

Abstract
Cardiovascular disease is the leading cause of death worldwide. As such, there is great interest in identifying novel mechanisms that govern the cardiovascular response to disease-related stress. First described in failing hearts, autophagy within the cardiovascular system has been widely characterized in cardiomyocytes, cardiac fibroblasts, endothelial cells, vascular smooth muscle cells, and macrophages. In all cases, a window of optimal autophagic activity appears to be critical to the maintenance of cardiovascular homeostasis and function; excessive or insufficient levels of autophagic flux can each contribute to heart disease pathogenesis. In this Review, we discuss the potential for targeting autophagy therapeutically and our vision for where this exciting biology may lead in the future.
Introduction
Cardiovascular disease is the leading cause of mortality throughout the developed world (1). Research identifying novel risk factors, innovative diagnostic modalities, and therapeutic interventions, coupled with education of clinicians and patients, have led to dramatic improvements in age-adjusted cardiovascular death rates (2). Nevertheless, changes in lifestyle-related factors and the associated worldwide obesity epidemic have fostered continued growth in the prevalence of cardiovascular disease. As such, despite robust successes in recent decades, much work remains to identify novel targets of therapeutic intervention.
Macroautophagy is an intracellular process that mediates protein degradation, organelle turnover, and recycling of cytoplasmic components during nutrient starvation or cellular stress. The process commences with formation of the autophagosome, a double-membrane structure of reticular origin that sequesters cytoplasmic components and ultimately fuses with a lysosome, where engulfed cargo is degraded by lysosome-derived acid hydrolases. Materials degraded within the autolysosome are recruited to anabolic reactions to sustain energy levels and to provide macromolecules for synthesis of higher-order structures (e.g., nucleic acids, proteins, and organelles). Thus, macroautophagy helps cells adapt to changing nutritional and energy demands by repurposing existing cellular components to sustain cellular metabolism, homeostasis, and survival (3).
Two other types of autophagy have been described. Microautophagy denotes direct engulfment of cytoplasmic material by lysosomes via inward invaginations of the lysosomal membrane. Chaperone-mediated autophagy involves a chaperone complex and lysosomal-associated membrane protein (LAMP) type 2A to degrade cytosolic proteins harboring a specific targeting motif. Whereas we recently reported that ryanodine receptor 2 can be specifically degraded by chaperone-mediated autophagy (4), our focus here is on the much more extensively characterized macroautophagy (hereafter termed autophagy).
Despite the key beneficial role of autophagy, excessive or insufficient autophagic activity can each contribute to cell death. Indeed, diverse studies have shown that autophagic flux contributes to the pathogenesis of cardiovascular diseases, diabetes, inflammatory disorders, infection, and cancer (5). Indeed, the term “autophagic cell death” was coined to denote morphologic changes observed by electron microscopy, in which a dying cell is characterized by abundant autophagic vacuoles in the cytoplasm (6). However, despite considerable evidence linking autophagic activity to heart failure (HF) progression, uncertainty remains regarding whether increased autophagy is an epiphenomenon or a causative factor in the dying cardiomyocyte.
Autophagic activity in all cardiovascular cell types
Autophagic activity has been reported in each of the diverse tissues and cell types that constitute the circulatory system. Abundant evidence indicates that this response participates in a wide range of cellular responses to both physiologic and disease-related events (Figure 1).
Figure 1. Autophagy in the cardiovascular system.
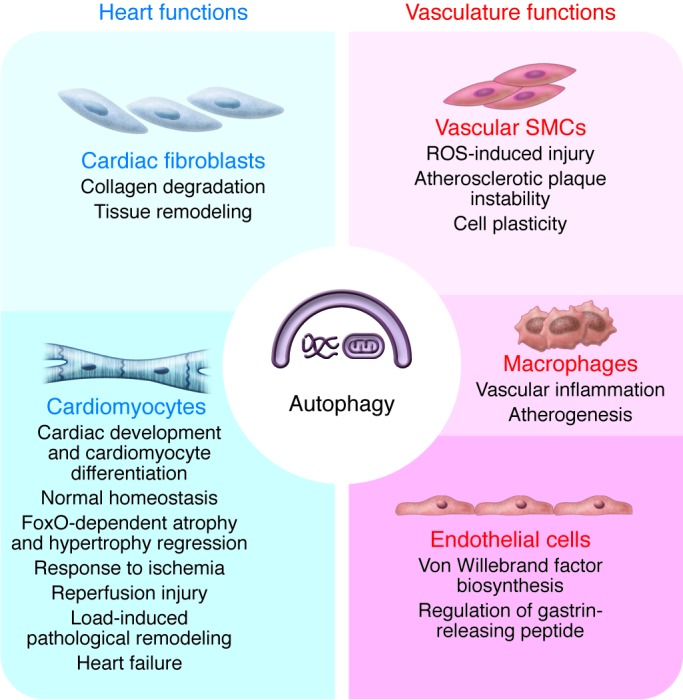
Autophagic activity occurs in all cell types within the cardiovascular system. This activity contributes to a wide range of cellular events in normal physiology, growth, and development and in disease-related pathophysiology.
Mammalian target of rapamycin (mTOR) is a key regulator of autophagic activity. In a generalized model, under nutrient-rich, proliferative conditions, a PI3K/Akt/mTOR signaling cascade inhibits autophagy. Many experimental scenarios employ starvation, or the mTOR inhibitor rapamycin, to activate autophagy. Changes in autophagic activity may be assessed ultrastructurally using microsopy, by monitoring the conversion of microtubule-associated protein 1 light chain 3 (LC3) to the active LC3-II form and by assessing changes in the abundance of autophagy-related proteins or degradation targets.
Autophagy in the vascular system
Endothelial cells.
Autophagy can be regulated in vascular endothelial cells by compounds circulating in the bloodstream or localized within the subendothelial layer of atherosclerotic plaque. For example, in cultured HUVECs, vitamin D increases beclin 1, a key component in the initiation of autophagy (7); oxidized LDL (oxLDL), advanced glycation end-products (AGEs), and C6-ceramide each promote the formation of autophagosomes (8–10). Clinorotation, a simulated model of microgravity, increases beclin 1 and promotes conversion of LC3-I to LC3-II in HUVECs (11). Endostatin, a potent inhibitor of neovascularization and tumor growth, triggers autophagic cell death in the human endothelial cell line EA.hy926 (12). The green tea polyphenol epigallocatechin gallate promotes formation of LC3-II and autophagosomes in primary bovine aortic endothelial cells (13). Moreover, mice harboring endothelial cell–specific silencing of autophagy-related gene 7 (Atg7) manifest normal vessel architecture and capillary density, yet exhibit impaired synthesis and release of von Willebrand factor (vWF), resulting in prolonged bleeding times (14). This suggests that, in this context, autophagy is not required for vessel development, but rather plays an important role in regulating the processing, maturation, and secretion of vWF. Interestingly, gastrin-releasing peptide (GRP), an inducer of tubule formation, decreases expression of proautophagic factors, including ATG5, beclin 1, and LC3, and can even attenuate rapamycin-induced autophagosome formation. Overexpression of ATG5 or beclin 1 significantly decreases GRP-induced tubule formation, whereas siRNA targeting of Atg5 or beclin 1 results in increased tubule formation in response to GRP (15). Taken together, these findings indicate that while autophagy may not be required for developmental patterning of vascular endothelial cells, its proper regulation is critical during fundamental adaptive responses such as secretion, cell proliferation, and tubule formation.
Vascular smooth muscle cells.
Electron microscopic studies from the early 1960s provided the first evidence of autophagic activity in vascular smooth muscle cells (VSMCs) (16). Structures consistent with autophagosomes were reported in VSMCs within atherosclerotic lesions (16) and atheromas (17) in humans and animal models. Subsequent studies in animal models of cardiovascular disease, using transgenic animals overexpressing labeled autophagic proteins (e.g., GFP-LC3) to monitor autophagic activity, similarly reported evidence of autophagy in VSMCs (18).
Macrophages.
