

Abstract
mTOR, a serine/threonine kinase, is a master regulator of cellular metabolism. mTOR regulates cell growth and proliferation in response to a wide range of cues, and its signaling pathway is deregulated in many human diseases. mTOR also plays a crucial role in regulating autophagy. This Review provides an overview of the mTOR signaling pathway, the mechanisms of mTOR in autophagy regulation, and the clinical implications of mTOR inhibitors in disease treatment.
Overview of mTOR signaling pathway
Nutrients, growth factors, and cellular energy levels are key determinants of cell growth and proliferation. mTOR, a serine/threonine kinase, is a master regulator of cellular metabolism and promotes cell growth in response to environmental cues. Deregulation of mTOR signaling has been implicated in many human diseases, including diabetes, neurodegenerative diseases, and cancer (1).
mTOR forms two distinct signaling complexes, mTOR complex 1 (mTORC1) and mTORC2, by binding with multiple companion proteins (Figure 1). mLST8, DEPTOR, and the Tti1/Tel2 complex exist in both mTORC1 and mTORC2 (2–5). On the other hand, RAPTOR and PRAS40 are specific to mTORC1 (6–11) whereas RICTOR, mSin1, and PROCTOR1/2 are specific to mTORC2 (10, 12–16). The two kinase complexes have specific substrate preferences and therefore elicit distinct downstream signaling events to modulate cellular function.
Figure 1. The mTOR signaling network.

mTOR forms two distinct signaling complexes, mTORC1 and mTORC2. mTORC1 integrates nutrient and growth factor signaling to promote anabolic metabolism, such as protein synthesis and lipid synthesis, and to inhibit catabolic pathways, such as lysosome biogenesis and autophagy. Growth factors activate RTKs and the downstream signaling cascades PI3K/AKT and Ras/Erk, resulting in inhibition of the TSC complex, which functions as a GAP for the mTORC1 upstream effector Rheb GTPase. In contrast, cellular stressors, such as low energy levels and hypoxia, activate the TSC complex via AMPK and REDD1. AMPK also directly inhibits mTORC1 activity by phosphorylating RAPTOR. In the presence of amino acids, Rag GTPases recruit mTORC1 to the lysosomal surface, where it can be activated by Rheb GTPase. Growth factor/RTK/PI3K signaling also activates mTORC2, which regulates cell survival, metabolism, and cytoskeletal organization via AGC family kinases. mTORC1 activation exerts feedback inhibition on RTK/PI3K/AKT signaling via the inhibition of IRS and activation of GRB10. S6K, an mTORC1 downstream effector, also inhibits IRS and mTORC2 via inhibitory phosphorylation.
One of the well-established roles of mTORC1 is to promote anabolic cellular metabolism to supply the necessary building blocks for cell growth and proliferation. mTORC1 integrates various stimuli and signaling networks to stimulate synthesis of protein, lipid, and nucleotides and block catabolic processes such as autophagy at the post-translational and transcriptional levels (reviewed in refs. 17, 18). The tuberous sclerosis (TSC) tumor suppressor complex (TSC1/TSC2) is arguably the most important upstream negative regulator of mTORC1. Genetic mutations in hamartin or tuberin (encoding TSC1 and TSC2, respectively) cause tumor development in various tissues such as angiofibromas, angiomyolipomas, lymphangioleiomyomatosis, and renal cell carcinoma. Loss-of-function mutations in either TSC1 or TSC2 lead to constitutive mTORC1 activation, which contributes to uncontrolled growth and underlies the TSC disease (19). These findings provide the scientific basis of using mTORC1 inhibitors for the treatment of TSC and related diseases such as cancer.
