPARα function in human disease is evident in kidney cortex biopsy tissue from patients with CKD. In a cohort of biopsies from patients with documented CKD and interstitial fibrosis, there were increased levels of tissue miR-21, decreased levels of PPARα compared with normal kidney, and significant negative correlation between miR-21 and PPARα in individual samples (Supplemental Figure 8).
Anti–miR-21 oligonucleotides reduce mitochondrial production of ROS.
MicroRNAs in general tend to target genes with related functions. In addition to PPARα, miR-21 directly targets other genes in kidney, coding for mitochondrial factors involved in detoxification, metabolism, and antioxidant functions including cytochrome B (Cytb), FA desaturase-6 (Fads6), and Mpv17-like (Mpv17L), respectively (Figure 7A and ref. 4). Treatment of Col4a3–/– mice with anti–miR-21 recapitulates miR-21 deficiency by enhancing expression of each of these target genes in the kidney (Figure 7, B and C, and Supplemental Figure 6D). Mpv17L, restricted to the proximal tubule, has been implicated in mitochondrial antioxidant and antiapoptotic functions (24). We predicted therefore that, in the setting of enhanced miR-21 activity, mitochondrial dysfunction (loss of mitochondrial biogenesis, FA metabolism, detoxification, and ATP production associated with loss of mitochondrial antioxidants and peroxisome activity) would occur and result in enhanced ROS generation. Mouse PTECs pretreated with anti–miR-21 produced far less mitochondrial ROS in response to TGF-β. Similarly, PTECs harvested from Col4a3–/– mouse kidneys treated with anti–miR-21 for 3 weeks produced less ROS than PTECs harvested from kidneys from control-treated mice. Moreover, tubules from the anti–miR-21–treated mice were resistant to the ROS-stimulating effects of TGF-β (Figure 7, D and E). In steady state, human primary PTECs produced little mitochondrial ROS, but in response to TGF-β, mitochondria rapidly generated ROS (Figure 7F and Supplemental Experimental Procedures). Collectively, these studies suggest that miR-21 directly augments mitochondrial ROS generation in stressed epithelium. We extended these findings to assess ROS generation in whole tissue. Using dihydroethidium (DHE) staining as a marker of tissue ROS generation, kidneys from Col4a3–/– mice treated with anti–miR-21 generated significantly less ROS than the vehicle-treated mice (Figure 7, G and H). Consistent with reduced ROS generation by the action of anti–miR-21, Col4a3–/– mice treated with anti–miR-21 had lower urinary hydrogen peroxide concentrations in the urine (Figure 7I).
Figure 7. Anti–miR-21 inhibits mitochondrial ROS generation in the kidney.

(A) Graph showing the effect of miR21 gene deficiency on levels of the epithelial mitochondrial antioxidant protein and anti–miR-21 target Mpv17-like in mouse PTECs in response to TGF-β. (B and C) (B) qPCR and (C) Western blot of MPV17L in kidney tissue at 9 weeks. Numbers indicate relative normalized band density (same experiment as in Figure 5C). (D and E) Images (D) and quantification (E) of mitochondrial ROS generation from primary PTEC cultures purified from 7-week-old Col4a3–/– mice treated with vehicle or anti–miR-21 in vivo. (F) Images showing the effect of TGF-β–induced mitochondrial ROS in human primary PTECs in the setting of pretreatment in vitro with anti–miR-21. (G and H) Images (G) and quantification (H) of DHE fluorescence indicative of ROS generation in kidney whole-tissue sections from Col4a3–/– at 9 weeks. (I) Quantification of urinary hydrogen peroxide in timed urine collections at 9 weeks. *P < 0.05, 1-way ANOVA or Mann-Whitney U test. n = 3–12/group. Scale bars: 25 μm.
Anti–miR-21 oligonucleotides normalize tubule secretion of uremic toxins.
