

Abstract
Autophagy (“self-eating”) constitutes one of the most spectacular yet subtly regulated phenomena in cell biology. Similarly to cell division, differentiation, and death, autophagy is perturbed in multiple diseases, in that excessive or deficient autophagy may contribute to pathogenesis. Numerous attempts have been launched to identify specific inducers or inhibitors of autophagy and to use them for the therapeutic correction of its deregulation. At present, several major disease categories (including but not limited to age-related, cardiovascular, infectious, neoplastic, neurodegenerative, and metabolic pathologies) are being investigated for pathogenic aberrations in autophagy and their pharmacologic rectification. Driven by promising preclinical results, several clinical trials are exploring autophagy as a therapeutic target.
Introduction
Macroautophagy, herein referred to as “autophagy,” is essential for cellular homeostasis and stress adaptation and constitutes a process that facilitates the lysosomal degradation of intracellular material sequestered within autophagosomes. Autophagy becomes insufficient in aging organisms, menacing their functionality and survival (1). Moreover, autophagy fails to maintain cellular functions in multiple chronic diseases including (but not limited to) infectious, neoplastic, and neurodegenerative diseases. This observation, which was first made in model organisms including rodents, has now been substantiated by clinical correlations in humans. Thus, polymorphisms in autophagy genes are associated with breast cancer and other malignancies, inflammatory bowel disease, mycobacterial infections, asthma, chronic obstructive pulmonary disease, systemic lupus erythematosus, and hereditary neurologic disorders (2). Logically, autophagy is now one of the most studied phenomena in cell biology and pathophysiology. Given its broad clinical implications, autophagy has become a major target for drug discovery and development, as discussed in the present series of Reviews on autophagy, which deal with the implications of autophagy in major, socioeconomically relevant diseases.
Pathophysiologic impact of autophagy
Neoplasia.
There are still some polemics on the importance of autophagy defects in the development of cancer. In mice, genetic defects of autophagy (such as liver-specific knockout of autophagy-related gene 7 [Atg7] or haploinsufficiency of the essential ATG Becn1, which codes for beclin 1) cause an increase in the incidence of cancers (3). Similarly, knockout of Atg5 or Atg7 can increase the incidence of p53-deficient, KRAS-induced pancreatic cancers (4), and knockout of Atg5 accelerates initial oncogenesis of KRAS-induced lung neoplasms (5). However, a preliminary study performed on human breast cancers suggested that the loss of functional beclin 1 is not associated with oncogenesis (6). Notwithstanding this latter result, there is mounting evidence that autophagy may play a dual role in cancer. While functional or genetic suppression of autophagy may accelerate early oncogenesis, autophagy often is essential for tumor progression, when cancer cells must be optimally fit to survive the challenge of surviving in hypoxic conditions or after detachment from their usual tropic support (3).
Cardiovascular disease.
The role of autophagy is also somehow controversial in the context of cardiac disease. Autophagy is required for the avoidance of multiple cardiovascular pathologies associated with aging, and autophagy is indispensable for ischemic preconditioning, i.e., the mechanism by which short, sublethal episodes of ischemia can protect the heart against massive infarction induced by protracted ischemia. Moreover, deficiencies in autophagy in different cell types precipitate arteriosclerosis and other cardiovascular diseases. However, autophagy may also play a pathogenic role in cardiac remodeling induced by pressure overload, arguing in favor of an ambiguous role of autophagy in cardiac physiology (7).
Immune-related diseases.
Autophagy mediates the removal of intracellular pathogens (such as Mycobacterium tuberculosis) and as such constitutes one of the most primitive mechanisms of immune defense. It is important to note that parts of the autophagic machinery are linked to phagocytosis, endocytosis, and non-canonical protein secretion, meaning that mutations in autophagy-relevant genes may affect other immune-relevant processes besides autophagy. As a result, autophagy (or proteins participating in the autophagic process) play multiple pleiotropic roles in inflammatory and immune responses. The preponderant role of autophagy in the immune system is to promote the clearance of intracellular bacteria, to mediate anti-inflammatory reactions, to improve antigen presentation, and to promote T cell memory (8).
Neurodegeneration.