Macrophage-localized autophagy has been reported in association with vascular disease (19, 20). During lesion formation, autophagic markers (sequestosome 1 [SQSTM1, also known as p62], and LC3) were detected in atherosclerotic plaques of apoE–/– mice, colocalizing primarily with monocytes/macrophages and leukocytes. In atherosclerotic aorta, p62 levels increased with increasing age and/or plaque burden, suggesting that autophagic flux may ultimately become compromised during disease progression, leading to accumulation of this linker protein. Although beclin 1 heterozygous–deficient mice on an apoE–/– background manifest degrees of atherosclerosis similar to those seen in apoE–/– mice wild type for beclin 1, complete abrogation of macrophage autophagy increases vascular inflammation and plaque formation (20).
Liao et al. (19) demonstrated that several proatherosclerotic stimuli induced autophagy in macrophages and that Atg5-deficient macrophages displayed enhanced apoptosis as well as increased NADPH oxidase–mediated oxidative stress. Macrophages in aortic root lesions of Ldlr–/– mice fed a high-fat diet (HFD) displayed a punctate pattern of GFP-LC3 transgene fluorescence indicative of autophagic activity, with the number of p62-positive macrophages increasing as lesions progressed. Suppression of macrophage-localized autophagy in Ldlr–/– mice increased apoptosis and oxidative stress in plaque macrophages, promoted plaque necrosis, and impaired lesional efferocytosis. Moreover, autophagy facilitated the mobilization of lipid droplet–associated cholesterol for reverse cholesterol transport in macrophage-derived foam cells (21). These data lend support for a protective role of macrophage autophagy in atherosclerosis.
Autophagy in the myocardium
Cardiac fibroblasts and myofibroblasts.
Cardiac fibroblasts synthesize collagens, fibronectins, and other interstitial elements to maintain the integrity of the cardiac extracellular matrix (22). In cultured adult cardiac fibroblasts, induction of autophagy by either serum withdrawal or β2-adrenergic stimulation correlates with enhanced degradation of collagen type I (23). Autophagy can eliminate misfolded, trimeric forms of type I pro-collagen that accumulate as aggregates in the endoplasmic reticulum of Hsp47–/– fibroblasts (24). These data suggest that autophagy can affect cardiac remodeling in part via its role in intracellular collagen degradation. Indeed, kidneys from mice deficient in beclin 1 exhibit a profibrotic phenotype with increased collagen deposition (25). However, no studies as yet have examined cardiac fibrosis in animals with a fibroblast-specific autophagy deficiency.
Cardiomyocytes.
Autophagy is required for normal cardiac development (ref. 26 and Figure 2). Using zebrafish expressing a GFP-LC3 reporter, autophagic activity was observed in multiple tissues during embryonic development, including the heart (27). Morpholino knockdown of essential autophagy genes resulted in increased cell death, reduced survival, and defects in morphogenesis. Abnormal cardiac development included defects in cardiac looping, abnormal chamber and valve morphologies, and ectopic expression of critical transcription factors (27). Similarly, Atg5-deficient mice displayed abnormal Tbx2 expression and defects in valve development and chamber septation (27). Thus, autophagy plays an essential and highly conserved role in cardiac morphogenesis during vertebrate development (27).
Figure 2. Autophagy in the heart.
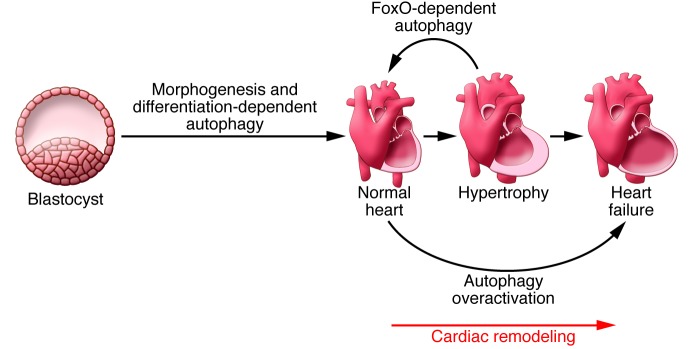
Autophagy is required for normal development of cardiac tissue and for cardiomyocyte terminal differentiation. Autophagy is also required for normal cardiac plasticity. Indeed, in the heart under normal conditions, autophagy plays a key role in regulating cardiomyocyte size as well as global cardiac structure and function. In the setting of disease, overactivation of autophagic flux can contribute to a transition to heart failure (HF).
Autophagy is also required for terminal cardiomyocyte differentiation. Autophagy was increased in cardiac progenitor cells deprived of FGF (28); induction of autophagy was followed by enhanced cardiomyocyte differentiation, suggesting that FGF prevents premature differentiation of cardiac progenitor cells by suppressing autophagic activity. Disruption of this process may contribute to congenital heart defects.
Autophagy in cardiovascular disease
Autophagy is essential for normal maintenance, repair, and adaptation of the heart over the course of a lifetime. Evidence of autophagy in human heart disease was first reported in tissue samples from patients with dilated cardiomyopathy (29). Analysis of hearts from patients with end-stage HF indicated that cardiomyocytes died by a variety of mechanisms including necrosis, apoptosis, and autophagy, with autophagy reported as the most prominent (30).
Autophagy during ischemia/reperfusion.
During ischemia, nutrient and oxygen supplies to the myocardium are limited, a state reminiscent of starvation. In this context autophagy can be adaptive, meeting cellular metabolic needs and eliminating damaged mitochondria, which could otherwise release damaging ROS and initiate apoptosis (31). Consistent with this, pharmacologic inhibition of autophagy in ischemia-mimicking conditions increases cardiomyocyte death, suggesting that autophagy functions as a pro-survival mechanism (32). During mild ischemic stress, activation of autophagy depends on AMPK-mediated inhibition of mTOR and is cardioprotective (33, 34). In the setting of chronic ischemia, autophagic activity can inhibit apoptosis and mitigate tissue damage (35).
In most instances, an infarct-related artery recanalizes, either spontaneously or by therapeutic intervention. When oxygen and nutrients are restored to the previously ischemic tissue, cardiomyocyte autophagy is upregulated dramatically in vivo (e.g., in rat [ref. 36], rabbit [ref. 37], and swine [ref. 35]), and in cells maintained in culture (e.g., in HL-1 cells [ref. 38] and primary neonatal cardiomyocytes [refs. 32, 33]). However, detailed time-course analyses indicate that reperfusion-triggered autophagy is transient such that autophagic flux eventually declines below baseline levels (39).
Reperfusion elicits a response that differs vastly from ischemia. Studies suggest that cardiac autophagy in response to reperfusion can be either adaptive or detrimental and involves beclin 1 activation independent of the AMPK/mTOR pathway (33). Indeed, whether changes in autophagic activity elicited by reperfusion are adaptive or maladaptive is the subject of debate, with experimental evidence supporting either point of view. In cultured neonatal cardiomyocytes exposed to simulated ischemia/reperfusion (I/R), chemical suppression of autophagy with 3-methyladenine enhances cell viability (32). In contrast, other studies report that autophagy is protective in simulated I/R (40, 41). Furthermore, short repetitive ischemic episodes, which elicit beneficial preconditioning effects, also induce autophagy, and when autophagy is suppressed, the protective effects of preconditioning are lost (35, 41). In HL-1 cells, I/R impairs autophagic flux at the level of both induction and processing, while enhancing autophagy protects against I/R injury (38). Moreover, autophagosomes have been detected in surviving cardiomyocytes in chronic stages of myocardial infarction, suggesting that autophagy may aid in survival. Exposure to the autophagy inhibitor bafilomycin A1 exacerbates cardiac dysfunction and remodeling (42). At present, the extent to which these discrepancies derive from differing cell types, model systems, or experimental paradigms is unclear.
Novel responses by beclin 1 and LAMP2 in I/R injury were recently reported (43). While beclin 1 is essential for initiation of autophagy, it can also inhibit vesicle processing late in the autophagic cascade. Cardiomyocyte autophagy was upregulated in response to I/R injury, but autophagosome clearance was defective, leading to cell death. Reperfusion increased beclin 1 levels, causing impairment of autophagosome processing and reduced levels of LAMP2, a protein critical for autophagosome-lysosome fusion. Partial beclin 1 knockdown restored autophagic processing and protected from I/R injury–induced cell death, whereas complete beclin 1 knockdown impaired autophagosome formation and increased cell death (43). Thus, beclin 1 abundance can be an important determinant of autophagic activity, either ensuring survival or triggering cell death (44). Although it is well documented that haploinsufficiency of beclin 1 protects the heart in transverse aortic constriction–induced (TAC-induced) hypertrophy (45) or I/R injury (33, 43), the actions of beclin 1 at both early and late points of the autophagic cascade complicate the interpretation of these studies.