A major but underappreciated role of the proximal tubule is to secrete into the tubule lumen protein-bound uremic toxins that are not filtered by the glomerulus, particularly those generated by gut flora (25). This tubule secretory function is critical in removing toxins from the body that otherwise directly cause morbidity and mortality in renal insufficiency (25). This process requires mitochondrial detoxification enzymes, FA metabolism, and SLC high-affinity transporters, including the OAT family of basolateral transporters. The detoxification and tubular secretion processes were identified as potential targets of anti–miR-21 as a part of the mitochondrial dysfunction pathway and detoxification pathways (ref. 4, Figure 5, Supplemental Figure 5). OAT-1 (Slc22a6) is a seed-matched target gene for miR-21 (4). It is silenced in Alport nephropathy and enhanced by anti–miR-21 treatment (Supplemental Figure 9A), and it is specifically derepressed in primary PTEC cultures treated with anti–miR-21 (Supplemental Figure 9B). The RNA-sequencing analysis indicated that additional basolateral detoxifying transporters were also derepressed by anti–miR-21, including OCTN1 and OAT7 (not shown), both implicated in proximal tubule excretion. Indoxyl sulfate (IS) is a uremic toxin specifically secreted by proximal tubule OAT1 into the urine, and its level in human plasma is associated closely with vascular disease and mortality in patients with kidney disease (26). Plasma IS levels, evaluated by liquid chromatograph–mass spectrometry (LC-MS), were highly elevated in Col4a3–/– mice, but Col4a3–/– mice treated with anti–miR-21 had normal levels (Supplemental Figure 9C).
miR-21 directly enhances myofibroblast activation.
Transcriptional analysis of whole kidney indicated that fibroblast activation pathways were significantly downregulated by anti–miR-21 (Figure 5). Resident mesenchymal cells known as pericytes (PCs) and resident fibroblasts, derived from FOXD1 transcription factor expressing nephrogenic progenitor cells, represent a major precursor population of kidney myofibroblasts (27, 28). Previous studies have shown that miR-21 is upregulated in these kidney myofibroblast precursors (fibroblasts/PCs) when they differentiate into myofibroblasts (27–30). To understand whether reduced myofibroblast activation was a consequence of reduced epithelial injury exclusively or whether miR-21 directly regulates myofibroblast activity, miR21–/– or WT fibroblast/PC and myofibroblast cultures were generated and studied in vitro (Figure 8A). miR21–/– fibroblast/PC cultures exhibited reduced stress-fiber formation in 2D culture detected by phalloidin staining (Figure 8A) and lower levels of collagen I production (Figure 8B). In keeping with miR-21 regulating mitochondrial functions, miR21–/– fibroblasts/PCs had increased mitochondrial content (Figure 8C) in resting culture compared with WT cells. Mitochondrial ROS was also reduced in miR21–/– fibroblasts/PCs and not enhanced by TGF-β stimulation (Figure 8D). Consistent with the link between miR-21 and PPARα function, Ppara and the mitochondrial biogenesis factor Ppargc1a were expressed at higher levels in resting cultures (Figure 8, E and F). These characteristics were preserved after treatment with TGF-β, which stimulates myofibroblast differentiation in vitro (Figure 8A). miR21–/– fibroblasts/PCs and myofibroblasts generated fewer proinflammatory cytokines and chemokines and migrated less in assays compared with controls (Figure 8, G–I), supporting a primary connection between miR-21 function and PC/myofibroblast activation. Since Ppara is a target gene for miR-21 and is consistently overexpressed in miR21–/– fibroblast/PCs, we evaluated the effect of Ppara deficiency on fibroblast/PC and myofibroblast functions (Figure 8, A, B, H, and I). Ppara–/– fibroblast/PC and myofibroblasts showed enhanced stress-fiber formation, migration, cytokine production, and collagen production. These characteristics were opposite those of miR-21 deficiency and support a direct link between miR-21 activity and PPARα in the regulation of resident fibrogenic cells. PPARα has been reported to have direct antiinflammatory and antifibrotic functions, which are mediated through activation of the NF-κB inhibitor IκB-α and through binding to and inhibiting the transcription factor C-JUN. Consistent with this, IκB-α is expressed at higher levels in activated miR21–/– fibroblast/PC (Figure 8J). To establish whether anti–miR-21 treatment recapitulated miR-21 deficiency in human fibroblast/PCs directly, primary human cultures were treated with anti–miR-21 oligos or control oligos. PPARA and PPARGC1A were derepressed in the setting of TGF-β signaling. Moreover, known C-JUN gene targets, downstream of PPARα activity, including collagen α2 type I (COL1A2) and integrin α3, were repressed (Figure 8, K and L).
Figure 8. miR-21 activity regulates fibroblast/PC activation directly.