A well-established upstream regulator of mTORC1 is the growth factor/PI3K/AKT signaling pathway. Growth factors such as insulin and IGF activate their cognate receptors (receptor tyrosine kinases [RTKs]) and subsequently activate the PI3K/AKT signaling axis. Activated AKT directly phosphorylates and thereby inhibits TSC1/2, a GTPase-activating protein (GAP) for the Ras homolog enriched in brain (Rheb) GTPase (19–23). The AKT-dependent phosphorylation results in dissociation of TSC1/2 from lysosome, where Rheb is localized, promoting Rheb activation (24). Since GTP-bound Rheb is a potent mTORC1 activator, inhibition of TSC1/2 by AKT-dependent phosphorylation results in mTORC1 activation (25, 26). Additionally, AKT directly phosphorylates and inhibits PRAS40, an mTORC1 component that negatively regulates the complex’s kinase activity, leading to mTORC1 activation (8–11). Furthermore, the activated RTK also stimulates the Ras/Erk/p90 ribosomal S6 kinase 1 (RSK1) signaling axis, which directly phosphorylates TSC2 to inactivate its GAP activity (27, 28). In contrast, cellular stressors such as low cellular energy levels or hypoxia activate TSC1/2 to inhibit mTORC1 activation. AMPK is a sensor of cellular energy levels and is activated by a high AMP/ATP ratio. AMPK phosphorylates TSC2 and presumably increases the TSC1/2 GAP activity (29). Moreover, AMPK directly phosphorylates RAPTOR, resulting in decreased mTORC1 activity through allosteric inhibition (30). These studies show a critical role of AMPK in linking cellular energy level to mTORC1 regulation. Low cellular oxygen levels also inhibit mTORC1 by upregulating DNA damage response 1 (REDD1), which may modulate TSC2 activity to inhibit mTORC1 (31, 32). Current studies reveal the TSC1/2 tumor suppressor as a key signaling hub receiving a diverse array of signals to control mTORC1 activity and thus cell growth.
Amino acids, which are essential components for protein synthesis, are also crucial regulators of mTORC1 (reviewed in refs. 33, 34). A genetic screen using a shRNA library targeting small GTPases and biochemical analysis using immunoaffinity-purified mTORC1-interacting proteins independently identified Rag proteins (RagA/B/C/D), a family of Ras-like small GTPases, as key mediators of amino acid–induced mTORC1 activation (35, 36). RagA and RagB have high amino acid homology and are functionally redundant, and RagC and RagD are homologous and functionally redundant when they are expressed in the same cell. RagA (or RagB) forms a heterodimer with RagC (or RagD) (37), and this dimer is tethered to the lysosome by the lysosomal protein complex Ragulator (38). In the presence of amino acids, the Rag GTPases are activated and recruit mTORC1 to the surface of the lysosome, where the kinase complex encounters its upstream effector Rheb, which activates the kinase complex through an undefined mechanism (38). Of note, only the GTP-bound form of Rheb is capable of activating mTORC1. Rag GTPases bind to RAPTOR when RagA/B and RagC/D are bound to GTP and GDP, respectively. Amino acids modulate Rag guanine nucleotide binding, therefore controlling the interaction between Rag GTPases and mTORC1. In addition, it has been shown that the insulin/AKT signaling axis regulates lysosomal localization of TSC1/2, demonstrating that the lysosome is the signaling hub of mTORC1 activation (24). Recent studies suggest that other subcellular compartments such as stress granule and peroxisome also play important roles in mTORC1 signaling regulation in response to oxidative stress (39, 40), although the interplay of those subcellular compartments with lysosome in mTORC1 activation is not fully elucidated. Moreover, despite recent findings of a guanine nucleotide exchange factor (GEF) and GAPs for the Rag GTPases and the requirement of vacuolar H+-ATPase (v-ATPase, a proton pump for lysosomal acidification) in mTORC1 activation (41–45), the signaling mechanism from amino acids to a Rag GEF and/or GAP is still unknown. A recent knockout mouse study confirms a role of Rag GTPases in mTORC1 activation during embryonic development (46). However, another study using conditional knockout mice shows that loss of RagA and RagB in cardiomyocytes causes lysosomal dysfunction due to deregulated lysosomal v-ATPase localization even though mTORC1 activation is not substantially affected (47). Further studies are needed to determine whether there is a Rag GTPase-independent mechanism in amino acid–induced mTORC1 activation.