With regard to human disease, analysis of tissue samples from patients with ischemic heart disease is typically confounded by co-existing HF (29). Autophagy was described in human right atrial appendages collected before cardioplegic arrest and again after reperfusion. Perioperative I/R upregulated 11 ATGs and downregulated three (of 84 examined). Elevated autophagic activity was also confirmed through observations of increased LC3-I levels and LC3-II/LC3-I ratios (46).
Autophagy in cardiac hypertrophy.
Cardiac hypertrophy is a major risk factor for adverse cardiovascular events (47). Initial phases of hypertrophic growth of the myocardium are thought to be adaptive, serving to normalize ventricular wall stress and oxygen demand; however, this concept has never been tested. Cardiac hypertrophy involves increases in the size of individual cardiomyocytes. When it occurs in response to physiologic cues such as exercise or pregnancy, it is not associated with HF. By contrast, hypertrophy elicited by disease-related stresses such as hypertension, obesity, valvular disease, or infarction is pathological and correlates with abnormal metabolic, structural, and functional alterations, including changes in metabolic substrate utilization, disorganization of the sarcomere, alterations in Ca2+ handling, changes in contractility, cardiomyocyte loss, systolic and/or diastolic dysfunction, and electrical remodeling (48).
Remodeling of any tissue involves alterations in the steady-state equilibrium between protein synthesis and protein degradation. A number of studies have focused on the relationship between catabolic pathways and myocardial remodeling, with autophagy being one of the most extensively studied. The first report of autophagy in cardiac hypertrophy dates from 1983, when reduced numbers of autophagic vacuoles were noted in ventricular hypertrophy stemming from supravalvular aortic constriction, suggesting that the degradation of cytoplasmic components was inhibited (49). Cardiomyocyte-specific conditional deletion of ATG5 uncovered a key role for autophagy in the maintenance of cardiac structure and function both in the basal state and under hemodynamic stress (50). In TAC-induced pressure overload, autophagic activity increased as early as 24 hours after surgery, and load-induced pathologic remodeling was blunted in beclin 1 haploinsufficient mice (45). Conversely, cardiomyocyte-restricted overexpression of beclin 1 substantially amplified adverse remodeling (45). Subsequent studies identified protein aggregation is a proximal trigger of load-induced cardiomyocyte autophagy (51).
As in the case of I/R, cardiomyocyte autophagy triggered by increased afterload appears to have both adaptive and maladaptive features. This dual nature of autophagy is a recurring theme in other organ systems and disease states (52). Indeed, we have postulated that the physiologic impact of autophagy is a continuum, and a window of optimal autophagic activation is critical to the maintenance of cellular homeostasis and function (53).
A shift in the balance between the oxidized and reduced forms of nicotinamide adenine dinucleotide (NAD+ and NADH) has been shown to influence autophagic flux. The prevention of downregulation of nicotinamide phosphoribosyltransferase, a rate-limiting enzyme in the mammalian NAD+ salvage pathway (54), protects against myocardial injury by increasing NAD+ and ATP levels, inhibits apoptosis, and stimulates autophagic flux (55). NAD+ acts in part by activating sirtuin 1 (Sirt1), which then induces autophagy through nuclear localization of FoxO1 (56). In Sirt1-knockout mice, beclin 1 levels are diminished, consistent with the involvement of Sirt1 in regulation of autophagy (57). In addition, exogenous NAD+ can block phenylephrine- and angiotensin II–induced cardiac hypertrophy through activation of the Sirt3/AMPK pathway (58). Thus, cellular events that govern NAD+ levels may emerge as therapeutic targets in cardiac remodeling.
In a hypertensive double-transgenic rat model harboring copies of both the human renin and angiotensinogen genes (dTGRs), caloric restriction decreased mortality, cardiomyocyte hypertrophy, vascular inflammation, cardiac damage, cardiac fibrosis, cardiomyocyte apoptosis, and transcript levels of atrial natriuretic peptide (59). These effects were independent of changes in blood pressure and were linked to increased autophagy (59).
The number of cellular processes regulated by microRNAs (miRNAs) has grown to include critical elements of cardiac biology, including cell size regulation, survival, action potentials, mitochondrial function, and energetics (60). Recently, Ucar et al. showed that the miRNA-212/132 family regulates both cardiac hypertrophy and autophagy by targeting FoxO3 (61), adding to the growing body of evidence for a crucial role of autophagy in the control of hypertrophy. Furthermore, because autophagy is controlled by a variety of tractable mechanisms, including histone deacetylases (HDACs), sirtuins, and miRNAs, autophagy has emerged as a promising “druggable” target in hypertrophic heart disease.
Autophagy in HF.
The initial response of the heart to increases in afterload is hypertrophic growth (48). If the afterload stress persists, the heart becomes dilated, contractile function declines, and HF ensues (62). Indeed, this disease progression commonly occurs in patients with hypertension or ischemic heart disease (ref. 63 and Figure 2).
In a model of pressure overload, we found that the degree of autophagic activity correlated with the magnitude of hypertrophic growth and the rate of transition to HF (45). Consistent with these findings, transgenic mice with cardiomyocyte-restricted overexpression of beclin 1 amplified the pathologic remodeling response (45, 64). Conversely, beclin 1 haploinsufficiency halved the stress-induced autophagic response and partially rescued the HF phenotype (45). Collectively, these data suggest that autophagy can be maladaptive under conditions of severe pressure overload, a common clinical scenario.
It is somewhat counterintuitive that autophagy, a mechanism of protein degradation, should facilitate hypertrophic growth. However, the natural history of HF involves substantial remodeling of ventricular morphology from concentric hypertrophy to eccentric hypertrophy, a structural change that would necessitate catabolism and recycling of existing cellular structures. Because of this, autophagy is a potential target for therapeutic intervention in afterload-induced HF. To test this, we recently employed small-molecule inhibitors of HDACs, which have previously been shown to suppress pathologic cardiac remodeling (65). We hypothesized that maladaptive autophagy is HDAC dependent and that the beneficial effects of HDAC inhibitors occur due to their ability to suppress autophagy. Consistent with this model, we found that HDAC inhibition was, in fact, capable of profoundly suppressing load-induced cardiomyocyte autophagy and thereby blunted the pathologic growth response (64).
As discussed earlier, although excessive cardiomyocyte autophagy can be maladaptive, complete abrogation of autophagy is similarly maladaptive and can accelerate the progression to HF. For example, inactivation of Atg5 in adult heart is sufficient to trigger rapid-onset HF (50). Similarly, systemic inactivation of the gene encoding the lysosomal enzyme cysteine endopeptidase cathepsin L (Ctsl) leads to the accumulation of large dysmorphic vesicles in the cardiomyocyte cytoplasm and dilated cardiomyopathy (66), consistent with the notion that basal levels of cardiomyocyte autophagy are required for cellular proteostasis. Given this, we favor a model in which titration of cardiomyocyte autophagy within an optimal, adaptive zone is a therapeutic goal of interest (53). Importantly, HDAC inhibition suppresses, but does not eliminate, the autophagic response to stress and hence is an attractive strategy worthy of additional investigation (39, 64).
Analysis of human samples has afforded additional evidence that autophagic cell death contributes to HF pathogenesis (30, 67). In patients with isolated aortic valve stenosis and varying degrees of left ventricular systolic dysfunction, cell loss, mainly due to autophagy and oncosis, was associated with the progression of left ventricular systolic dysfunction (5). More recently, analyses of biopsy samples of left ventricular myocardium from nine patients with idiopathic dilated cardiomyopathy obtained at the time of both implantation and explantation of a left ventricular assist device showed that mechanical unloading of the failing human heart was associated with decreased markers of autophagy (68). It is not known whether mechanical unloading restores autophagy to basal levels or establishes a new set point. Furthermore, as these were static endpoint studies, it is possible that initial increases in autophagic activity accompanied unloading to facilitate atrophic remodeling.