(A) Phase contrast or phalloidin-stained images of primary mouse kidney fibroblast (fibro/PC) cultures showing morphology and stress=fiber formation in control or TGF-β–activated conditions. Note that miR21–/– fibro/PCs have fewer stress fibers in both conditions, whereas Ppara–/– fibro/PCs have elongated morphology and increased lamellipodia formation (arrowheads). (B) Images showing mitochondrial density in miR21–/– fibro/PCs compared with WT fibro/PCs, detected by MitoTracker. (C) Images showing mitochondrial ROS generation in WT fibro/PCs in control or TGF-β–activated conditions compared with miR21–/– fibro/PCs. (D) Col1a1 transcript levels in primary fibro/PC cultures from mutant mouse kidneys or strain-matched control (WT). (E and F) Graphs showing expression of Ppara and Ppargc1a in mouse fibro/PCs in control or TGF-β–activated conditions. (G and H) Cytokines and growth factor secretion by fibro/PCs from (G) WT or miR21–/– kidneys or (H) WT or Ppara–/– kidneys. (I) Migration of WT, miR21–/–, Ppara–/– fibro/PCs and myofibroblasts in a wound-healing assay in control conditions. Note that miR21–/– cells are hypomigratory. (J) Western blot showing levels of the Ppar gene target IκB-α at rest and in response to TGF-β. (K) Graphs showing the effect of anti–miR-21 on transcript levels in human fibro/PCs in control or TGF-β–stimulated conditions. (L) Western blots showing effect of anti–miR-21 on human fibro/PC expression of C-JUN target genes: collagen I protein (60 kDa) and integrin-α3 (130 kDa), in control or TGF-β–stimulated conditions. Numbers indicate relative normalized band density. *P < 0.05; **P < 0.01; ***P < 0.001, 1-way ANOVA or Mann-Whitney U test. n = 3–5/group. Scale bars: 25 μm.
Discussion
The studies described herein show clearly that inhibition of miR-21 by anti–miR-21 oligonucleotides administered s.c. improved kidney function, improved disease pathology, and increased the median life span of mice that had a relentlessly progressive kidney disease by more than 40 percent. As markers of this improved function, anti–miR-21 reduces blood BUN and albuminuria and preserves the secretion of uremic toxins, including IS, by the kidney tubule. As such, anti–miR-21 represents an approach for new therapeutics to treat human Alport nephropathy patients and potentially to treat patients with other forms of CKD. The mouse model of Alport nephropathy has been used successfully for the preclinical development of other therapies in clinical practice or currently in clinical trials, including blockade of the TGF-β–signaling pathway (31), TNF-α antagonism (32), chemokine receptor blockade (33), and inhibition of angiotensin activity (20, 34). It therefore provides a robust model for human disease, and since the mechanisms of CKD progression are believed increasingly to represent stereotyped responses to injury of the kidney (35), the efficacy of miR-21 blockade in this mouse model has broad implications as a potential therapy in other forms of CKD.
Current and previous strategies to design and develop therapeutics to treat kidney diseases have focused heavily on reducing blood pressure and targeting overactivity of the immune response (36). More recently, there has been increasing interest in targeting fibrogenic processes, which are an intrinsic component of many forms of human kidney disease and widely thought to accelerate functional decline. miR-21, identified as a factor in kidney fibrosis, is upregulated particularly in fibrogenic cells (4, 8). But in addition to fibrogenic cells, miR-21 is also highly expressed in the epithelium. Mechanistically, it now appears that miR-21 exerts a negative influence in CKDs by inhibiting metabolic functions in fibrogenic cells as well as the kidney tubule epithelium and, shown here for what we believe is the first time, glomerular epithelial cells, including podocytes. In the normal proximal tubule, the metabolic function of FA oxidation, occurring in peroxisomes and mitochondria, is required to maintain the high metabolic demands of an array of transporters involved in reabsorption from, and secretion into, the tubular lumen as well as detoxification of endogenous and exogenous molecules. Suppression of this important pathway appears to be central to miR-21 function, which serves to enhance epithelial cell stress and dysfunction. In diabetic kidney disease, mitochondrial dysfunction created by the metabolic imbalance from the diabetes has recently been reported to be the driving force for progression (37). Also, recent large genome-wide association studies have identified polymorphisms predominantly in metabolic genes that associate with development and progression of human CKDs (38). Collectively, these reports support the postulate that metabolic dysfunction is an important problem driving the progression of CKD, including Alport nephropathy. The current studies were performed in Col4A3–/– mice on a 129X1/SvJ background; these progress rapidly and die by week 10 to 11. Expression of this mutation on a C57BL/6 background results in slower disease progression, and F1 progeny of this cross have intermediate disease progression (39). Dependence of the Alport syndrome phenotype disease progression on strain has also been observed in Col4a4 mutations (40). The efficacy described here is not limited to the 129X1/SvJ strain, as it has also been observed in the Col4A3 F1 progeny of 129X1/SvJ crossed with C57BL/6 mice (41). Similarly, studies performed previously in the ureteral obstruction and ischemic injury models used mice on the C57BL/6 background (4).