The molecular mechanism of mTORC2 regulation by upstream effectors is largely unknown. The only known upstream activator is the growth factor/PI3K signaling axis. One study proposes that PI3K signaling activates mTORC2 by promoting the association of the kinase complex with ribosomes (48). Again, the mechanism of how ribosomal association activates mTORC2 has yet to be revealed. Moreover, this model needs to be verified in other systems. A subset of AGC family kinases (PKA, PKG, and PKC) has been shown to be phosphorylated by mTORC2, thereby regulating cell survival, metabolism, and cytoskeletal organization (reviewed in ref. 49). mTORC2 phosphorylates both the turn motif and hydrophobic motif of the AGC family kinases, thereby leading to stabilization and activation of these kinases, respectively. Of note, the hydrophobic motif of AKT (Ser473) is a well-characterized substrate of mTORC2, and its phosphorylation is critical for maximal activity of AKT (50). Therefore, AKT mediates positive crosstalk from mTORC2 to mTORC1 because AKT is an upstream stimulator of mTORC1 (as discussed above).
There are multiple negative feedback circuits from mTORC1 to mTORC2 via downregulation of the RTK/PI3K signaling pathway. Ribosomal protein S6 kinase 1 (S6K1), a downstream effector of mTORC1, negatively regulates the insulin receptor substrate 1 (IRS1), which is a positive mediator of RTK/PI3K signaling (51, 52). It has been also shown that S6K1 phosphorylates RICTOR, thereby affecting mTORC2 activity (53). Furthermore, mTORC1 directly inhibits RTK/PI3K signaling by phosphorylating IRS1 and growth factor–bound protein 10 (GRB10) (54–56). Therefore, the kinetics of mTORC1 and mTORC2 signaling are finely tuned to respond to dynamic changes in cellular metabolism and environmental cues.
Regulation of autophagy by mTOR
Autophagy is the major cellular digestion process that removes damaged macromolecules and organelles. In addition, autophagy is critical to providing energy and molecular building blocks by recycling macromolecules in response to nutrient and environmental stress (reviewed in ref. 57). The discovery by electron microscopy of vesicle structures containing amorphous materials and cytoplasmic organelles in the kidneys of newborn mice led to the introduction of autophagy in the late 1950s (58, 59). About 20 years after that initial discovery, it was observed that amino acid deprivation is a potent autophagy inducer in cultured mammalian cells and in perfused rat livers (60, 61). As previously mentioned, amino acids are key regulators of mTORC1 activation. Furthermore, most if not all autophagy induction conditions such as nutrient or growth factor deprivation and low cellular energy levels have been shown to inhibit mTORC1 activity. This suggests a tight, inverse coupling of autophagy induction and mTORC1 activation.
Autophagy induction by genetic or pharmacologic inhibition of mTORC1 (TORC1 in yeast) was first demonstrated in yeast (62) and later in Drosophila (63). However, the mechanistic understanding of how mTORC1 regulates autophagy in mammalian cells is fairly recent (Figure 2 and reviewed in refs. 64, 65). Three groups independently demonstrated that mTORC1 inhibits the autophagy-initiating UNC-5 like autophagy activating kinase (ULK) complex by phosphorylating complex components including autophagy related gene 13 (ATG13) and ULK1/2 (66–68). Inhibition of mTORC1 results in increased ULK1/2 kinase activity. ULK1/2 then phosphorylates ATG13 and FIP200, which are critical subunits of the ULK1/2 kinase complex (66–68). In yeast, TORC1 inhibits the autophagy-initiating kinase ATG1 (the mammalian ULK1/2 homolog) by phosphorylating ATG13 and disrupting the ATG1 and ATG13 interaction (69). In mammalian cells, mTORC1 phosphorylates Ser758 (Ser757 in mouse) of ULK1, preventing the interaction and phosphorylation of ULK1 by AMPK, which is essential for ULK1 activation (70). Thus, the initiation of autophagy by ULK is reciprocally regulated by mTORC1 and AMPK in response to dynamic changes in cellular nutrients and energy levels. In addition, another layer of ULK1 regulation by mTORC1 has been suggested in which mTORC1 inhibits ULK1 stability by inhibitory phosphorylation of autophagy/beclin 1 regulator 1 (AMBRA1) (71). Furthermore, AMPK and mTORC1 also regulate the VPS34 complex, a class III PI3K whose activity is crucial for autophagosome formation. VPS34 forms multiple complexes and has critical roles in cellular vesicle trafficking and autophagy induction. The ATG14L-associated VPS34 complex is specifically involved in autophagy regulation. In response to nutrient stress, AMPK activates the proautophagy VPS34 complex by phosphorylating Beclin 1, whereas it simultaneously inhibits the nonautophagy VPS34 complex via phosphorylation of Thr163/Ser165 in VPS34 (72). In contrast, mTORC1 phosphorylates ATG14L in the VPS34 complex and inhibits the lipid kinase activity of VPS34, providing another mTORC1-mediated mechanism in autophagy inhibition (73).
