

Abstract
Life and health span can be prolonged by calorie limitation or by pharmacologic agents that mimic the effects of caloric restriction. Both starvation and the genetic inactivation of nutrient signaling converge on the induction of autophagy, a cytoplasmic recycling process that counteracts the age-associated accumulation of damaged organelles and proteins as it improves the metabolic fitness of cells. Here we review experimental findings indicating that inhibition of the major nutrient and growth-related signaling pathways as well as the upregulation of anti-aging pathways mediate life span extension via the induction of autophagy. Furthermore, we discuss mounting evidence suggesting that autophagy is not only necessary but, at least in some cases, also sufficient for increasing longevity.
Introduction
One hallmark of aging is the accumulation of various forms of molecular damage, embodied in malfunctioning organelles, defective enzymes, proteinaceous aggregates, or DNA mutations (1). At the same time, the incidence of chronic diseases such as neurodegeneration, type II diabetes, or cancer rises with age concomitantly with accumulating cellular damage. Therefore, one of the main challenges for future medicine is the development of strategies to prolong health span (i.e., maintaining a healthy state without necessarily extending the maximum life span) by subverting the etiology of age-related disorders rather than solely providing symptomatic treatments. One of the most promising toeholds in the ascension toward a causal treatment of aging is autophagy, a cellular program for the removal of damaged cellular components and the digestion of cell-intrinsic macromolecules in the context of dwindling nutritional resources. Since lysosomes were first described in the 1950s (2, 3), a spectrum of different lysosome-mediated degradation pathways has been discovered. Macroautophagy (hereafter referred to as autophagy) constitutes a mechanism through which cytoplasmic organelles or cytosolic molecules are sequestered in double-membrane vesicles, so-called autophagosomes, that subsequently fuse with lysosomes for bulk digestion of the autophagic cargo (4). Besides its role in mobilizing endogenous macromolecules for meeting the cell’s energetic demands when extracellular nutrients are scarce, autophagy contributes to the maintenance of organelle homeostasis and the avoidance of proteotoxic stress, thereby attenuating or avoiding age-associated processes and mediating cytoprotection (5).
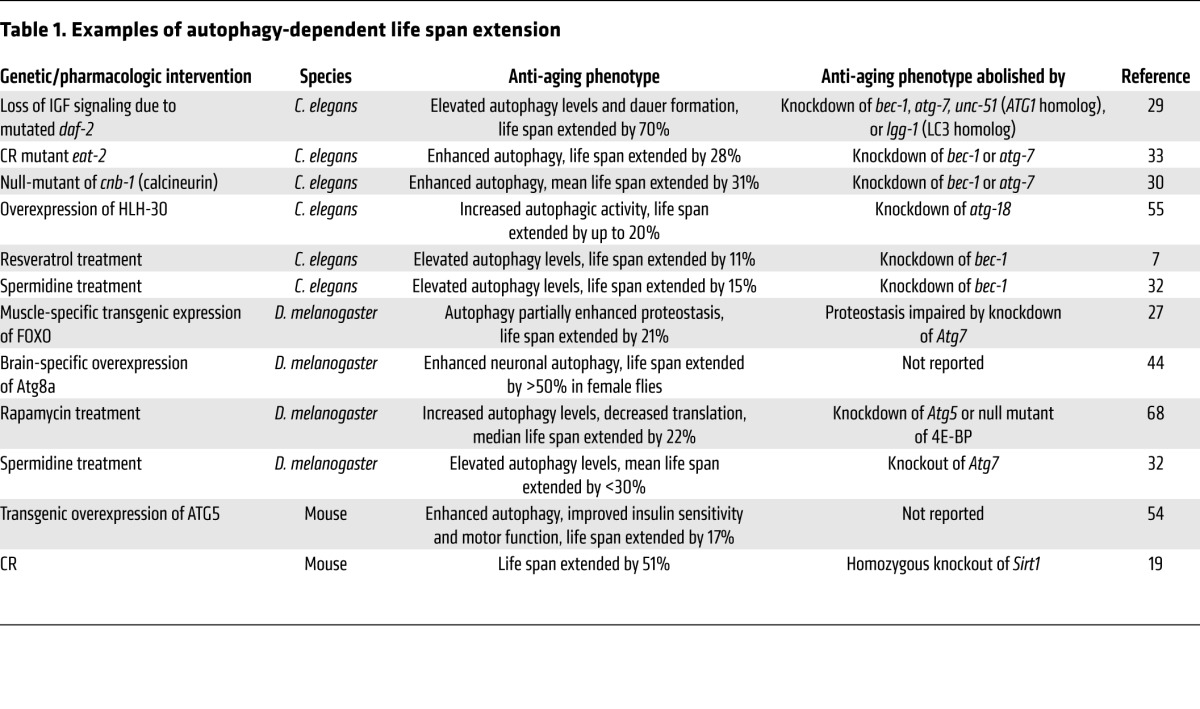
In this Review we discuss recent evidence indicating that nutritional, pharmacologic, and genetic manipulations to extend life span and/or health span often (or possibly always) stimulate autophagy, and that increasing autophagic flux mechanistically contributes to the extension of longevity (Table 1).
Table 1.
Examples of autophagy-dependent life span extension
Nutrient and growth hormone signaling are master regulators of autophagy
The discovery of different genetic and pharmacologic interventions affecting autophagy has been facilitated by a detailed molecular description of the autophagic machinery, which is highly conserved among a wide spectrum of eukaryotic model organisms, ranging from the unicellular yeast Saccharomyces cerevisiae to metazoans including the nematode Caenorhabditis elegans, the fruitfly Drosophila melanogaster, and mammals (6). The initial identification of autophagy-related genes (ATGs) was accomplished in S. cerevisiae (4), in which autophagy is essential for the response to nutrient starvation.