Systemic administration of anti-miRNA oligonucleotides results in functional inhibition of target miRNA in multiple organs, with greatest effects in kidney and liver. In kidney, oligonucleotides concentrate extensively within proximal tubule epithelium. Previous studies suggested that healthy glomerular cells did not concentrate systemic anti-miRNAs (4, 19). One unexpected finding in the Col4a3–/– mice was the robust effects of anti–miR-21 on glomerular pathology. The studies here support a direct role for anti–miR-21 on glomerular pathology as well as tubulointerstitial pathology. We found clear accumulation of anti–miR-21 oligonucleotides in all glomerular cell types in vivo during disease, and we provide clear evidence that miR-21 is upregulated in both the tubule and glomeruli and show in vitro that anti–miR-21 specifically and directly derepresses target genes and reduces the TGF-β–mediated stress responses in podocytes. Recent studies have implicated mitochondrial dysfunction, ROS production, and cell-stress pathways in podocytes directly in the development of glomerulosclerosis (37, 42), suggesting that miR-21 is an important target in glomerular disease.
The investigations into mechanism of action of anti–miR-21 reinforce and build on previous genetic studies where we showed that miR-21 silenced genes associated with metabolism and FA oxidation (4). Those studies employed 2 short-term models of interstitial kidney fibrosis. PPARα and its downstream FA oxidation pathways were key targets for miR-21 function in those models. Genetic deletion of PPARα abrogated the effects of anti–miR-21, and overexpression of PPARα in the renal tubule epithelium phenocopied the inhibitory effects of anti–miR-21, suggesting that PPARα was a critical target for miR-21 (4, 43). It is striking, therefore, that in the Alport nephropathy model reported here, we again identified metabolic functions and PPARα signaling as critical pathways derepressed by anti–miR-21. Taken together, these data strongly support a preeminent role for PPARα and its downstream antiinflammatory function, FA oxidation, detoxification, and mitochondrial/peroxisomal biogenic functions as relevant therapeutic approaches in Alport nephropathy and CKD progression. PPARα not only stimulates healthy mitochondrial functions, but also plays diverse antiinflammatory roles by inhibiting NF-κB signaling, stress signaling, and TGF-β signaling (44). The studies reported here indicate that, in fibroblasts and glomerular cells, regulation of PPARα by anti–miR-21 directly inhibits inflammatory and profibrotic signaling. Several reports have indicated that synthetic ligands for PPARα are antifibrotic in kidney disease models, and fibrates that are weak ligands for PPARα have also been reported to have antiinflammatory effects in the kidney (43, 45). Moreover, transgenic overexpression of PPARα in the proximal tubule alone is sufficient to reduce inflammation and fibrosis in models of kidney fibrosis. Therefore, anti–miR-21 is a potent, nontoxic mechanism for enhancing PPARα functions in the kidney.
Increased activity of miR-21 in the kidney also appears to be a key driver of ROS production (4, 46). While much focus of pathological ROS production in chronic disease has been on nonmitochondrial NADPH oxidases, the studies here provide strong evidence that mitochondrial ROS is generated under the control of miR-21. miR-21 directly silences antioxidant proteins in the mitochondria, including MPV17-like and alkylglycerone phosphate (Agps); it also inhibits mitochondrial and peroxisome biogenesis. Reduced biogenesis results in dysfunctional mitochondria producing increased ROS through reduced superoxide dismutase, glutathione, nuclear factor erythroid 2-related factor 2, and other pathways (4). miR-21 also silences mitochondrial UCP enzymes, which regulate uncoupling of respiration and therefore prevent ROS generation (47). Peroxisomes, also under the regulation of miR-21, play critical roles in ROS detoxification by the conversion of ROS to hydrogen peroxide, thence to water (48). Strikingly, tubular epithelial cells cultured ex vivo from diseased kidneys retain mitochondrial dysfunction, suggesting the dysfunction imposed by disease is not transient and may persist due to mitochondrial DNA mutations or epigenetic changes (37, 49). Therefore, exaggerated miR-21 provokes ROS generation at multiple levels, an effect that can be effectively monitored by urinary hydrogen peroxide production.