Figure 2. Regulation of autophagy by mTORC1.
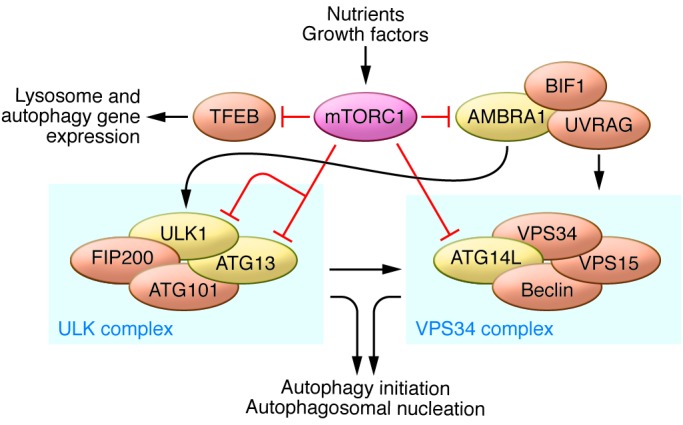
Activation of mTORC1 by nutrients and growth factors leads to inhibition of autophagy through the phosphorylation of multiple autophagy-related proteins, such as ULK1, ATG13, AMBRA1, and ATG14L, which promote autophagy initiation and autophagosome nucleation. mTORC1 also phosphorylates and prevents nuclear localization of the transcription factor TFEB, a master regulator of lysosomal and autophagy gene expression. Proper lysosome function is essential for autophagy completion.
mTORC1 also regulates autophagy at the transcriptional level by modulating localization of transcription factor EB (TFEB), a master transcriptional regulator of lysosomal and autophagy genes (reviewed in ref. 74). The transcriptional activity of TFEB is regulated by nutrient and phosphorylation-dependent cytoplasm-to-nucleus shuttling (75). Although other kinases may also phosphorylate TFEB, it has been shown that mTORC1 directly phosphorylates TFEB at Ser142 and Ser211, and these phosphorylation events result in cytoplasmic sequestration of TFEB (76, 77). As a key signal transducer of amino acids, the Rag GTPases can bind and sequester TFEB in the lysosome, thereby inhibiting TFEB activity (78). Thus, TFEB is constitutively activated regardless of nutrient availability in RagA and RagB deficient cells (47). In conclusion, mTORC1 coordinates both anabolism and catabolism to meet the needs of cell growth. In growing cells, high mTORC1 activity promotes biomolecule synthesis and simultaneously inhibits autophagy. mTORC1 tightly regulates autophagy by suppressing autophagy induction via phosphorylation-dependent inhibition of ULK1/2 and the VPS34 complex and by preventing global expression of lysosomal and autophagy genes through TFEB phosphorylation.
Autophagy flux denotes the sum total of autophagic molecular events, from the induction of autophagy and autophagosome formation to the autolysosomal degradation and reformation of lysosome. Interestingly, although mTORC1 is inactivated during autophagy initiation, the kinase complex is reactivated by energy supplies generated by the degradation of autolysosomal products at the end of autophagy flux. Its reactivation is required for the reformation of functional lysosomes, indicating the critical role of mTORC1 in the completion of autophagy flux (79).
As discussed above, it is not fully understood how mTORC2 activity is regulated. However, it was proposed that insulin/PI3K signaling activates mTORC2 by promoting its interaction with ribosome, and subsequently mTORC2 phosphorylates AKT at the turn motif site, Thr450, during translation (48, 80). In addition, mTORC2 phosphorylates AKT at the hydrophobic motif site, Ser473, which can lead to the activation of the AKT/mTORC1 signaling axis. Therefore, mTORC2 may indirectly suppress autophagy by activating mTORC1. Further studies are needed to determine whether mTORC2 can directly regulate autophagy.