Mitophagy.
Mitophagy is the selective sequestration of mitochondria by autophagosomes and their subsequent delivery to lysosomes for degradation. This process is important for myocardial homeostasis and stress adaptation. Destruction of damaged mitochondria by mitophagy induces cardiomyocyte preconditioning and reduces cell death during I/R (69, 70). Oka et al. showed that impaired mitophagy leads to TLR9-mediated inflammatory responses in cardiomyocytes and induces myocarditis and dilated cardiomyopathy (71). Hoshino et al. showed that cytosolic p53 impairs autophagic degradation of damaged mitochondria, triggering mitochondrial dysfunction and HF in mice (72).
Autophagy in other cardiomyopathies
Protein aggregation–related cardiomyopathy.
Defective autophagy contributes to disease progression in certain genetic forms of cardiomyopathy and skeletal myopathy linked to protein aggregation (73) and muscle wasting (74). Autophagic protection of cardiomyocytes from proteotoxic stress has also been demonstrated in experimental mouse models of proteinopathy (75–77). Autophagic activity was elevated in a model of proteotoxicity, and the blunting of autophagy accelerated progression to HF (75). Conversely, increasing autophagic flux by elevating ATG7 levels decreased pathology and prolonged survival (78).
Glycogen storage disease–related cardiomyopathy.
Glycogen storage disease can present as hypertrophic cardiomyopathy (73, 79, 80). This is particularly the case for Danon disease, a condition characterized by defective autophagosome-lysosome fusion owing to a mutation in LAMP2. In a mouse model of Pompe disease, a disorder marked by defective metabolism of glycogen due to insufficiency of lysosomal acid α-glucosidase, the suppression of autophagy through ATG7 inactivation facilitated successful enzyme replacement therapy (81). A late-onset, LAMP2-positive dilated cardiomyopathy resembling Danon disease has also been reported, characterized by increased autophagic vacuoles, but not caused by a mutation in LAMP2 (82).
Diabetic cardiomyopathy.
Cardiac autophagic flux was inhibited in a model of type 1 diabetes (T1D) (83), and T1D-induced cardiac damage was substantially attenuated in beclin 1– and Atg16-deficient mice. In contrast, cardiac damage was exacerbated in a dose-dependent manner by beclin 1 overexpression in the same model of T1D. Taken together, these findings suggest that decreased autophagy is an adaptive response in T1D that helps to limit cardiac dysfunction (83). T1D is an autoimmune disease characterized by the destruction of pancreatic β cells by autoreactive T cells. Some evidence suggests that autophagy favors this autoimmune process (84). Therefore, decreased autophagic activity in the pancreas might also protect β cells, limiting T1D progression and subsequent cardiac damage.
Autophagy has been implicated in the development of insulin resistance and T2D. Genetic ablation of Atg7 in pancreatic β cells resulted in degeneration of islets, impaired glucose tolerance, and reduced insulin secretion (85, 86). Moreover, cardiomyocytes isolated from T2D db/db mice and HFD-induced obese mice exhibited reduced autophagic activity (87, 88). However, some recent reports conflict with these findings. Mellor et al. reported that increased myocardial autophagic flux in fructose diet–induced T2D mice resulted in pathologic remodeling of the heart (89). Upregulation of autophagy was also found in human T2D pancreatic β cells (90). These results suggest that in T2D, increased autophagy may serve as a compensatory response to insulin resistance by providing cellular components essential for maintaining normal cellular architecture and function.
Atrial fibrillation.
Postoperative atrial fibrillation (POAF) is a common surgical complication. Electron micrographs of atrial tissue from patients with POAF revealed significant accumulation of autophagic vesicles and lipofuscin deposits. Total protein ubiquitination was similar in patients with or without POAF, but LC3B processing was markedly reduced in those with POAF, suggesting that selective impairment of autophagic flux contributes to atrial remodeling in patients developing POAF (91).
Aging.
Aging is often accompanied by geometric and functional changes in the heart. Aging induces cardiac hypertrophy, fibrosis, and decreased cardiac contractility. Levels of beclin 1 and ATG5 and LC3-II/LC3-I ratios are decreased in aged hearts, and levels of p62 are increased, which suggests an impairment of autophagy (92). Rapamycin reduces aging-induced cardiomyocyte contractile and intracellular Ca2+ dysfunction (92).
Anticancer drug–induced cardiomyopathy.
Cancer chemotherapy, particularly with anthracyclines, has long been associated with significant cardiotoxicity (93). The majority of human studies suggest that doxorubicin increases autophagy and likely contributes to cellular dysfunction and apoptosis (94). Notably, many preclinical studies are based on models of high-dose doxorubicin exposure, which triggers lethargy, anorexia, and weight loss, thereby confounding the interpretation of cardiomyocyte autophagy. In some instances, autophagic flux was never evaluated. Doxorubicin may stimulate autophagy in the heart via depletion of GATA binding protein 4 and activation of ribosomal protein S6 kinase 1 (S6K1), which in turn may regulate essential autophagy genes (94). Rat models of doxorubicin-induced cardiomyopathy further implicate cardiomyocyte autophagy in the progression to HF (95). Drug-related cardiotoxicity is suspected in cancer patients treated with the reversible proteasome inhibitor bortezomib (96). Rats exposed to bortezomib develop HF, endoplasmic reticulum stress, and increased autophagy (96).
Autophagy in atherosclerosis
The role of autophagy in atherosclerosis has been investigated extensively, with a particular focus on VSMCs and endothelial cells. Transmission electron microscopy of VSMCs in the fibrous cap of experimental or human plaques revealed features of autophagy (97), and Western blot analysis of advanced human plaques showed elevated levels of LC3-II (98).
Several autophagy triggers are present within the atherosclerotic plaque, such as inflammatory mediators (99), ROS (100), oxLDL (101, 102), TNF-α (99), osteopontin, and AGEs (103, 104). In aortic smooth muscle cells (SMCs), increases in LC3 and autophagy were detected during lipid peroxidation (105). Autophagic activity confers cytoprotection from aldehyde-induced death via removal of aldehyde-modified proteins (106). Moreover, mild oxidative stress activates autophagy to facilitate the removal of damaged organelles (107). Treatment of VSMCs in culture with 7-ketocholesterol, one of the major oxysterols present in atherosclerotic plaques, not only triggers oxidative damage but also extensive autophagic activity (108). In these models, autophagy promotes survival of VSMCs. However, SKF 96365 (an inhibitor of transient receptor potential calcium channels) induces apoptosis and autophagy in the VSMC line A7r5 (109). TNF-α stimulates autophagic VSMC death (99). Thus, in these last two models, SKF 96365 and TNF-α–induced autophagy are involved in the promotion of cell death rather than in cytoprotection.
VSMCs within the fibrous cap are surrounded by a thick layer of basal lamina, resulting in local hypoxia due to inadequate vascularization (110), as well as nutrient and growth factor deprivation, conditions that induce autophagy. Dying VSMCs in the fibrous cap of advanced plaques in humans harbor ubiquitinated inclusions in the cytoplasm and may undergo autophagic death (108, 111). Yet there is general consensus that basal autophagy is protective against oxidative stress (107). A protective role for autophagy was demonstrated in vitro by showing that statin-induced VSMC death could be attenuated by 7-ketocholesterol (112), which induced autophagy through NADPH oxidase 4 and ATG4 (113). Similarly, exposure of endothelial cells in culture to oxLDL or AGEs (102, 114) induced autophagy, which protected against endothelial cell injury (115). Moreover, verapamil can inhibit vascular injury–induced neointima formation by promoting autophagy (116). Conversely, excessive autophagic activity can provoke plaque destabilization, lesional thrombosis, and acute clinical events (98).
Autophagy in vascular remodeling
Evidence has been documented of both pro- and antiatherosclerotic autophagic functions. Hu et al. showed that proatherosclerotic AGEs can induce VSMC proliferation via ERK and AKT pathway–mediated induction of autophagy (103). The developmental morphogen, sonic hedgehog, is re-expressed in atherosclerotic lesions, stimulating both autophagy and proliferation of VSMCs through an AKT-dependent pathway (117). PDGF-BB, which promotes development of the proliferative VSMC phenotype, is a robust inducer of autophagy (118).