To conclude, inhibition of miR-21 with blocking oligonucleotides delivered s.c. is a simple, effective approach for retarding the downstream consequences of mutations in type IV collagen genes that ultimately drive progression of Alport nephropathy. Such an approach may potentially inhibit progression of other forms of CKD. The conservation of miR-21 gene-target engagement across species, the consistent finding of chronically upregulated miR-21 in human CKD, and the reproducibility of miR-21 biology in human primary culture suggest miR-21 blockade is an attractive target for human disease. A biologically important action of anti–miR-21 in the diseased kidney is derepression of PPARα-regulated signaling pathways in FA oxidation, detoxification, mitochondrial biogenesis, and antiinflammatory signaling. miR-21 has many gene targets in the kidney, including multiple regulators of metabolism in mitochondria and the cytosol, and many of these gene targets are derepressed by anti–miR-21. In addition to enhanced mitochondrial functions, anti–miR-21 markedly reduces mitochondrial ROS generation. Finally, miR-21 appears to provoke stress responses, not only in the epithelium, but also in glomerular cells, including podocytes and fibroblasts, suggesting its broad role as an amplifier of chronic disease.
Methods
Animal studies and anti–miR-21 administration.
Col4a3–/–, Col4a3+/–, and Col4a3+/+ mice on a 129X1/SvJ genetic background were obtained from heterozygous breeding after mutant mice on the SJL background were crossed to 129X1/SvJ mice for 9 generations (39, 50). To mitigate minor genetic variations, animals from the same litter were utilized for different treatments. In some experiments, Col4a3–/– mice were used on a pure 129sv background (129-Col4a3tm1Dec/J). miR21–/– mice and littermate controls were described previously and genotyped by methods described (4). The anti–miR-21 oligonucleotides used in these studies are chemically modified single-stranded RNA molecules with full-sequence complementarity to miR-21 that were synthesized at Regulus Therapeutics. The anti-miRs have phosphorothioate linkages to protect from nuclease activity in vivo and enhance plasma-protein binding to reduce renal clearance (51, 52). The anti-miRs also have modified sugar moieties (2′MOE and cEt) to enhance hybridization affinity to miR-21, thereby blocking its ability to interact with target genes (53). Multiple anti–miR-21 compounds have been tested in the Col4a3-deficient mouse with similar results and also in other miR-21–dependent models (4). For some studies, a Cy3 moiety was attached to the 5′ end of the oligonucleotide to allow visualization of compound distribution. Mice received anti–miR-21 (25 mg/kg) or control anti-miR (25 mg/kg) in PBS by interscapular s.c. injection twice per week. In some experiments, mice received a range of doses, from 12.5 mg/kg once a week to 50 mg/kg once a week. Anti–miR-21 is a high-affinity oligonucleotide complementary to the active site of miR-21. Mice received injections starting at 24 days (3.5 weeks) after birth and ending at 5, 7, 9, or 16 weeks after birth, depending on the study objectives. In some experiments, a 1:20 dilution of Cy3-labeled anti–miR-21 was included at week 6 or week 8 to allow visualization of the compound delivery locations in the diseased kidney. Kidney tissues were analyzed 48 hours after s.c. delivery to study cellular distribution.
Bioinformatic analysis.
All original microarray data were deposited in the NCBI’s Gene Expression Omnibus (GEO GSE62295) and were quality controlled for outliers using principal component analysis (PCA). Differential expression analysis between transcriptome profiles of experimental groups was performed using the R/Bioconductor package limma (Linear Models for Microarray Data), which includes methods for RNA-Seq data analysis. A threshold of absolute fold-change of greater than 1.2 (P < 0.05) was used to select genes for pathway analysis. IPA (Ingenuity Systems) and MetaCore (Thomson Reuters), version 6.15, were used for pathway analyses, and pathway images were generated using MetaCore software. Multispecies alignment of miR-21 sequences was done in CLC Genomics Workbench, version 5.5.1.