There is ample evidence that autophagy defects can contribute to a number of neurodegenerative diseases including but not limited to Alzheimer’s disease, Parkinson’s disease, and ALS (9). As a recurrent leitmotif, in neurons, which are non-proliferating cells, accumulating protein aggregates cannot be “diluted” by cell division and must be eliminated by autophagy, meaning that either an increase in the generation of misfolded protein or a decrease in their autophagic removal is pathogenic. There are several familial forms of Parkinson’s disease in which genes affecting mitophagy, i.e., the autophagic removal of dysfunctional mitochondria, are mutated, underscoring a direct genetic link between autophagy and neurodegeneration. Such a direct genetic link is also observed in familial ALS, in which autophagic adaptor proteins such as sequestosome 1 and optineurin are affected by loss-of-function mutations (9).
Aging.
In old age, autophagy is insufficient, either because the autophagic flux is diminished or because there is too much cargo resulting from chronic cellular damage. Maneuvers designed to induce autophagy at the level of the whole body, for instance by dietary interventions (such as general caloric restriction, specific protein restriction, or restriction of single amino acids) or by pharmacologic agents (such as supplementation with spermidine or rapamycin) can reduce the incidence of age-related pathologies and extend life span in model organisms ranging from yeast to mammals including rodents and macaques (1, 10). Importantly, there is extensive evidence that any kind of nutritional, pharmacologic, or genetic manipulation resulting in an extension of longevity also induces an increase in autophagy, which — at least in yeast, nematodes, and flies — is essential for favorable effects on life span (11).
Therapeutic manipulation of autophagy
Gene therapy for autophagy induction.
In rodent models, gene therapeutic approaches designed to induce autophagy have had positive effects on a variety of disease models including aging (when ATG5 is overexpressed in the entire organism), obesity (when ATG7 or the pro-autophagic transcription factor EB [TFEB] are overexpressed in hepatocytes), cancer (when beclin 1 is expressed in KRAS-induced lung adenomas), neurodegeneration induced by β-amyloid or α-synuclein or toxicity (when TFEB or beclin 1 are overexpressed in the brain or when cystatin B, an inhibitor of lysosomal cysteine proteases, is knocked out), myodegenerative conditions (when TFEB or beclin 1 are targeted to the skeletal muscle), and chronic lung inflammation caused by cystic fibrosis (when beclin 1 is expressed in the lung) (2).
Pharmacologic inducers of autophagy.
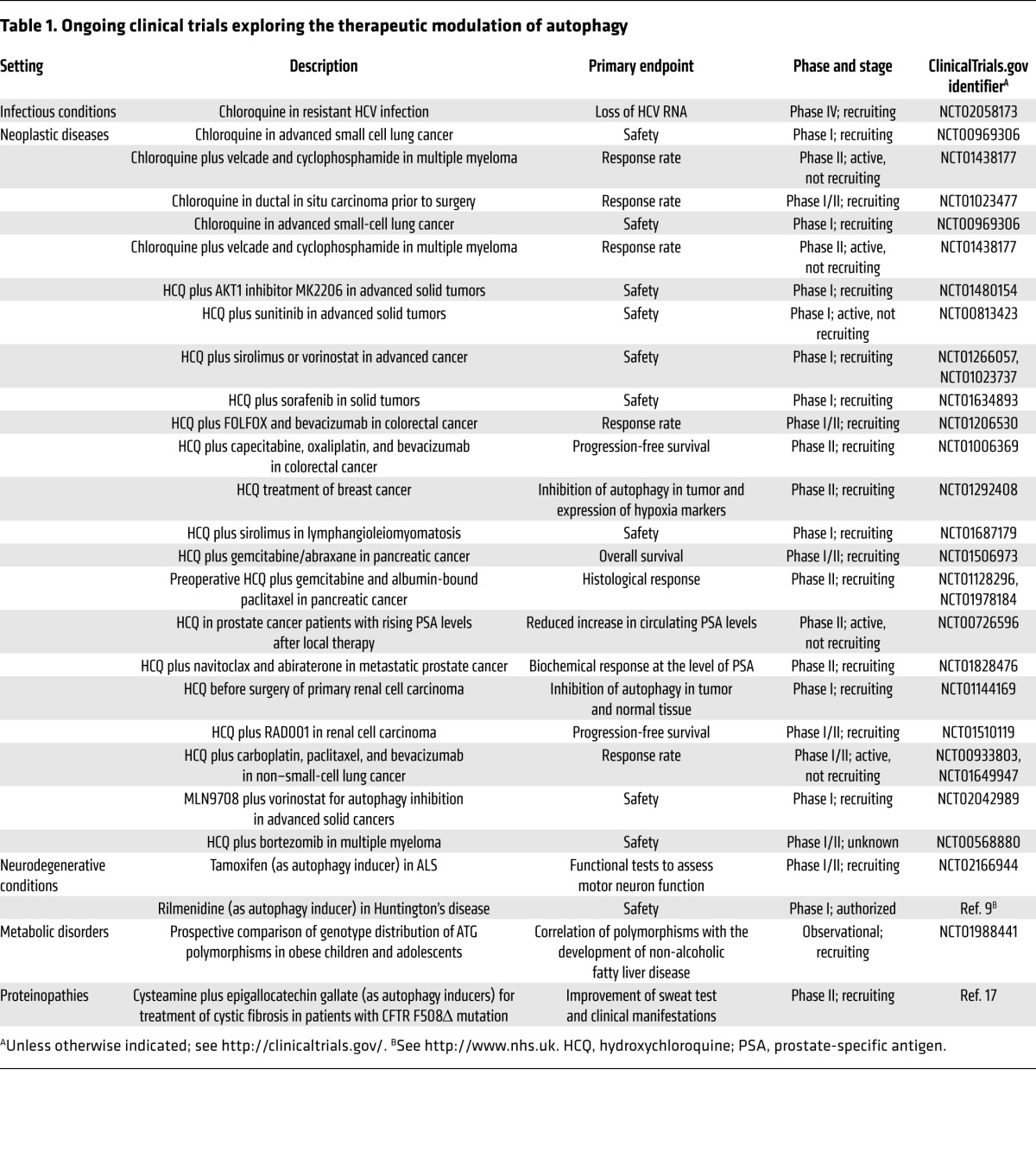