The process of autophagy can be broken down into three steps: regulation, phagophore formation, and ultimately cargo loading and degradation (Figure 1). First, extracellular signals are transmitted to the autophagic core machinery. In all organisms tested so far, the autophagic response to dwindling nutrients is regulated by upstream signaling cascades that encompass the nutrient sensor kinases TOR, ribosomal protein S6 kinase (S6K), and RAC-α serine/threonine-protein kinase (Akt). Intuitively, downregulation of these pathways (e.g., under nutrient or growth factor depletion) results in an upregulation of autophagic activity. In turn, other kinases such as the cellular energy status–sensing AMPK and the deacetylase sirtuin 1 (SIRT1) are positive regulators of autophagy (Figure 2 and refs. 5, 7).
Figure 1. Molecular mechanism of autophagosome formation and disposal of cellular material.
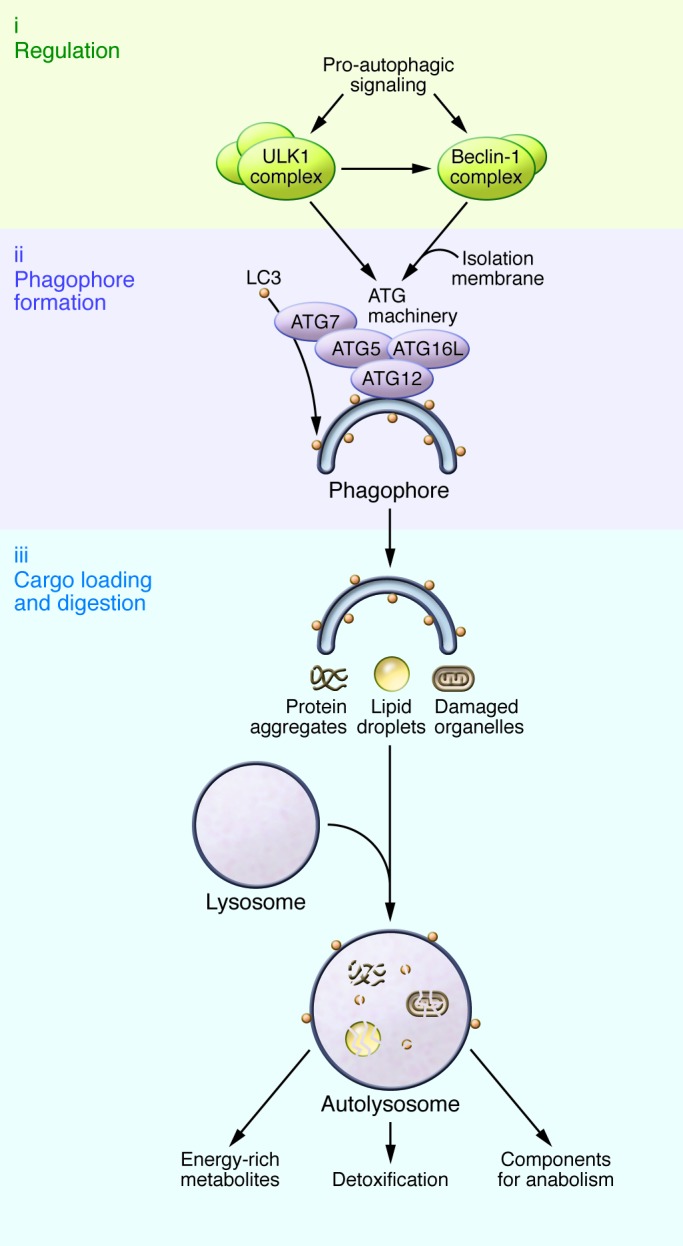
Pro-autophagic signals such as nutrient or growth factor depletion activate regulatory components of the autophagic machinery, such as the ULK1 and Beclin-1 complexes. Isolation membranes form at ER-mitochondrial interfaces or recycling endosomes under the guidance of the ATG machinery, which, among other components, consists of ATG7, ATG5-ATG12, and ATG16L. ATG7 drives the lipidation of LC3, which incorporates into the incipient phagophore to mediate the recognition and loading of cargo, such as protein aggregates, damaged organelles, or lipid droplets. The mature autophagosomes (not shown) fuse with lysosomes to form autolysosomes, through which the engulfed cellular material gets digested. As a result, potentially harmful misfolded protein aggregates or damaged organelles are detoxified, and their degradation products can be used to replenish cellular energy reserves and anabolic reactions.
Figure 2. Nutrient and growth factor signaling regulates autophagy induction.

Growth factor binding to IGFR and/or nutrient availability stimulates the TORC1 pathway, which in turn deactivates the pro-autophagic ULK1 complex and activates the autophagy inhibitory kinase S6K. Cellular energy resources are also detected by AMPK, which initiates pro-autophagic signaling when the AMP/ATP ratio rises. Recent data have underscored the importance of post-translational protein acetylation (Ac), which is controlled by HATs such as EP300, HDACs such as SIRT1, and levels of acetyl-CoA. In addition to epigenetic regulation via histone acetylation and methylation, transcription factors such as FOXO3A or TFEB also influence transcription of pro-autophagic genes (e.g., ATGs).
Caloric restriction (CR) is the most effective strategy to induce autophagy, as it activates multiple regulatory pathways. For example, CR results in the inhibition of TOR complex 1 (TORC1) and activation of AMPK, which in turn activates the autophagy-promoting Unc-51 like autophagy activating kinase 1 (ULK1) complex (Figure 2 and ref. 5), as well as the acetyltransferase MEC-17, which stimulates the cellular microtubule transport machinery that is indispensable for autophagy (8). Furthermore, CR stimulates SIRT1, which deacetylates and thereby activates essential autophagic proteins (9). This interplay between acetylation and deacetylation seems to be a leitmotif of autophagy control that we discuss in some detail below.