These results clearly contrast with the observation that both serum starvation and nutrient deprivation, standard inducers of autophagy, inhibit VSMC proliferation (119, 120). Moreover, treatment of VSMC with the cortisone reductase inhibitor emodin induces growth arrest and cell death by autophagy (121). Rapamycin blocks VSMC migration and proliferation in vitro and intimal hyperplasia in vivo, and induces VSMC differentiation in culture (122). Rapamycin also increases the activity of the cyclin-dependent kinase inhibitor p27Kip1 (123, 124), as well as Rb, to attenuate VSMC proliferation (125). Furthermore, rapamycin and its analogs are used clinically to limit VSMC proliferation, such as in drug-eluting coronary artery stents (126, 127). Sirolimus from drug-eluting stents induces autophagy in vascular endothelial cells suppressing reendothelialization and revascularization (128).
The roles of mTOR and AKT and their relationship to autophagic activity in VSMCs are complex. Rapamycin induces VSMC differentiation by specific activation of the Akt2 isoform, promoting expression of contractile proteins (129). In fact, arachidonic acid, TGF-β, FK506, and leptin induce VSMC proliferation through PI3K-dependent activation of mTOR (130–133). Thus, autophagy in VSMCs can confer both adaptive and maladaptive actions depending on the context. Currently, many studies are underway to parse “good” autophagy from “bad” and to define underlying mechanisms.
Pharmacologic modulation of autophagy in cardiovascular diseases
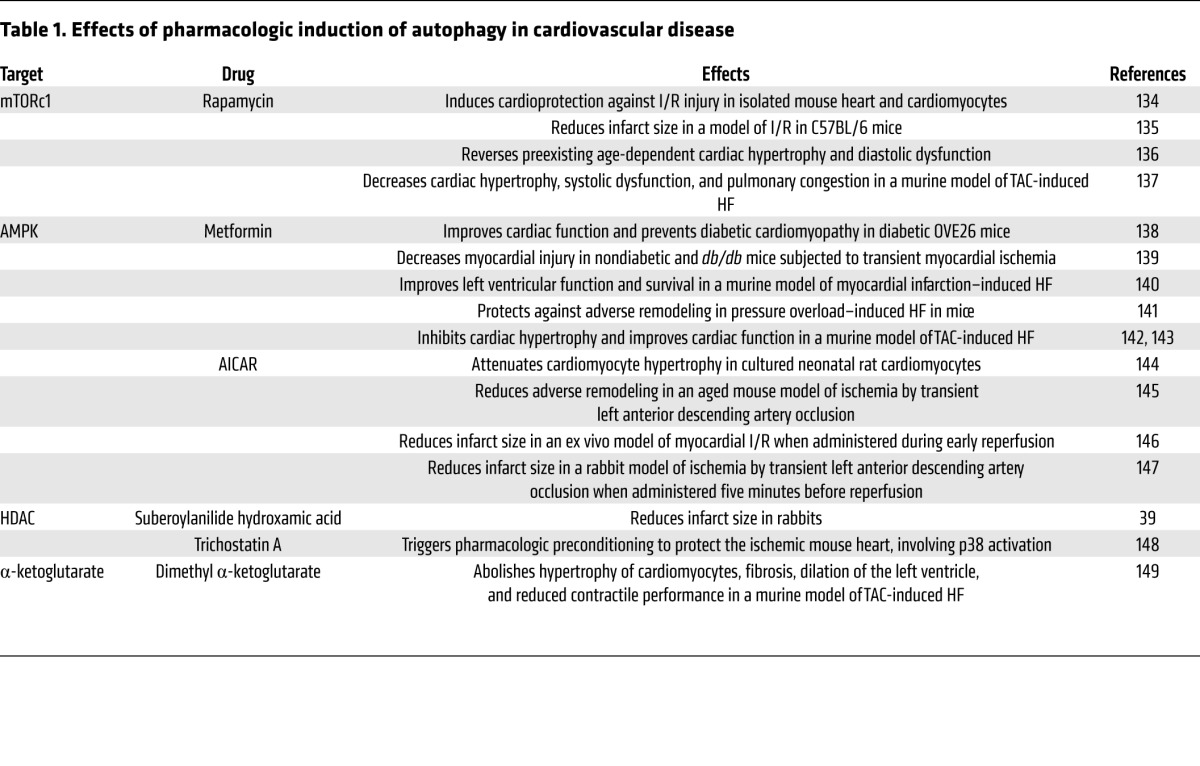
Several pharmacologic agents have been identified that modulate autophagy. Many of them manifest efficacy in the treatment of cardiovascular disorders (Table 1). The only clinical trial related to manipulation of autophagy in cardiovascular disease involved the use of sirolimus after cardiac allograft (ClinicalTrials.gov identifier NCT01889992). Insights gleaned from such work raise the exciting prospect of tuning the autophagic response for therapeutic gain. However, considering the dual role of autophagy in cytoprotection and cell death, specific targeting and careful titration will be required.
Table 1.
Effects of pharmacologic induction of autophagy in cardiovascular disease
Challenges for the future
Autophagic flux participates in cardiovascular physiology and pathophysiology in myriad ways and in essentially all cell types. Recent studies reveal that this process is druggable, such that it could be manipulated for therapeutic gain. As such, it is possible that this evolutionarily conserved cellular mechanism will emerge as a therapeutic target; indeed, recent work suggests that this is already the case (39). To bring this to fruition, however, a number of major challenges must be addressed.
In many instances, the role of autophagic flux in cardiovascular pathophysiology is unclear: whether changes in autophagic flux are causal, secondary, or occur as epiphenomena remains to be determined. Is the response a mechanism contributing to disease progression? Is it an adaptive cellular response to withstand and resist disease-related stress? Is the response unrelated to disease pathogenesis? Also, it will be critical to differentiate the role(s) of autophagic flux in normal cellular homeostasis, development, aging, and responses to environmental stimuli (e.g., exercise) from events occurring in the context of disease.
Are the differences between good and bad autophagy merely quantitative, such that too little or too much autophagy is harmful? Or are the differences qualitative, such that the cellular elements that are targeted differ or are processed differently? When is autophagy in the heart non-selective in terms of its catabolic targets, and when does it target specific molecular species or organelles? What are the relative contributions of defects in autophagosome initiation, processing, lysosomal fusion, cargo degradation, and release of catabolic end-products to human disease?
A significant limitation in this field is the lack of noninvasive means of gauging autophagic flux in the myocardium. A knowledge of circulating biomarkers or the availability of imaging modalities that allow for determination and quantification of autophagic activity in the heart would be a major advance. Further, biomarkers that distinguish maladaptive from adaptive autophagic flux would help to propel this field to clinical translation.
Another major challenge is the development of efficacious and well-tolerated means of regulating autophagic flux for therapeutic gain. It will be important to effectively target those strategies to specific cells and tissues, as globally altered autophagic flux might be beneficial in one tissue (e.g., heart) and deleterious elsewhere in the body.
Conclusion and perspective
Changes in the level of autophagic flux are seen in essentially all forms of heart disease and in all cell types. In some instances, that response is beneficial; in other cases, it is maladaptive, promoting disease progression. Despite formidable challenges we are optimistic that targeting autophagic flux in the cardiovascular system will emerge as a therapeutic target of clinical relevance. Much work remains to decipher this fascinating biology, but patients with heart disease are likely to benefit.
Acknowledgments
This work was supported by funds from the NIH (grants HL-120732, HL-100401, and HL-097768), American Heart Association Strategically Focused Research Network (14SFRN20740000), Cancer Prevention Research Institute of Texas (grant RP110486P3), Leducq Foundation (grant 11CVD04), and Comision Nacional de Investigacion Cientifica y Tecnologica de Chile (FONDAP grant 15130011 to S. Lavandero and M. Chiong, Redes grant 120003 to S. Lavandero and J.A. Hill, ACT grant 1111 to S. Lavandero and M. Chiong, FONDECYT grants 1120212 to S. Lavandero and 1140329 to M. Chiong).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(1):55–64. doi:10.1172/JCI73943.