Encouraged by the health-promoting effects of genetic autophagy induction, a number of industrial and academic laboratories are attempting to develop small molecules for autophagy stimulation. Induction of autophagy can be achieved by activating pro-autophagic enzymes such as AMPK or the protein deacetylase sirtuin 1 via the disruption of inhibitory interactions that usually restrain pro-autophagic proteins, or by inhibiting negative regulators of autophagy including mTOR complex 1 (mTORC1) (12), certain deubiquitinating enzymes, and the acetyl transferase EP300 (13). Yet another strategy for inducing autophagy consists of mimicking the broad biochemical effects of starvation, resulting in the depletion of acetyl coenzyme A. Hence, the inhibition of enzymatic reactions required for the biosynthesis of acetyl coenzyme A constitutes an efficient, non-toxic approach for autophagy stimulation (14, 15). Moreover, autophagy can be triggered by many kinds of sublethal (hormotic) stress. Thus, several FDA-approved or over-the-counter drugs stimulate autophagy, but mostly have pleiotropic actions, making it difficult to parse out the contribution of increased autophagic flux in their therapeutic actions on patients (2). Some of these agents are currently being investigated in clinical trials (Table 1).
Table 1.
Ongoing clinical trials exploring the therapeutic modulation of autophagy
Gene therapy for autophagy inhibition.
The genetic inhibition of autophagy has been used in several mouse models to have a positive impact on disease outcome. Thus, inactivation of autophagy by knockout of essential autophagy genes (such as Atg5 and Atg7) within prospective tumor cells can prevent the progression of KRASG12D- or BRAFV600E-induced lung adenomas to adenocarcinomas (5, 6) and prevent the full manifestation of KRASG12D pancreatic ductal adenocarcinoma (4). These genetic manipulations appear to be particularly efficient when the tumors lack p53 expression. Knockdown of essential autophagy genes can also sensitize tumor cells to chemotherapy or radiotherapy, both in vitro and vivo, supporting the idea of therapeutic autophagy inhibition for chemo- or radiosensitization (6). When the essential autophagy gene Atg7 is irreversibly inactivated at the whole body level in mice, the animals suffer a sudden, potentially lethal defect in their immune defense as well as major metabolic alterations (within days) before they develop degenerative processes in virtually all organs (within weeks) and finally succumb to brain dysfunction (within two months) (6). These results clearly indicate that full inhibition of autophagy for a protracted period cannot constitute a realistic therapeutic goal. Rather, partial or intermittent inhibition of autophagy should be attempted.
Pharmacologic inhibitors of autophagy.