In contrast to nutrient starvation, cellular growth signals (along with nutrient opulence) such as receptor binding of IGF-1 suppress autophagy via the activation of Akt, which inhibits the TORC1 repressor TSC1/2 (in addition to a variety of other targets), resulting in the activation of TORC1. Active TORC1 inhibits the autophagy-relevant ULK1 complex (Figure 2) and in parallel de-represses protein translation via inactivation of S6K and inhibition of the translational repressor translation initiation factor 4E-binding protein 1 (4E-BP1) (10).
The second step of autophagy involves a panel of ATG proteins that dictate the generation of double-membrane vesicles to ingest cytoplasmic material (Figure 1). Prevailing evidence suggests that these phagophores derive from ER-mitochondrial interfaces (11). In addition, recycling endosomes may deliver parts of the plasma membrane that are marked by ATG16L1 to the core autophagic machinery (12). Phagophore elongation involves ATG6 (also known as Beclin-1) that, via its interaction with different binding partners, also constitutes a platform for the integration of programmed cell death pathways and autophagy (13). Several ATG16L1-interacting ATG proteins, such as ATG5 and ATG12, contribute to stabilizing incipient phagophores (Figure 1 and ref. 14).
Finally, as a third step, cellular material designated for degradation is enclosed by phagophores, forming vesicular autophagosomes that fuse with lysosomes for cargo digestion. The loading of autophagic cargo is promoted by the ATG8 family of ubiquitin-like proteins, including microtubule-associated protein 1 light chain 3 (LC3) (4). Following its lipidation under the guidance of ATG7, LC3 incorporates into the nascent phagophore and guides the engulfment of cargo designated for degradation by recognizing proteins containing LC3-interacting region (LIR) motifs. One prominent LIR-containing protein is sequestosome 1 (also referred to as p62), which recognizes damaged, polyubiquitinated proteins (15). LC3 also binds to externalized cardiolipin at the outer membrane of defective mitochondria in neurons and thereby targets them for selective autophagy (referred to as mitophagy) (16), which contributes to cytoprotection by autophagy (Figure 1 and ref. 17).
The enemy inside: protein acetylation interferes with the autophagic response
There is rising evidence that posttranslational modifications including protein acetylation represent another layer of autophagy regulation. In addition to the well-known involvement of the sirtuin family of deacetylases in autophagy-dependent life span regulation (18, 19), two recent studies have shown that the nucleocytosolic concentration of acetyl-CoA, the sole acetyl group donor for protein acetylation, inversely correlates with the rate of autophagy in human cells, yeast, flies, and mice (20, 21). Common autophagy inducers such as rapamycin, CR, or deletion of the yeast Akt/S6K homolog SCH9 failed to induce autophagy in cells in which nucleocytosolic acetyl-CoA was artificially elevated, and general protein acetylation was enhanced. These findings underscore the central role of acetyl-CoA as a repressor of autophagy. In yeast, histone H3 mutants mimicking constitutive acetylation of lysine residues (by substitution of glutamine for lysine) exhibited a low level of autophagy, while H3 mutants mimicking constitutive deacetylation (by substitution of arginine for lysine) demonstrated high constitutive levels of autophagy, presumably due to the epigenetic control of ATG gene expression. In human cells and mice, various strategies for depleting acetyl-CoA reduced the overall acetylation of cytoplasmic proteins commensurate with the initiation of autophagy (Figure 2). Depletion of acetyl-CoA reduced the enzymatic activity of the acetyltransferase EP300 in cell-free assays, and EP300 was required for the suppression of autophagy by high acetyl-CoA levels in cells (17).
Taken together, these findings indicate that depletion of acetyl-CoA (which can be achieved physiologically by CR or fasting) elicits autophagy via protein deacetylation. During short-term starvation, such pro-autophagic deacetylation reactions mostly affect cytoplasmic proteins such as ATG proteins, as demonstrated by the fact that depletion of acetyl-CoA or inhibition of acetyltransferases can stimulate autophagy in enucleated human cells (21). However, in long-term CR, transcriptional regulation is likely to contribute to the pro-autophagic effects of acetyl-CoA depletion (22). Although we surmise that general deacetylation reactions (such as those stimulated by acetyl-CoA depletion) favor autophagy, there are individual proteins whose acetylation by defined acetyltransferases favors autophagy. Thus, H4K16 acetylation by the histone acetyltransferase KAT8 stimulates the expression of lethal levels of autophagy-relevant gene products (23). Nonetheless, mice lacking histone deacetylases (HDACs) such as HDAC1, HDAC2, or HDAC6 have impaired autophagy (24, 25). Whether this impaired autophagy is based on epigenetic effects mediated by histone (de)acetylation has not been studied. In addition to epigenetic surveillance, autophagy is controlled by several specific transcription factors including FOXO3A (Figure 2), which in turn is controlled by several deacetylases including SIRT1 and HDAC1. Hematopoietic stem cells lacking FOXO3A exhibit a reduced autophagic response to metabolic changes in the bone marrow microenvironment, compromising their long-term function and precipitating premature aging (26). Additionally, muscle-specific overexpression of FOXO in Drosophila enhances muscle proteostasis and longevity (27).