Statistics.
Data are presented as mean ± SEM. Differences between multiple groups were assessed by 1-way ANOVA with Dunnett’s post hoc test or Mann-Whitney U test for 2 groups. Differences in survival were assessed by Gehan-Breslow-Wilcoxon test. P < 0.05 was considered significant.
Study approval.
Human kidney biopsy samples were obtained from patients who received a kidney transplant and were undergoing biopsy for clinically indicated reasons under IRB DR-1 LCID-2010-044 at the Lahey Clinic (Burlington, Massachusetts, USA), as described previously (4). All patients provided written informed consent. All biopsies were evaluated by a pathologist for features of CKD and fibrosis, and all patients had a reduction in glomerular filtration rate (GFR) or had developed proteinuria. Kidney tissue was fixed in RNAlater immediately after collection. Human primary kidney cells were from discarded human tissue (IRB447773EA at the University of Washington). All animal studies were performed under protocols approved by the Institutional Animal Care and Use Committee at the University of Washington or Regulus Therapeutics.
Supplementary Material
Acknowledgments
These studies were funded by NIH grants (DK087389, DK093493, DK094768, TR000504), an American Heart Association grant (12040023), the University of Washington, and a sponsored research agreement with Regulus Therapeutics. D. Portilla was funded by DK75976 and a Veterans Affairs Merit Award. We thank the Lynn and Mike Garvey Microscopy Suite for assistance with imaging; Essence Underwood, Rong Tian, Steve Standage, Kimberly Muczinski, Giovanni Ligresti, Kelly Hudkins, and Charles Alpers (University of Washington) for assistance or advice or providing reagents; Erwin Bottinger (Mount Sinai School of Medicine, New York, New York, USA) for anti-MPV17l antibodies; and Bill Stallcup (Burnham Institute, UCSD, La Jolla, California, USA) for anti-PDGFRβ antibodies.
Footnotes
Conflict of Interest: Jeremy S. Duffield and Deidre A. MacKenna have submitted a patent for use of anti–miR-21. All authors from Regulus Therapeutics have stock options in the company. Jeremy S. Duffield has served on the Scientific Advisory Board at Regulus Therapeutics, and the Duffield lab has had a sponsored research agreement with Regulus Therapeutics. Jeremy S. Duffield is on the Scientific Advisory Board at, and has stock options in, Promedior Inc.
Reference information:J Clin Invest. 2015;125(1):141–156. doi:10.1172/JCI75852.
Ivan G. Gomez, Bryce G. Johnson, Allie M. Roach, Shuyu Ren, and Jeremy S. Duffield’s present address is: Biogen Idec, Cambridge, Massachusetts, USA.
References
- 1.Godwin JG, Ge X, Stephan K, Jurisch A, Tullius SG, Iacomini J. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc Natl Acad Sci USA. 2010;107(32):14339–14344. doi: 10.1073/pnas.0912701107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kato M, Natarajan R. MicroRNA circuits in transforming growth factor-β actions and diabetic nephropathy. Semin Nephrol. 2012;32(3):253–260. doi: 10.1016/j.semnephrol.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thum T, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456(7224):980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 4.Chau BN, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. 2012;4(121):121ra18. doi: 10.1126/scitranslmed.3003205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5(167):167sr1. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 6.Denby L, et al. miR-21 and miR-214 are consistently modulated during renal injury in rodent models. Am J Pathol. 2011;179(2):661–672. doi: 10.1016/j.ajpath.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong X, et al. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia. 2013;56(3):663–674. doi: 10.1007/s00125-012-2804-x. [DOI] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee R, Feinbaum R, Ambros V. A short history of a short RNA. Cell. 2004;116(2 suppl):S89–S92. doi: 10.1016/s0092-8674(04)00035-2. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh AK, Quaggin SE, Vaughan DE. Molecular basis of organ fibrosis: potential therapeutic approaches. Exp Biol Med (Maywood). 2013;238(5):461–481. doi: 10.1177/1535370213489441. [DOI] [PubMed] [Google Scholar]