Pharmacologic regulation of mTOR and autophagy
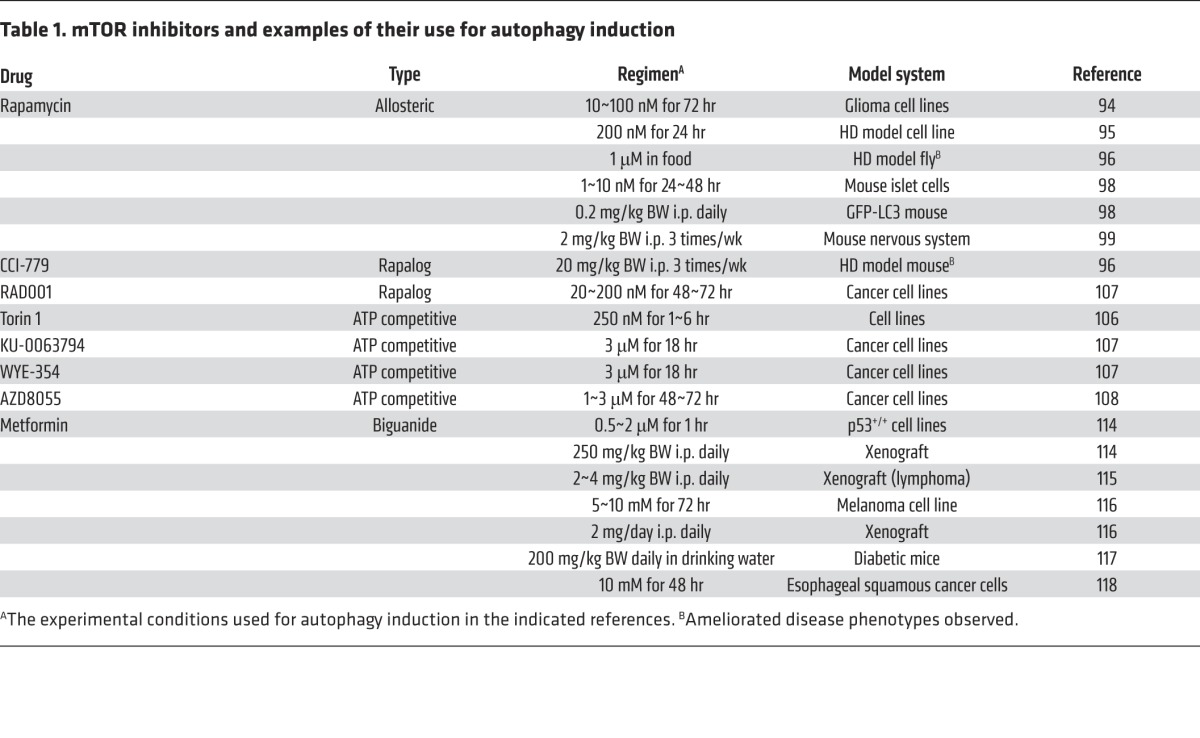
Autophagy is a cellular process essential for development and tissue homeostasis. Autophagy is implicated in various physiologic and pathologic processes (including exercise, metabolic adaptation, and disorders such as neurodegenerative diseases, infectious diseases, cardiovascular diseases, cancer, and aging), and thus pharmacologic modulation of autophagy is of great interest (reviewed in refs. 81, 82). As a master regulator of cellular metabolism and autophagy, mTORC1 is an appealing pharmacologic target to manipulate autophagy. In fact, deregulation of mTORC1 has been implicated in diseases that are associated with autophagy defects (1), and there are mTOR inhibitors already in clinical trials or approved for treatment of these diseases (reviewed in refs. 83–86). There are also pharmacologic molecules that can induce or inhibit autophagy via mTOR-independent mechanisms (81). For example, agents such as bafilomycin A1 and hydroxychloroquine that increase lysosomal pH can block autophagy flux by inhibiting autolysosomal formation. Such inhibitors could be combined with mTOR inhibitors to finely modulate autophagy flux. In this section we discuss currently available mTOR inhibitors and their effects on autophagy. We also summarize some mTOR inhibitors and their use for autophagy induction in preclinical studies (Table 1).