Autophagy is an integral mediator of life span extension
In laboratory conditions, the life span of model organisms such as yeast, C. elegans, and Drosophila can be extended by a plethora of nutritional, pharmacologic, or genetic manipulations. Such longevity-increasing manipulations usually fail if autophagy is inactivated (Table 1). For example, the markedly extended life span of C. elegans lacking functional insulin-like signaling (due to mutated daf-2) (28) is abrogated when the Beclin-1 ortholog BEC-1 is depleted (29). Increased longevity due to calcineurin deficiency in nematodes is reversed upon knockdown of bec-1 or the ATG7 ortholog atg-7 (30). Similarly, life span extension by depletion of the p53 ortholog CEP-1 (31), genetic or pharmacologic activation of the SIRT1 ortholog SIR-2.1 (7), or treatment with the acetyltransferase inhibitor spermidine (32) fails if essential autophagy genes are inactivated. CR-driven life span extension in C. elegans depends on the expression of SIR-2.1 and functional autophagy (33). In mice, CR correlates with enhanced autophagic activity and prolongs life span depending on the presence of SIRT1 (19). Results from a University of Washington (UW) study indicated that CR in primates extends life span (34); however, results from a study by the National Institute on Aging (NIA) indicated that CR in primates extends health span (35). This discrepancy in the observed effects on life span might be due to the different control diets of the studies, which might either reflect an unhealthy diet (UW study) or a mild CR diet (NIA study) (36). Nevertheless, both the UW and NIA studies corroborated that CR promotes health span. Similarly, fasting regimens in humans have been associated with improved health span, including beneficial effects on metabolic syndrome, cardiovascular diseases, and cancer incidence (37, 38). Still future studies will need to clarify whether these effects on health span are autophagy dependent. Intriguingly, low protein consumption in humans and mice is associated with reduced risk for age-related diseases and mortality, probably through reduced signaling of IGF-1, which is an important negative regulator of autophagy (38). Reduced supply of the amino acid methionine increases longevity across species, possibly by activation of a CR-like state, and in yeast, methionine restriction extends life span in an autophagy-dependent manner (39).
Unfortunately, it is not possible to investigate the causal involvement of autophagy on life span extension in mice because the constitutive inactivation of essential autophagy genes either causes lethal developmental abnormalities or a fatal defect in the adaptation from intrauterine to postnatal metabolism (40, 41). In D. melanogaster, however, loss of Atg7 does not interfere with metamorphosis and rather affects physiologic neuronal functions in the adult animal (42), meaning that the effects of autophagy defects on longevity can be studied. There are indeed several reports demonstrating that autophagy-deficient flies fail to respond to longevity-extending regimens that are successful in autophagy-competent strains (Table 1).
A plethora of studies report that reduced levels of key autophagy regulators such as ULK1, ATG7, LC3/ATG8, or Beclin-1 accelerate aging in yeast, nematodes, flies, and rat pancreatic cells, accompanied by age-associated pathologies such as damaged mitochondria, reduced cardiac performance, lipid accumulation, and muscle degeneration (43–45). Deficient autophagosome formation resulting from a loss-of-function mutation of ATG16L1 has been linked to the pathogenesis of Crohn’s disease (46). Furthermore, ATG16L1 deficiency led to increased levels of the pro-inflammatory cytokine IL-1β in mice (47). Autophagy is also involved in immunogenic signaling by ATP replenishment and subsequent ATP release into the extracellular space (48), underscoring the relevance of autophagy for the regulation of inflammatory processes (17). Although the genome-wide association studies conducted thus far have failed to demonstrate any reproducible link between human longevity and polymorphisms in autophagy-relevant genes, some studies suggest a link between autophagy and age-related diseases, such as frontotemporal dementia (49), breast cancer (50), and colorectal cancer (51). These findings indicate that effects of autophagy gene polymorphisms on (decreased) longevity might be preceded and possibly concealed by associated fatal pathologies, corroborating the significance of autophagy for health span extension. Given the importance of autophagy for embryonic development (52), it is tempting to speculate that many loss-of-function mutations in autophagy-relevant genes are not detectable due to prenatal lethality.
Autophagy induction suffices to promote longevity
Logically, the demonstration that autophagy deficiency can accelerate aging suggests that enhanced autophagic activity should prolong health span, especially if normal physiologic autophagy levels were insufficient to counteract age-associated cellular damage (53). Genetic manipulations solely designed to increase autophagy are able to extend life span in several species. For instance, transgene-enforced overexpression of the pro-autophagic protein ATG5 enhanced the life span of mice and improved several features of aging, including leanness, insulin sensitivity, and impaired motor function. Furthermore, embryonic fibroblasts originating from ATG5 transgenic mice showed higher stress resistance, which was dependent on functional autophagy (54). Consistently, overexpression of HLH-30, the homolog of the mammalian transcription factor EB (TFEB), which was recently identified as an autophagy stimulator, was found to extend life span in C. elegans (55). Overexpression of other key elements of the autophagic machinery such as ATG12 or LC3/ATG8 contributed to improved life span and mitochondrial maintenance in a human in vitro model of aging (56), which suggests that the beneficial effects of sustained expression of pro-autophagic proteins can be corroborated in the human system, or at least in human cells. There is also evidence that induction of autophagy in the nervous system is particularly important, as brain-specific overexpression of LC3/ATG8 in D. melanogaster is sufficient to prolong life span (44) and mice lacking the polyglutamine stretch of huntingtin display elevated autophagy levels in the brain that were coupled to increased longevity (57). However, it is unknown whether the pathologically expanded polyglutamine stretch of mutant huntingtin associated with Huntington’s disease is responsible for the commonly observed autophagy defects (58).
CR is a simple nutritional intervention that potently stimulates autophagy, but it is generally judged to be contraindicated for broad clinical application because it causes weight loss, compromises wound healing, and generates discomfort (59). Therefore, pharmacologic induction of autophagy has been pursued extensively in the last decade for the treatment of age-associated pathologies, in particular neurodegenerative diseases. The TOR inhibitor rapamycin is a potent autophagy inducer (via inhibition of TORC1) that extends life span in a variety of organisms (Figure 3 and refs. 60–64) yet provokes severe side effects such as insulin resistance (65), glucose intolerance (66), testicular degeneration, and cataracts (63). Most of these side effects have been attributed to the chronic inhibition of TORC2 (67). Therefore, much effort is being invested in the discovery of TORC1-specific rapalogs, which should cause fewer side effects. Importantly, longevity extension by rapamycin in Drosophila is abrogated by deleting either 4E-BP1 or Atg5 (68). Therefore, the question arises whether (ATG5-dependent) autophagy and (4E-BP1–dependent) translation control different TORC1-mediated longevity pathways, or whether autophagy and translation converge on a common pathway that might affect proteostasis.