References
- 1.Go AS, et al. Heart disease and stroke statistics — 2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366(1):54–63. doi: 10.1056/NEJMra1112570. [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 4.Pedrozo Z, et al. Cardiomyocyte ryanodine receptor degradation by chaperone-mediated autophagy. Cardiovasc Res. 2013;98(2):277–285. doi: 10.1093/cvr/cvt029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14(7):1237–1243. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 7.Uberti F, et al. Vitamin D protects Human Endothelial Cells from oxidative stress through the autophagic and survival pathways. J Clin Endocrinol. 2013;Metab(4):jc20132103. doi: 10.1210/jc.2013-2103. [DOI] [PubMed] [Google Scholar]
- 8.Zhang YL, et al. The autophagy-lysosome pathway: a novel mechanism involved in the processing of oxidized LDL in human vascular endothelial cells. Biochem Biophys Res Commun. 2010;394(2):377–382. doi: 10.1016/j.bbrc.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 9.Xie Y, et al. Protective role of autophagy in AGE-induced early injury of human vascular endothelial cells. Mol Med Rep. 2011;4(3):459–464. doi: 10.3892/mmr.2011.460. [DOI] [PubMed] [Google Scholar]
- 10.Bansode RR, Ahmedna M, Svoboda KR, Losso JN. Coupling in vitro and in vivo paradigm reveals a dose dependent inhibition of angiogenesis followed by initiation of autophagy by C6-ceramide. Int J Biol Sci. 2011;7(5):629–644. doi: 10.7150/ijbs.7.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang YC, et al. Clinorotation enhances autophagy in vascular endothelial cells. Biochem Cell Biol. 2013;91(5):309–314. doi: 10.1139/bcb-2013-0029. [DOI] [PubMed] [Google Scholar]
- 12.Chau YP, Lin SY, Chen JH, Tai MH. Endostatin induces autophagic cell death in EAhy926 human endothelial cells. Histol Histopathol. 2003;18(3):715–726. doi: 10.14670/HH-18.715. [DOI] [PubMed] [Google Scholar]
- 13.Kim HS, Montana V, Jang HJ, Parpura V, Kim JA. Epigallocatechin gallate (EGCG) stimulates autophagy in vascular endothelial cells: a potential role for reducing lipid accumulation. J Biol Chem. 2013;288(31):22693–22705. doi: 10.1074/jbc.M113.477505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torisu T, et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med. 2013;19(10):1281–1287. doi: 10.1038/nm.3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim KW, Paul P, Qiao J, Chung DH. Autophagy mediates paracrine regulation of vascular endothelial cells. Lab Invest. 2013;93(6):639–645. doi: 10.1038/labinvest.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geer JC, McGill HC, Jr, Strong JP. The fine structure of human atherosclerotic lesions. Am J Pathol. 1961;38:263–287. [PMC free article] [PubMed] [Google Scholar]
- 17.Buck RC. Lamellae in the spindle of mitotic cells of Walker 256 carcinoma. J Biophys Biochem Cytol. 1961;11:227–236. doi: 10.1083/jcb.11.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salabei JK, Hill BG. Implications of autophagy for vascular smooth muscle cell function and plasticity. Free Radic Biol Med. 2013;65:693–703. doi: 10.1016/j.freeradbiomed.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao X, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15(4):545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Razani B, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15(4):534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13(6):655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber KT, Sun Y, Diez J. Fibrosis: a living tissue and the infarcted heart. J Am Coll Cardiol. 2008;52(24):2029–2031. doi: 10.1016/j.jacc.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Aranguiz-Urroz P, et al. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim Biophys Acta. 2011;1812(1):23–31. doi: 10.1016/j.bbadis.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Ishida Y, et al. Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Biol Cell. 2009;20(11):2744–2754. doi: 10.1091/mbc.E08-11-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J Biol Chem. 2012;287(15):11677–11688. doi: 10.1074/jbc.M111.308460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, et al. FRS2α-mediated FGF signals suppress premature differentiation of cardiac stem cells through regulating autophagy activity. Circ Res. 2012;110(4):e29–e39. doi: 10.1161/CIRCRESAHA.111.255950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee E, et al. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy. 2014;10(4):572–587. doi: 10.4161/auto.27649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Liu J, Liu L, McKeehan WL, Wang F. The fibroblast growth factor signaling axis controls cardiac stem cell differentiation through regulating autophagy. Autophagy. 2012;8(4):690–691. doi: 10.4161/auto.19290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimomura H, Terasaki F, Hayashi T, Kitaura Y, Isomura T, Suma H. Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Jpn Circ J. 2001;65(11):965–968. doi: 10.1253/jcj.65.965. [DOI] [PubMed] [Google Scholar]
- 30.Kostin S, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92(7):715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Valentim L, et al. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006;40(6):846–852. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]
- 33.Matsui Y, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 34.Troncoso R, et al. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 2012;93(2):320–329. doi: 10.1093/cvr/cvr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan L, et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102(39):13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang C, et al. Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. Am J Physiol Heart Circ Physiol. 2010;298(2):H570–H579. doi: 10.1152/ajpheart.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol. 1980;98(2):425–444. [PMC free article] [PubMed] [Google Scholar]
- 38.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281(40):29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 39.Xie M, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129(10):1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2009;13(2):373–387. doi: 10.1111/j.1582-4934.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang C, et al. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res. 2010;3(4):365–373. doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanamori H, et al. The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc Res. 2011;91(2):330–339. doi: 10.1093/cvr/cvr073. [DOI] [PubMed] [Google Scholar]
- 43.Ma X, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125(25):3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Diwan A. Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy. 2012;8(9):1394–1396. doi: 10.4161/auto.21036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu H, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117(7):1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh KK, et al. Autophagy gene fingerprint in human ischemia and reperfusion. J Thorac Cardiovasc Surg. 2014;147(3):1065–1072.e1061. doi: 10.1016/j.jtcvs.2013.04.042. [DOI] [PubMed] [Google Scholar]
- 47.Drazner MH, et al. Increased left ventricular mass is a risk factor for the development of a depressed left ventricular ejection fraction within five years: the Cardiovascular Health Study. J Am Coll Cardiol. 2004;43(12):2207–2215. doi: 10.1016/j.jacc.2003.11.064. [DOI] [PubMed] [Google Scholar]
- 48.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358(13):1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 49.Dammrich J, Pfeifer U. Cardiac hypertrophy in rats after supravalvular aortic constriction. II. Inhibition of cellular autophagy in hypertrophying cardiomyocytes. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983;43(3):287–307. doi: 10.1007/BF02932962. [DOI] [PubMed] [Google Scholar]
- 50.Nakai A, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13(5):619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 51.Tannous P, et al. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117(24):3070–3078. doi: 10.1161/CIRCULATIONAHA.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–477. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 53.Rothermel BA, Hill JA. Autophagy in load-induced heart disease. Circ Res. 2008;103(12):1363–1369. doi: 10.1161/CIRCRESAHA.108.186551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imai S. The NAD World: a new systemic regulatory network for metabolism and aging — Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem Biophys. 2009;53(2):65–74. doi: 10.1007/s12013-008-9041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsu CP, Hariharan N, Alcendor RR, Oka S, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through autophagy in cardiomyocytes. Autophagy. 2009;5(8):1229–1231. doi: 10.4161/auto.5.8.10275. [DOI] [PubMed] [Google Scholar]