Autophagy is an extremely complex process in which multiple enzymatic complexes have to participate in a temporally and spatially coordinated manner. Thus, this pathway offers the possibility to pharmacologically target a number of essential enzymatic reactions to inhibit autophagy, including but not limited to protein kinase, lipid kinase, and protease activities (13, 16). As a word of caution, it is important to note that even totally specific inhibition of kinases and proteases by yet-to-be-discovered pharmacologic agents would also affect other processes that are not related to autophagy, with the consequent risk of major undesired side effects, as none of these putative drug targets is exclusively involved in autophagy. There are multiple clinical trials in which therapeutic inhibition of autophagy is attempted by means of relatively nonspecific lysosomotropic agents, in particular chloroquine and 3-hydroxychloroquine, for the treatment of neoplasia (Table 1).
Perspectives
The pharmacologic development of autophagy modulators is in its infancy, and it can be easily anticipated that novel, relatively specific inducers and inhibitors will soon become available for preclinical and clinical characterization. Paradoxically, the current clinical armamentarium is endowed with a larger arsenal of autophagy inducers than of autophagy inhibitors. At the level of the whole body, autophagy can be easiest stimulated by starvation, at least in mice, and it remains to be seen whether it will be possible to create autophagy inducers that have a better safety profile than caloric restriction or rapamycin. An alternative stratagem might consist in engineering autophagy inducers that are targeted to specific cell types or tissues, thus allowing for a new kind of specific therapeutic intervention. At present, a large panel of clinical trials is devoted to the therapeutic inhibition of autophagy in the context of cancer using rather nonspecific agents that had been previously used for the treatment of malaria and rheumatoid arthritis (Table 1). It remains to be seen, however, whether such agents, if successful, truly mediate their antineoplastic effects through autophagy inhibition in cancer cells, or whether they instead act through other cytotoxic or immunomodulatory effects. It will be important to develop truly specific autophagy inhibitors before further therapeutic breakthroughs can be envisaged. Thus, an entire community of basic and clinical investigators is awaiting the advent of highly specific autophagy modulators with great expectations.
Acknowledgments
G. Kroemer is supported by Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR), Projets blancs, ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC), Cancéropôle Ile-de-France, Institut National du Cancer (INCa), Fondation Bettencourt-Schueller, Fondation de France, Fondation pour la Recherche Médicale (FRM), European Commission (ArtForce), European Research Council (ERC), LabEx Immuno-Oncology, SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE), SIRIC Cancer Research and Personalized Medicine (CARPEM), and the Paris Alliance of Cancer Research Institutes (PACRI).
Footnotes
Conflict of interest: Guido Kroemer receives income and research support from Bayer Healthcare for his work related to cancer but not autophagy. He also receives research support from GlaxoSmithKline.
Reference information:J Clin Invest. 2015;125(1):1–4. doi:10.1172/JCI78652.
References
- 1.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125(1):14–24. doi: 10.1172/JCI73938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang X, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest. 2015;125(1):47–54. doi: 10.1172/JCI73942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenfeldt MT, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 5.Rao S, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 6.White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46. doi: 10.1172/JCI73941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest. 2015;125(1):55–64. doi: 10.1172/JCI73943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deretic V, Kimura T, Timmins G, Moseley P, Chauhan S, Mandell M. Immunologic manifestations of autophagy. J Clin Invest. 2015;125(1):75–84. doi: 10.1172/JCI73945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frake RA, Ricketts T, Menzies FM, Rubinsztein DC.Autophagy and neurodegeneration. J Clin Invest. 2015125165–74 10.1172/JCI73944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015;125(1):85–93. doi: 10.1172/JCI73946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YC, Guan K-L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vakifahmetoglu-Norberg H, Xia H, Yuan J. Pharmacologic agents targeting autophagy. J Clin Invest. 2015;125(1):5–13. doi: 10.1172/JCI73937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisenberg T, et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 2014;19(3):431–444. doi: 10.1016/j.cmet.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marino G, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014;53(5):710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Fernández ÁF, López-Otín C. The functional and pathologic relevance of autophagy proteases. J Clin Invest. 2015;125(1):33–41. doi: 10.1172/JCI73940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Stefano D, et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy. doi: 10.4161/15548627.2014.973737. doi: 10.4161/15548627.2014.973737. [DOI] [PMC free article] [PubMed] [Google Scholar]