Figure 3. Autophagy inducers and functions of autophagy that prolong life span.

Several pharmacologic and dietary interventions activate autophagy signaling and thereby promote beneficial effects at the cellular and organismal levels, contributing to prolonged life span and health span.
The natural phenol resveratrol, an activator of deacetylases (in particular SIRT1), induces autophagy and extends the life span of yeast, flies, and nematodes (Figure 3 and refs. 18, 69). Although resveratrol does not promote longevity in mice fed standard chow, it can improve the health and increase the life span of mice fed a high-fat, Western-style diet (70, 71). Interestingly, administration of (polyphenol-rich) coffee to mice has also been shown to induce autophagy with a concomitant decrease in protein acetylation (72). Naturally occurring polyamines, which are inhibitors of acetyltransferases, also mediate beneficial longevity-extending effects. The polyamine spermidine is a potent acetyltransferase inhibitor and likely acts by inhibiting protein acetylation in cytoplasmic and nuclear proteins (Figure 3 and ref. 73). Thus, spermidine prolongs life span of yeast, flies, and nematodes in an autophagy-dependent fashion (32). Increased dietary polyamine intake also extends the life span of short-lived mouse strains and the health span of long-lived mouse strains (74). Genetic engineering of the gut microbiome to increase polyamine synthesis in the gut also prolongs the life span of mice (75). The brain concentration of endogenous spermidine positively correlates with memory capacity in Drosophila. Moreover, external supply of spermidine can avoid the memory impairment that usually is associated with aging in flies. The positive effect of spermidine on cognitive decline is abolished by knockout of Atg7, which indicates that these effects are autophagy dependent (76). Taken together, these results suggest that agents that favor protein deacetylation either by stimulating deacetylases (such as resveratrol) or by inhibiting acetyltransferases (such as spermidine) may extend longevity of model organisms. Whether this concept is broadly applicable remains to be explored. Moreover, the concept that deacetylase activators and acetyltransferase inhibitors can synergistically induce autophagy in cultured human cells and in mice (73) remains to be confirmed in experiments that address the anti-aging effects of such combination treatments.
Mechanisms that mediate the pro-survival effects of autophagy
Proteostasis.
The principal cytoprotective function of autophagy is believed to be related to its capacity to remove proteotoxic aggregates that accumulate with aging (77). Several age-related neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease, and ALS are linked to the accumulation of proteinaceous debris (78). Cellular programs that favor a euproteomic state, such as the protein-degrading ubiquitin-proteasome system or the ER-localized unfolded protein response, only eliminate relatively small protein oligomers. In contrast, autophagy is endowed with the capacity to remove larger protein aggregates (79). Prolonged rapamycin treatment retards the clinical manifestation of AD in mouse models (80), and various autophagy inducers such as trehalose, rapamycin, and spermidine slow disease progression in mouse models of ALS (81, 82). Moreover, maintenance of a juvenile level of autophagy postpones age-related memory impairment in Drosophila (76).
Metabolism.
Besides its canonical role in proteostasis, increasing evidence substantiates the involvement of autophagy in metabolism beyond a solely replenishing function. For example, it has been shown that lipid droplets, cellular organelles that regulate lipid homeostasis, can be degraded by the autophagic machinery by a process called lipophagy. Since autophagy levels have been shown to decrease with age, this impairment of bulk mobilization of lipid stores might contribute to age-related hepatic lipid accumulation and related pathologies, including hepatic steatosis (83). Of note, the lipophagic response to starvation in mice and C. elegans is transcriptionally activated by TFEB and its homolog HLH-30, respectively (84). Mice with liver-specific overexpression of TFEB were rescued from dietary-induced obesity and metabolic syndrome, but only when autophagy was functional (85). Interestingly, recent evidence from studies in D. melanogaster suggests that spermidine treatment alters organismal lipid profiles in an autophagy-dependent manner (86). It remains to be investigated whether a nonspecific change in lipid flux or selective modulation of some lipid species, e.g., by shifting saturation states (87), contributes to the beneficial effects of autophagy.
Vice versa, excessive lipid levels — in particular, excessive levels of free fatty acids — have been shown to inhibit autophagy by impeding the fusion of autophagosomes with lysosomes (88). Similarly, dietary lipids were reported to alter lysosomal lipid profiles and to thereby interfere with autophagic pathways (89). Accordingly, mice fed a high-fat diet depend on adiponectin-mediated activation of autophagy in muscle cells to overcome insulin resistance (89).
Importantly, lipid-driven disturbance of autophagy might be strictly tissue specific, as some studies have demonstrated elevated levels of autophagy in adipose tissue from obese patients (90, 91). In turn, the resulting mobilization of triglycerides might block lipophagy in hepatocytes and thus contribute to obesity-associated maladies in trans (83, 92). Furthermore, inhibition of adipocyte lipophagy provokes elevated pro-inflammatory cytokine levels, which strengthens the model of a compensatory autophagic response in adipose tissue (90). Therefore, tissue-specific (e.g., liver-specific) activation of autophagy might be a possible strategy to prolong health span in obesity-related disorders.
Whether similar autophagy-dependent mechanisms also influence other metabolic fluxes, either directly by targeting specific organelles or indirectly by balancing enzyme levels, remains to be investigated.
Programmed cell death.