- 56.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010;107(12):1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. 2014;114(2):368–378. doi: 10.1161/CIRCRESAHA.113.300536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pillai VB, et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285(5):3133–3144. doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finckenberg P, et al. Caloric restriction ameliorates angiotensin II-induced mitochondrial remodeling and cardiac hypertrophy. Hypertension. 2012;59(1):76–84. doi: 10.1161/HYPERTENSIONAHA.111.179457. [DOI] [PubMed] [Google Scholar]
- 60.Latronico MV, Condorelli G. Therapeutic use of microRNAs in myocardial diseases. Curr Heart Fail Rep. 2011;8(3):193–197. doi: 10.1007/s11897-011-0068-2. [DOI] [PubMed] [Google Scholar]
- 61.Ucar A, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078. doi: 10.1038/ncomms2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hein S, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107(7):984–991. doi: 10.1161/01.CIR.0000051865.66123.B7. [DOI] [PubMed] [Google Scholar]
- 63.St John Sutton MG, Plappert T, Rahmouni H. Assessment of left ventricular systolic function by echocardiography. Heart Fail Clin. 2009;5(2):177–190. doi: 10.1016/j.hfc.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 64.Cao DJ, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108(10):4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kong Y, et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation. 2006;113(22):2579–2588. doi: 10.1161/CIRCULATIONAHA.106.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dennemarker J, et al. Impaired turnover of autophagolysosomes in cathepsin L deficiency. Biol Chem. 2010;391(8):913–922. doi: 10.1515/BC.2010.097. [DOI] [PubMed] [Google Scholar]
- 67.Saijo M, et al. Cardiomyopathy with prominent autophagic degeneration, accompanied by an elevated plasma brain natriuretic peptide level despite the lack of overt heart failure. Intern Med. 2004;43(8):700–703. doi: 10.2169/internalmedicine.43.700. [DOI] [PubMed] [Google Scholar]
- 68.Kassiotis C, et al. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009;120(11):S191–S197. doi: 10.1161/CIRCULATIONAHA.108.842252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andres AM, et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal. 2014;21(14):1960–1973. doi: 10.1089/ars.2013.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One. 2011;6(6):e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoshino A, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 73.Nishino I, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000;406(6798):906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 74.Fukuda T, et al. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol Ther. 2006;14(6):831–839. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tannous P, et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2008;105(28):9745–9750. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109(2):151–160. doi: 10.1161/CIRCRESAHA.110.237339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109(3):296–308. doi: 10.1161/CIRCRESAHA.111.244707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bhuiyan MS, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123(12):5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tanaka Y, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406(6798):902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 80.Arad M, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352(4):362–372. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 81.Raben N, et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder — murine Pompe disease. Autophagy. 2010;6(8):1078–1089. doi: 10.4161/auto.6.8.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sugimoto S, Shiomi K, Yamamoto A, Nishino I, Nonaka I, Ohi T. LAMP-2 positive vacuolar myopathy with dilated cardiomyopathy. Intern Med. 2007;46(11):757–760. doi: 10.2169/internalmedicine.46.6265. [DOI] [PubMed] [Google Scholar]
- 83.Xu X, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288(25):18077–18092. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fierabracci A. The putative role of proteolytic pathways in the pathogenesis of Type 1 diabetes mellitus: the ‘autophagy’ hypothesis. Med Hypotheses. 2014;82(5):553–557. doi: 10.1016/j.mehy.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 85.Ebato C, et al. Autophagy is important in islet homeostasis and compensatory increase of β cell mass in response to high-fat diet. Cell Metab. 2008;8(4):325–332. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 86.Fujitani Y, Ebato C, Uchida T, Kawamori R, Watada H. β-Cell autophagy: a novel mechanism regulating β-cell function and mass: Lessons from β-cell-specific Atg7-deficient mice. Islets. 2009;1(2):151–153. doi: 10.4161/isl.1.2.9057. [DOI] [PubMed] [Google Scholar]
- 87.Marsh BJ, et al. Regulated autophagy controls hormone content in secretory-deficient pancreatic endocrine β-cells. Mol Endocrinol. 2007;21(9):2255–2269. doi: 10.1210/me.2007-0077. [DOI] [PubMed] [Google Scholar]
- 88.Sciarretta S, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125(9):1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol. 2011;50(6):1035–1043. doi: 10.1016/j.yjmcc.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 90.Masini M, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia. 2009;52(6):1083–1086. doi: 10.1007/s00125-009-1347-2. [DOI] [PubMed] [Google Scholar]
- 91.Garcia L, et al. Impaired cardiac autophagy in patients developing postoperative atrial fibrillation. J Thorac Cardiovasc Surg. 2012;143(2):451–459. doi: 10.1016/j.jtcvs.2011.07.056. [DOI] [PubMed] [Google Scholar]
- 92.Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM, Ren J. Chronic Akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol. 2011;106(6):1173–1191. doi: 10.1007/s00395-011-0222-8. [DOI] [PubMed] [Google Scholar]
- 93.Zbinden G, Bachmann E, Holderegger C. Model systems for cardiotoxic effects of anthracyclines. Antibiot Chemother (1971). 1978;23:255–270. doi: 10.1159/000401489. [DOI] [PubMed] [Google Scholar]
- 94.Dirks-Naylor AJ. The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sci. 2013;93(24):913–916. doi: 10.1016/j.lfs.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 95.Lu L, Wu W, Yan J, Li X, Yu H, Yu X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int J Cardiol. 2009;134(1):82–90. doi: 10.1016/j.ijcard.2008.01.043. [DOI] [PubMed] [Google Scholar]
- 96.Nowis D, et al. Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am J Pathol. 2010;176(6):2658–2668. doi: 10.2353/ajpath.2010.090690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kockx MM, De Meyer GR, Buyssens N, Knaapen MW, Bult H, Herman AG. Cell composition, replication, and apoptosis in atherosclerotic plaques after 6 months of cholesterol withdrawal. Circ Res. 1998;83(4):378–387. doi: 10.1161/01.RES.83.4.378. [DOI] [PubMed] [Google Scholar]
- 98.Martinet W, De Meyer GR. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. 2009;104(3):304–317. doi: 10.1161/CIRCRESAHA.108.188318. [DOI] [PubMed] [Google Scholar]
- 99.Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol. 2006;84(5):448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 100.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36(1):30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 101.Muller C, Salvayre R, Negre-Salvayre A, Vindis C. Oxidized LDLs trigger endoplasmic reticulum stress and autophagy: prevention by HDLs. Autophagy. 2011;7(5):541–543. doi: 10.4161/auto.7.5.15003. [DOI] [PubMed] [Google Scholar]
- 102.Nowicki M, Zabirnyk O, Duerrschmidt N, Borlak J, Spanel-Borowski K. No upregulation of lectin-like oxidized low-density lipoprotein receptor-1 in serum-deprived EA.hy926 endothelial cells under oxLDL exposure, but increase in autophagy. Eur J Cell Biol. 2007;86(10):605–616. doi: 10.1016/j.ejcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 103.Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med. 2012;29(4):613–618. doi: 10.3892/ijmm.2012.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zheng YH, et al. Osteopontin stimulates autophagy via integrin/CD44 and p38 MAPK signaling pathways in vascular smooth muscle cells. J Cell Physiol. 2012;227(1):127–135. doi: 10.1002/jcp.22709. [DOI] [PubMed] [Google Scholar]
- 105.Lee SJ, et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. 2011;183(5):649–658. doi: 10.1164/rccm.201005-0746OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hill BG, Haberzettl P, Ahmed Y, Srivastava S, Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem J. 2008;410(3):525–534. doi: 10.1042/BJ20071063. [DOI] [PubMed] [Google Scholar]
- 107.Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal. 2006;8(1):152–162. doi: 10.1089/ars.2006.8.152. [DOI] [PubMed] [Google Scholar]
- 108.Martinet W, De Bie M, Schrijvers DM, De Meyer GR, Herman AG, Kockx MM. 7-ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24(12):2296–2301. doi: 10.1161/01.ATV.0000146266.65820.a1. [DOI] [PubMed] [Google Scholar]