- 11.Gomez IG, Grafals M, Portilla D, Duffield JS. MicroRNAs as potential therapeutic targets in kidney disease. J Formos Med Assoc. 2013;112(5):237–243. doi: 10.1016/j.jfma.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anker MC, Arnemann J, Neumann K, Ahrens P, Schmidt H, König R. Alport syndrome with diffuse leiomyomatosis. Am J Med Genet A. 2003;119A(3):381–385. doi: 10.1002/ajmg.a.20019. [DOI] [PubMed] [Google Scholar]
- 13.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N Engl J Med. 2003;348(25):2543–2556. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- 14.Heidet L, Gubler M-C. The renal lesions of Alport syndrome. J Am Soc Nephrol. 2009;20(6):1210–1215. doi: 10.1681/ASN.2008090984. [DOI] [PubMed] [Google Scholar]
- 15.Temme J, et al. Outcomes of male patients with Alport syndrome undergoing renal replacement therapy. Clin J Am Soc Nephrol. 2012;7(12):1969–1976. doi: 10.2215/CJN.02190312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruegel J, Rubel D, Gross O. Alport syndrome — insights from basic and clinical research. Nat Rev Nephrol. 2013;9(3):170–178. doi: 10.1038/nrneph.2012.259. [DOI] [PubMed] [Google Scholar]
- 17.Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrol Dial Transplant. 2013;28(12):2946–2960. doi: 10.1093/ndt/gft253. [DOI] [PubMed] [Google Scholar]
- 18. Crooke ST. Antisense Drug Technology: Principles, Strategies, and Applications. 2nd ed. Boca Raton, Florida, USA: CRC Press; 2007. [Google Scholar]
- 19.Masarjian L, de Peyster A, Levin AA, Monteith DK. Distribution and excretion of a phosphorothioate oligonucleotide in rats with experimentally induced renal injury. Oligonucleotides. 2004;14(4):299–310. doi: 10.1089/oli.2004.14.299. [DOI] [PubMed] [Google Scholar]
- 20.Gross O, et al. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63(2):438–446. doi: 10.1046/j.1523-1755.2003.00779.x. [DOI] [PubMed] [Google Scholar]
- 21.Heilig CW, et al. GLUT1 regulation of the pro-sclerotic mediators of diabetic nephropathy. Am J Nephrol. 2013;38(1):39–49. doi: 10.1159/000351989. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez ML, Distefano JK. The role of non-coding RNAs in diabetic nephropathy: potential applications as biomarkers for disease development and progression. Diabetes Res Clin Pract. 2013;99(1):1–11. doi: 10.1016/j.diabres.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Zhou T-B, Drummen GPC, Jiang Z-P, Long Y-B, Qin Y-H. Association of peroxisome proliferator-activated receptors/retinoic acid receptors with renal diseases. J Recept Signal Transduct Res. 2013;33(6):349–352. doi: 10.3109/10799893.2013.838786. [DOI] [PubMed] [Google Scholar]
- 24.Krick S, et al. Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc Natl Acad Sci USA. 2008;105(37):14106–14111. doi: 10.1073/pnas.0801146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeung CK, Shen DD, Thummel KE, Himmelfarb J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int. 2014;85(3):522–528. doi: 10.1038/ki.2013.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niwa T, Shimizu H. Indoxyl sulfate induces nephrovascular senescence. J Ren Nutr. 2012;22(1):102–106. doi: 10.1053/j.jrn.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 27.Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest. 2014;124(6):2299–2306. doi: 10.1172/JCI72267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asada N, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest. 2011;121(10):3981–3990. doi: 10.1172/JCI57301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Humphreys BD, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schrimpf C, Duffield JS. Mechanisms of fibrosis: the role of the pericyte. Curr Opin Nephrol Hypertens. 2011;20(3):297–305. doi: 10.1097/MNH.0b013e328344c3d4. [DOI] [PubMed] [Google Scholar]
- 31.Molteni A, et al. Cytostatic properties of some angiotensin I converting enzyme inhibitors and of angiotensin II type I receptor antagonists. Curr Pharm Des. 2003;9(9):751–761. doi: 10.2174/1381612033455396. [DOI] [PubMed] [Google Scholar]
- 32.Ryu M, Mulay SR, Miosge N, Gross O, Anders H-J. Tumour necrosis factor-α drives Alport glomerulosclerosis in mice by promoting podocyte apoptosis. J Pathol. 2012;226(1):120–131. doi: 10.1002/path.2979. [DOI] [PubMed] [Google Scholar]
- 33.Ninichuk V, et al. Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J Am Soc Nephrol. 2005;16(4):977–985. doi: 10.1681/ASN.2004100871. [DOI] [PubMed] [Google Scholar]