Upon autophagy activation, Beclin-1 also mediates crosstalk to the apoptotic machinery by liberating the anti-apoptotic protein Bcl-2. This mechanism might ensure that cellular damage can be cleared by autophagy before it induces unwarranted cell death, especially in post-mitotic cells like neurons (93). As described above, the autophagic machinery can also detect and dispose damaged mitochondria that otherwise release pro-apoptotic factors and harmful reactive oxygen species, thereby offering cellular self-rescue (94). This mechanism might contribute to the commonly observed increase of autophagy in skeletal muscle myopathies. However, skeletal muscle–specific knockout of ATG5 or ATG7 in mice does not ameliorate but instead aggravates myopathy phenotypes (95, 96). Therefore, autophagy might act as a maintenance mechanism for muscle mass and counteract age-related muscle atrophy (97). Along these lines, exercise, which is generally believed to protect against a variety of age-related diseases, reportedly induces autophagy in skeletal muscle by a mechanism controlled by Bcl-2 (presumably through interaction with Beclin-1) (98).
Hormesis.
Hormesis is a commonly observed phenomenon in which exposure to low doses of stressors induces resistance to subsequent higher doses of the same stressor. Although the beneficial effects of hormesis are well known and already taken advantage of, e.g., in case of ischemic preconditioning, its molecular basis is still poorly described (99). Autophagy has been suggested to mediate this protective state following some forms of hormetic preconditioning, such as low-dose treatment with agents that liberate Bcl-2 from Beclin-1 or the apoptosis inducer zerumbone (100, 101). Thereby, autophagy might govern preemptive cellular detoxification that could also render the cell more resistant to age-related stresses such as protein aggregates or reactive oxygen species (99).
Inflammation.
At the organismal level, the age-related reduction in autophagy correlates with a variety of archetypal senescence-related symptoms including a general increase in smoldering inflammation that is referred to as “inflammaging” (102). Autophagy might facilitate the clearance of damaged (and potentially harmful) cells by maintaining effective efferocytosis as it reduces the propensity of leukocytes to produce pro-inflammatory cytokines (103–105).
Oncogenesis.
Autophagy defects have also been connected to cancer development, as this has been extensively documented in mice expressing reduced levels of autophagy-relevant genes such as those encoding Beclin-1 or its interactors UV radiation resistance–associated gene protein (UVRAG) and Bcl-2–modifying factor 1 (BMF1) (106). Whether a general age-related defect in autophagy may contribute to the increase in tumor incidence observed in the elderly remains to be explored.
Conclusions and outlook
Although there is little doubt that autophagy has potent anti-aging properties, it remains mostly unknown how such positive effects can be achieved in mechanistic terms. We speculate that three major functions may contribute to cytoprotection by autophagy: (a) buffering of cellular stress in conditions of fluctuating nutrient availability by enhanced provision of substrates for bioenergetic metabolism and anabolic reactions, (b) removal of dysfunctional and harmful organelles, including uncoupled mitochondria, and (c) clearance of aggregate-prone, potentially toxic proteins. On the organismal level, these functions may be expanded by the immunoregulatory properties of autophagy as well as by the cell-autonomous and the immunosurveillance-mediated suppression of oncogenesis (Figure 3).
An important objective for autophagy research in forthcoming years will be the identification of causal (instead of correlative) connections between autophagy and aging. We surmise that this goal will be facilitated by the identification of specific, highly potent pharmacologic activators or inhibitors of autophagy, as well as by the generation of sophisticated mouse models in which autophagy can be genetically switched on and off at will, in a spatially and temporarily controlled fashion. Future studies should also encompass epigenetic and posttranslational effects, as they might have a crucial impact on the regulation and functional outcome of autophagy (20, 21, 23). Additionally, it will be necessary to assess which potential autophagy inducers are effective and applicable to humans. As we advance along these lines, we may be able to conceive novel strategies for exploiting the broad health-improving and life span–extending effects of autophagy.
Acknowledgments
We thank Sabrina Schroeder for helpful discussion and manuscript preparation. The authors acknowledge support from BioTechMed-Graz and NAWI Graz. This work was supported by Austrian Science Fund FWF (grants LIPOTOX, I1000, P23490-B12, and P24381-B20 to F. Madeo). This work was also supported by grants to G. Kroemer from Ligue Contre le Cancer (Équipe Labelisée), Agence National de la Recherche (ANR), Association Pour la Recherche sur le Cancer (ARC) of the Cancéropôle Ile-de-France, AXA Chair for Longevity Research of the Institut National du Cancer (INCa), Fondation Bettencourt-Schueller, Fondation de France, Fondation pour la Recherche Médicale (FRM), European Commission (ArtForce), European Research Council (ERC), LabEx Immuno-Oncology, SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE), SIRIC Cancer Research and Personalized Medicine (CARPEM), and Paris Alliance of Cancer Research Institutes (PACRI).
Footnotes
Conflict of interest: Guido Kroemer receives income and research support from Bayer Healthcare. He also receives research support from GlaxoSmithKline.
Reference information:J Clin Invest. 2015;125(1):85–93. doi:10.1172/JCI73946.