- 109.Park EJ, et al. SK&F 96365 induces apoptosis and autophagy by inhibiting Akt-mTOR signaling in A7r5 cells. Biochim Biophys Acta. 2011;1813(12):2157–2164. doi: 10.1016/j.bbamcr.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 110.Sluimer JC, et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51(13):1258–1265. doi: 10.1016/j.jacc.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 111.Martinet W, De Meyer GR. Selective depletion of macrophages in atherosclerotic plaques: myth, hype, or reality? Circ Res. 2007;100(6):751–753. doi: 10.1161/01.RES.0000263397.14481.96. [DOI] [PubMed] [Google Scholar]
- 112.Martinet W, Schrijvers DM, Timmermans JP, Bult H. Interactions between cell death induced by statins and 7-ketocholesterol in rabbit aorta smooth muscle cells. Br J Pharmacol. 2008;154(6):1236–1246. doi: 10.1038/bjp.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.He C, et al. 7-Ketocholesterol induces autophagy in vascular smooth muscle cells through Nox4 and Atg4B. Am J Pathol. 2013;183(2):626–637. doi: 10.1016/j.ajpath.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6(2):239–247. doi: 10.4161/auto.6.2.11062. [DOI] [PubMed] [Google Scholar]
- 115.Schrijvers DM, De Meyer GR, Martinet W. Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol. 2011;31(12):2787–2791. doi: 10.1161/ATVBAHA.111.224899. [DOI] [PubMed] [Google Scholar]
- 116.Salabei JK, Balakumaran A, Frey JC, Boor PJ, Treinen-Moslen M, Conklin DJ. Verapamil stereoisomers induce antiproliferative effects in vascular smooth muscle cells via autophagy. Toxicol Appl Pharmacol. 2012;262(3):265–272. doi: 10.1016/j.taap.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li H, et al. Sonic hedgehog promotes autophagy of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2012;303(11):H1319–H1331. doi: 10.1152/ajpheart.00160.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Salabei JK, Cummins TD, Singh M, Jones SP, Bhatnagar A, Hill BG. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem J. 2013;451(3):375–388. doi: 10.1042/BJ20121344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hultgardh-Nilsson A, Krondahl U, Jiang WQ, Nilsson J, Ringertz NR. Endogenous activation of c-myc expression and DNA synthesis in serum-starved neonatal rat smooth muscle cells. Differentiation. 1993;52(2):161–168. doi: 10.1111/j.1432-0436.1993.tb00626.x. [DOI] [PubMed] [Google Scholar]
- 120.Zimmermann O, et al. Serum starvation and growth factor receptor expression in vascular smooth muscle cells. J Vasc Res. 2006;43(2):157–165. doi: 10.1159/000090945. [DOI] [PubMed] [Google Scholar]
- 121.Wang X, et al. Emodin induces growth arrest and death of human vascular smooth muscle cells through reactive oxygen species and p53. J Cardiovasc Pharmacol. 2007;49(5):253–260. doi: 10.1097/FJC.0b013e318033dfb3. [DOI] [PubMed] [Google Scholar]
- 122.Martin KA, et al. The mTOR/p70 S6K1 pathway regulates vascular smooth muscle cell differentiation. Am J Physiol Cell Physiol. 2004;286(3):C507–C517. doi: 10.1152/ajpcell.00201.2003. [DOI] [PubMed] [Google Scholar]
- 123.Sun J, Marx SO, Chen HJ, Poon M, Marks AR, Rabbani LE. Role for p27(Kip1) in vascular smooth muscle cell migration. Circulation. 2001;103(24):2967–2972. doi: 10.1161/01.CIR.103.24.2967. [DOI] [PubMed] [Google Scholar]
- 124.Tanner FC, Yang ZY, Duckers E, Gordon D, Nabel GJ, Nabel EG. Expression of cyclin-dependent kinase inhibitors in vascular disease. Circ Res. 1998;82(3):396–403. doi: 10.1161/01.RES.82.3.396. [DOI] [PubMed] [Google Scholar]
- 125.Lightell DJ, Lightell DJ, Jr, Moss SC, Woods TC. Loss of canonical insulin signaling accelerates vascular smooth muscle cell proliferation and migration through changes in p27Kip1 regulation. Endocrinology. 2011;152(2):651–658. doi: 10.1210/en.2010-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Park KW, et al. Safety and efficacy of everolimus- versus sirolimus-eluting stents: a systematic review and meta-analysis of 11 randomized trials. Am Heart J. 2013;165(2):241–250.e244. doi: 10.1016/j.ahj.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 127.Townsend JC, Rideout P, Steinberg DH. Everolimus-eluting stents in interventional cardiology. Vasc Health Risk Manag. 2012;8:393–404. doi: 10.2147/VHRM.S23388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hayashi S, et al. The stent-eluting drugs sirolimus and paclitaxel suppress healing of the endothelium by induction of autophagy. Am J Pathol. 2009;175(5):2226–2234. doi: 10.2353/ajpath.2009.090152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Martin KA, et al. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem. 2007;282(49):36112–36120. doi: 10.1074/jbc.M703914200. [DOI] [PubMed] [Google Scholar]
- 130.Cavet ME, et al. Gas6-axl receptor signaling is regulated by glucose in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2008;28(5):886–891. doi: 10.1161/ATVBAHA.108.162693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Giordano A, et al. FK506 can activate transforming growth factor-β signalling in vascular smooth muscle cells and promote proliferation. Cardiovasc Res. 2008;79(3):519–526. doi: 10.1093/cvr/cvn079. [DOI] [PubMed] [Google Scholar]
- 132.Neeli I, Yellaturu CR, Rao GN. Arachidonic acid activation of translation initiation signaling in vascular smooth muscle cells. Biochem. Biophys Res Commun. 2003;309(4):755–761. doi: 10.1016/j.bbrc.2003.08.066. [DOI] [PubMed] [Google Scholar]
- 133.Shan J, Nguyen TB, Totary-Jain H, Dansky H, Marx SO, Marks AR. Leptin-enhanced neointimal hyperplasia is reduced by mTOR and PI3K inhibitors. Proc Natl Acad Sci U S A. 2008;105(48):19006–19011. doi: 10.1073/pnas.0809743105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41(2):256–264. doi: 10.1016/j.yjmcc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 135.Chen HH, et al. Fluorescence tomography of rapamycin-induced autophagy and cardioprotection in vivo. Circ Cardiovasc Imaging. 2013;6(3):441–447. doi: 10.1161/CIRCIMAGING.112.000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dai DF, et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell. 2014;13(3):529–539. doi: 10.1111/acel.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bishu K, et al. Anti-remodeling effects of rapamycin in experimental heart failure: dose response and interaction with Angiotensin receptor blockade. PLoS One. 2013;8(12):e81325. doi: 10.1371/journal.pone.0081325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xie Z, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60(6):1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Calvert JW, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57(3):696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- 140.Gundewar S, et al. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. 2009;104(3):403–411. doi: 10.1161/CIRCRESAHA.108.190918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xu X, et al. Metformin protects against systolic overload-induced heart failure independent of AMP-activated protein kinase alpha2. Hypertension. 2014;63(4):723–728. doi: 10.1161/HYPERTENSIONAHA.113.02619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li HL, et al. Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J Cell Biochem. 2007;100(5):1086–1099. doi: 10.1002/jcb.21197. [DOI] [PubMed] [Google Scholar]
- 143.Meng RS, et al. Adenosine monophosphate-activated protein kinase inhibits cardiac hypertrophy through reactivating peroxisome proliferator-activated receptor-alpha signaling pathway. Eur J Pharmacol. 2009;620(1):63–70. doi: 10.1016/j.ejphar.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 144.Chen BL, et al. Activation of AMPK inhibits cardiomyocyte hypertrophy by modulating of the FOXO1/MuRF1 signaling pathway in vitro. Acta Pharmacol Sin. 2010;31(7):798–804. doi: 10.1038/aps.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cieslik KA, Taffet GE, Crawford JR, Trial J, Mejia Osuna P, Entman ML. AICAR-dependent AMPK activation improves scar formation in the aged heart in a murine model of reperfused myocardial infarction. J Mol Cell Cardiol. 2013;63:26–36. doi: 10.1016/j.yjmcc.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Paiva MA, Goncalves LM, Providencia LA, Davidson SM, Yellon DM, Mocanu MM. Transitory activation of AMPK at reperfusion protects the ischaemic-reperfused rat myocardium against infarction. Cardiovasc Drugs Ther. 2010;24(1):25–32. doi: 10.1007/s10557-010-6222-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kingma JG, Kingma JG, Jr, Simard D, Rouleau JR. Timely administration of AICA riboside reduces reperfusion injury in rabbits. Cardiovasc Res. 1994;28(7):1003–1007. doi: 10.1093/cvr/28.7.1003. [DOI] [PubMed] [Google Scholar]
- 148.Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res. 2007;76(3):473–481. doi: 10.1016/j.cardiores.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 149.Marino G, et al. Dimethyl α-ketoglutarate inhibits maladaptive autophagy in pressure overload-induced cardiomyopathy. Autophagy. 2014;10(5):930–932. doi: 10.4161/auto.28235. [DOI] [PMC free article] [PubMed] [Google Scholar]