- 34.Gross O, et al. Safety and efficacy of the ACE-inhibitor Ramipril in Alport Syndrome: The double-blind, randomized, placebo-controlled, multicenter phase III EARLY PRO-TECT Alport trial in pediatric patients. ISRN Pediatr. 2012;2012:436046. doi: 10.5402/2012/436046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snyder JJ, Foley RN, Collins AJ. Prevalence of CKD in the United States: a sensitivity analysis using the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis. 2009;53(2):218–228. doi: 10.1053/j.ajkd.2008.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satou R, Gonzalez-Villalobos RA. JAK-STAT and the renin-angiotensin system: The role of the JAK-STAT pathway in blood pressure and intrarenal renin-angiotensin system regulation. JAKSTAT. 2012;1(4):250–256. doi: 10.4161/jkst.22729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dugan LL, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123(11):4888–4899. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Köttgen A, et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat Genet. 2009;41(6):712–717. doi: 10.1038/ng.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang JS, et al. Loss of α3/α4(IV) collagen from the glomerular basement membrane induces a strain-dependent isoform switch to α5α6(IV) collagen associated with longer renal survival in Col4a3–/– Alport mice. J Am Soc Nephrol. 2006;17(7):1962–1969. doi: 10.1681/ASN.2006020165. [DOI] [PubMed] [Google Scholar]
- 40.Korstanje R, et al. A mouse Col4a4 mutation causing Alport glomerulosclerosis with abnormal collagen α3α4α5(IV) trimers. Kidney Int. 2014;85(6):1461–1468. doi: 10.1038/ki.2013.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boulanger JH, et al. Identification of the pathological role of miR-21 in Alport’s kidney disease [Abstract]. J Am Soc Nephrol. 2013;24:525A. [Google Scholar]
- 42.Inoki K, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121(6):2181–2196. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li S, et al. Proximal tubule PPARα attenuates renal fibrosis and inflammation caused by unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2013;305(5):F618–F627. doi: 10.1152/ajprenal.00309.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cimini A, Cerù MP. Emerging roles of peroxisome proliferator-activated receptors (PPARs) in the regulation of neural stem cells proliferation and differentiation. Stem Cell Rev. 2008;4(4):293–303. doi: 10.1007/s12015-008-9024-2. [DOI] [PubMed] [Google Scholar]
- 45.Boor P, et al. The peroxisome proliferator-activated receptor-α agonist, BAY PP1, attenuates renal fibrosis in rats. Kidney Int. 2011;80(11):1182–1197. doi: 10.1038/ki.2011.254. [DOI] [PubMed] [Google Scholar]
- 46.Kashihara N, Haruna Y, Kondeti VK, Kanwar YS. Oxidative stress in diabetic nephropathy. Curr Med Chem. 2010;17(34):4256–4269. doi: 10.2174/092986710793348581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mailloux RJ, Harper M-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med. 2011;51(6):1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 48.Modlinger PS, Wilcox CS, Aslam S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin Nephrol. 2004;24(4):354–365. doi: 10.1016/j.semnephrol.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124(6):2333–2340. doi: 10.1172/JCI72271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cosgrove D, et al. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;10(23):2981–2992. doi: 10.1101/gad.10.23.2981. [DOI] [PubMed] [Google Scholar]
- 51. Levin AA, Yu RZ, Geary RS. Basic principles of the pharmacokinetics of antisense oligonucleotide drugs. In: Crooke ST, ed. Antisense Drug Technology: Principles, Strategies, and Applications. 2nd ed. Boca Raton, Florida, USA: CRC Press; 2007:183–216. [Google Scholar]
- 52. Swayze EE, Bhat B. The medicinal chemistry of oligonucleotides. In: Crooke ST, ed. Antisense Drug Technology: Principles, Strategies, and Applications. 2nd ed. Boca Raton, Florida, USA: CRC Press; 2007:143–182. [Google Scholar]
- 53.Davis S, et al. Potent inhibition of microRNA in vivo without degradation. Nucleic Acids Res. 2009;37(1):70–77. doi: 10.1093/nar/gkn904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.