References
- 1.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Novikoff AB, Beaufay H, de Duve C. Electron microscopy of lysosome-rich fractions from rat liver. J Biophys Biochem Cytol. 1956;2(4 suppl):179–184. [PMC free article] [PubMed] [Google Scholar]
- 3.Clark SL. Cellular differentiation in the kidneys of newborn mice studied with the electron microscope. J Biophys Biochem Cytol. 1957;3(3):349–362. doi: 10.1083/jcb.3.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 5.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and Aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 6.Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24(1):9–23. doi: 10.1038/cr.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morselli E, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1(1):e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackeh R, et al. Reactive oxygen species, AMP-activated protein kinase, and the transcription cofactor p300 regulate α-tubulin acetyltransferase-1 (αTAT-1/MEC-17)-dependent microtubule hyperacetylation during cell stress. J Biol Chem. 2014;289(17):11816–11828. doi: 10.1074/jbc.M113.507400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee IH, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105(9):3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korets SB, Czok S, Blank SV, Curtin JP, Schneider RJ. Targeting the mTOR/4E-BP pathway in endometrial cancer. Clin Cancer Res. 2011;17(24):7518–7528. doi: 10.1158/1078-0432.CCR-11-1664. [DOI] [PubMed] [Google Scholar]
- 11.Hamasaki M, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495(7441):389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 12.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12(8):747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizushima N, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116(9):1679–1688. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 15.Birgisdottir ÅB, Lamark T, Johansen T. The LIR motif – crucial for selective autophagy. J Cell Sci. 2013;126(15):3237–3247. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- 16.Chu CT, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15(10):1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howitz KT, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 19.Mercken EM, et al. SIRT1 but not its increased expression is essential for lifespan extension in caloric-restricted mice. Aging Cell. 2014;13(1):193–196. doi: 10.1111/acel.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mariño G, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014;53(5):710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Eisenberg T, et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 2014;19(3):431–444. doi: 10.1016/j.cmet.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng F, Tang BL. Sirtuins’ modulation of autophagy. J Cell Physiol. 2013;228(12):2262–2270. doi: 10.1002/jcp.24399. [DOI] [PubMed] [Google Scholar]
- 23.Füllgrabe J, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500(7463):468–471. doi: 10.1038/nature12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lam HC, et al. Histone deacetylase 6–mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123(12):5212–5230. doi: 10.1172/JCI69636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moresi V, et al. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc Natl Acad Sci U S A. 2012;109(5):1649–1654. doi: 10.1073/pnas.1121159109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warr MR, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494(7437):323–327. doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143(5):813–825. doi: 10.1016/j.cell.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorman JB, Albinder B, Shroyer T, Kenyon C. The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics. 1995;141(4):1399–1406. doi: 10.1093/genetics/141.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meléndez A, et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301(5638):1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 30.Dwivedi M, Song H-O, Ahnn J. Autophagy genes mediate the effect of calcineurin on life span in C. elegans. Autophagy. 2009;5(5):604–607. doi: 10.4161/auto.5.5.8157. [DOI] [PubMed] [Google Scholar]
- 31.Tavernarakis N, Pasparaki A, Tasdemir E, Maiuri MC, Kroemer G. The effects of p53 on whole organism longevity are mediated by. Autophagy. 2008;4(7):870–873. doi: 10.4161/auto.6730. [DOI] [PubMed] [Google Scholar]
- 32.Eisenberg T, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11(11):1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 33.Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3(6):597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- 34.Colman RJ, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mattison JA, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489(7415):318–321. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Cabo R, Carmona-Gutierrez D, Bernier M, Hall MN, Madeo F. The search for antiaging interventions: from elixirs to fasting regimens. Cell. 2014;157(7):1515–1526. doi: 10.1016/j.cell.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willcox BJ, Willcox DC. Caloric restriction, caloric restriction mimetics, and healthy aging in Okinawa: controversies and clinical implications. Curr Opin Clin Nutr Metab Care. 2014;17(1):51–58. doi: 10.1097/MCO.0000000000000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levine ME, et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014;19(3):407–417. doi: 10.1016/j.cmet.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruckenstuhl C, et al. Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS Genet. 2014;10(5):e1004347. doi: 10.1371/journal.pgen.1004347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee E, et al. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy. 2014;10(4):572–587. doi: 10.4161/auto.27649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Juhász G, Érdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21(23):3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tóth ML, et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy. 2008;4(3):330–338. doi: 10.4161/auto.5618. [DOI] [PubMed] [Google Scholar]
- 44.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4(2):176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y, et al. Impaired autophagic function in rat islets with aging. Age (Dordr). 2013;35(5):1531–1544. doi: 10.1007/s11357-012-9456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murthy A, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506(7489):456–462. doi: 10.1038/nature13044. [DOI] [PubMed] [Google Scholar]
- 47.Joosten LAB, Netea MG, Dinarello CA. Interleukin-1β in innate inflammation, autophagy and immunity. Semin Immunol. 2013;25(6):416–424. doi: 10.1016/j.smim.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 48.Tasdemir E, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferrari R, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014;13(7):686–699. doi: 10.1016/S1474-4422(14)70065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qin Z, et al. Potentially functional polymorphisms in ATG10 are associated with risk of breast cancer in a Chinese population. Gene. 2013;527(2):491–495. doi: 10.1016/j.gene.2013.06.067. [DOI] [PubMed] [Google Scholar]
- 51.Nicoli ER, et al. Determination of autophagy gene ATG16L1 polymorphism in human colorectal cancer. Rom J Morphol Embryol. 2014;55(1):57–62. [PubMed] [Google Scholar]
- 52.Wada Y, Sun-Wada G-H, Kawamura N, Aoyama M. Role of autophagy in embryogenesis. Curr Opin Genet Dev. 2014;27:60–66. doi: 10.1016/j.gde.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 53.Adler MI, Bonduriansky R. Why do the well-fed appear to die young? BioEssays. 2014;36(5):439–450. doi: 10.1002/bies.201300165. [DOI] [PubMed] [Google Scholar]
- 54.Pyo J-O, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lapierre LR, et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun. 2013;4:2267. doi: 10.1038/ncomms3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mai S, Muster B, Bereiter-Hahn J, Jendrach M. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy. 2012;8(1):47–62. doi: 10.4161/auto.8.1.18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng S, et al. Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet. 2010;6(2):e1000838. doi: 10.1371/journal.pgen.1000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cortes CJ, La Spada AR. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov Today. 2014;19(7):963–971. doi: 10.1016/j.drudis.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dirks AJ, Leeuwenburgh C. Caloric restriction in humans: Potential pitfalls and health concerns. Mech Ageing Dev. 2006;127(1):1–7. doi: 10.1016/j.mad.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 60.Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19(3):373–379. doi: 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robida-Stubbs S, et al. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15(5):713–724. doi: 10.1016/j.cmet.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilkinson JE, et al. Rapamycin slows aging in mice: Rapamycin slows aging in mice. Aging Cell. 2012;11(4):675–682. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neff F, et al. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013;123(8):3272–3291. doi: 10.1172/JCI67674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Houde VP, et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59(6):1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang G-R, et al. Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol Toxicol. 2009;105(3):188–198. doi: 10.1111/j.1742-7843.2009.00427.x. [DOI] [PubMed] [Google Scholar]
- 67.Kennedy BK, Pennypacker JK. Drugs that modulate aging: the promising yet difficult path ahead. Transl Res. 2014;163(5):456–465. doi: 10.1016/j.trsl.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bjedov I, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11(1):35–46. doi: 10.1016/j.cmet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wood JG, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430(7000):686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 70.Pearson KJ, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8(2):157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baur JA, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444(7117):337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pietrocola F, et al. Coffee induces autophagy in vivo. Cell Cycle Georget Tex. 2014;13(12):1987–1994. doi: 10.4161/cc.28929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morselli E, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol. 2011;192(4):615–629. doi: 10.1083/jcb.201008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Soda K, Dobashi Y, Kano Y, Tsujinaka S, Konishi F. Polyamine-rich food decreases age-associated pathology and mortality in aged mice. Exp Gerontol. 2009;44(11):727–732. doi: 10.1016/j.exger.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 75.Kibe R, et al. Upregulation of colonic luminal polyamines produced by intestinal microbiota delays senescence in mice. Sci Rep. 2014;4:4548. doi: 10.1038/srep04548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gupta VK, et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat Neurosci. 2013;16(10):1453–1460. doi: 10.1038/nn.3512. [DOI] [PubMed] [Google Scholar]
- 77.Tanaka K, Matsuda N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim Biophys Acta. 2014;1843(1):197–204. doi: 10.1016/j.bbamcr.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 78.Son JH, Shim JH, Kim K-H, Ha J-Y, Han JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med. 2012;44(2):89–98. doi: 10.3858/emm.2012.44.2.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schreiber A, Peter M. Substrate recognition in selective autophagy and the ubiquitin–proteasome system. Biochim Biophys Acta. 2014;1843(1):163–181. doi: 10.1016/j.bbamcr.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 80.Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6(9):e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang X, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2014;10(4):22–36. doi: 10.4161/auto.27710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang I-F, Tsai K-J, Shen C-KJ. Autophagy activation ameliorates neuronal pathogenesis of FTLD-U mice: A new light for treatment of TARDBP/TDP-43 proteinopathies. Autophagy. 2013;9(2):239–240. doi: 10.4161/auto.22526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O’Rourke EJ, Ruvkun G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat Cell Biol. 2013;15(6):668–676. doi: 10.1038/ncb2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Settembre C, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15(6):647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Minois N, Rockenfeller P, Smith TK, Carmona-Gutierrez D. Spermidine feeding decreases age-related locomotor activity loss and induces changes in lipid composition. PLoS One. 2014;9(7):e102435. doi: 10.1371/journal.pone.0102435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mei S, et al. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339(2):487–498. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24(8):3052–3065. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rodriguez-Navarro JA, et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2012;109(12):E705–E714. doi: 10.1073/pnas.1113036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jansen HJ, et al. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology. 2012;153(12):5866–5874. doi: 10.1210/en.2012-1625. [DOI] [PubMed] [Google Scholar]
- 91.Cummins TD, et al. Metabolic remodeling of white adipose tissue in obesity. Am J Physiol Endocrinol Metab. 2014;307(3):E262–E277. doi: 10.1152/ajpendo.00271.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Christian P, Sacco J, Adeli K. Autophagy: Emerging roles in lipid homeostasis and metabolic control. Biochim Biophys Acta. 2013;1831(4):819–824. doi: 10.1016/j.bbalip.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 93.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 94.Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res. 2012;111(9):1198–1207. doi: 10.1161/CIRCRESAHA.112.268946. [DOI] [PubMed] [Google Scholar]
- 95.Raben N, et al. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. 2008;17(24):3897–3908. doi: 10.1093/hmg/ddn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Masiero E, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10(6):507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 97.Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584(7):1411–1416. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- 98.He C, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481(7382):511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zimmermann A, Bauer MA, Kroemer G, Madeo F, Carmona-Gutierrez D. When less is more: hormesis against stress and disease. Microb Cell. 2014;1(5):150–153. doi: 10.15698/mic2014.05.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Malik SA, et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene. 2011;30(37):3918–3929. doi: 10.1038/onc.2011.104. [DOI] [PubMed] [Google Scholar]
- 101.Ohnishi K, Nakahata E, Irie K, Murakami A. Zerumbone, an electrophilic sesquiterpene, induces cellular proteo-stress leading to activation of ubiquitin-proteasome system and autophagy. Biochem Biophys Res Commun. 2013;430(2):616–622. doi: 10.1016/j.bbrc.2012.11.104. [DOI] [PubMed] [Google Scholar]
- 102.Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging. 2012;4(3):166–175. doi: 10.18632/aging.100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu B, Zhang B, Guo R, Li S, Xu Y. Enhancement in efferocytosis of oxidized low-density lipoprotein-induced apoptotic RAW264.7 cells through Sirt1-mediated autophagy. Int J Mol Med. 2014;33(3):523–533. doi: 10.3892/ijmm.2013.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Teplova I, et al. ATG proteins mediate efferocytosis and suppress inflammation in mammary involution. Autophagy. 2013;9(4):459–475. doi: 10.4161/auto.23164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity. 2013;39(2):211–227. doi: 10.1016/j.immuni.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 106.Edinger AL, Thompson CB. Defective autophagy leads to cancer. Cancer Cell. 2003;4(6):422–424. doi: 10.1016/S1535-6108(03)00306-4. [DOI] [PubMed] [Google Scholar]