Table 1.
mTOR inhibitors and examples of their use for autophagy induction
Rapamycin and rapalogs.
Rapamycin was originally isolated from the soil bacterium Streptomyces hygroscopicus as an antifungal compound in 1975, and was later shown to be a strong immunosuppressant with broad anti-proliferative effects in mammalian cells (reviewed in ref. 84). About 16 years after the isolation of rapamycin, an elegant yeast genetic screen to identify rapamycin-resistant genes led to the discovery of TOR1 and TOR2 (87). mTOR was identified in the mid-1990s through biochemical isolation using a rapamycin-FKBP12 complex and by yeast two-hybrid analysis (88–91). Rapamycin forms a complex with FKB12 in the cell, and this complex specifically binds to mTORC1 and allosterically inhibits its kinase activity. Although the rapamycin-FKB12 complex does not bind directly to mTORC2, it has been reported that long-term incubation with rapamycin can decrease mTORC2 activity (reviewed in ref. 92). Because of its anti-proliferation effects, rapamycin has been thoroughly evaluated as a therapeutic drug for cancer. In addition, rapamycin has been used as an immunosuppressant for organ transplantation and a cell growth inhibitor for preventing restenosis. In order to improve the pharmacokinetics of rapamycin, several derivative compounds (RAD001, CCI-779, and AP23573, collectively called rapalogs) have been developed. These rapalogs have a similar ability to inhibit mTORC1 with fewer immunosuppressive effects (93).
Induction of autophagy by rapamycin or rapalogs has been tested in various model systems. Treatment of rapamycin strongly induces autophagy in yeast even in the presence of nutrients (62); however, the effectiveness of rapamycin on inducing autophagy in mammalian cells is dependent on the cell type. Thus, in a panel of glioma cell lines, rapamycin effectively induces autophagy in U87-MG and T98G cells but is not sufficient to induce autophagy in U373-MG cells, although rapamycin in combination with a PI3K or AKT inhibitor sensitizes the cells to autophagy induction (94). In contrast, rapamycin or CCI-779 treatment reduces protein aggregates through autophagy induction, thereby ameliorating symptoms of Huntington’s disease (HD) in both an in vitro cell culture model and in vivo fly and mouse models (95, 96). In addition, there are reports suggesting that rapamycin-induced autophagy may sensitize cancer cells to radiotherapy. Treatment of PTEN-null prostate cancer cells with the rapamycin derivative RAD001 (also called everolimus) induces autophagy and sensitizes the cells to radiotherapy. Moreover, prolonging autophagy with rapamycin causes radioresistant cancer cells to enter senescence and inhibits the growth of cancer cells in a xenograft model (97). Furthermore, through the use of in vivo reporter systems, rapamycin treatment has been shown to induce autophagy in both the nervous system and pancreatic β cells of mice (98, 99). Although further studies are necessary to determine whether the beneficial effects seen in these model systems can be attributed to increased autophagy, these studies demonstrate that rapamycin or rapalogs can induce autophagy in vitro and in vivo. Of note, a study using a mouse model of TSC showed that autophagy is prosurvival for TSC tumorigenesis, suggesting that rapamycin or rapalog treatment is not effective, but autophagy inhibition is beneficial for the treatment of TSC (100). Additionally, the rapalog RAD001 has been approved by FDA for the treatment of certain cancers such as subependymal giant cell astrocytoma, advanced hormone receptor–positive and HER2-negative breast cancer, progressive neuroendocrine tumors of pancreatic origin, and renal cell carcinoma (101).
ATP-competitive mTOR inhibitors (mTOR kinase inhibitors).
Despite the high expectations for rapamycin and rapalogs as anticancer agents, the outcomes from clinical trials have not been uniformly positive except for certain types of cancers such as renal cell carcinoma and TSC-associated angiomyolipoma (83, 84). This is possibly due in part to the loss of the negative feedback circuit from mTORC1 to the PI3K/AKT/mTORC2 signaling axis, which mediates cell survival. Thus, mTORC1 inhibition by rapamycin treatment may lead to activation of PI3K/AKT/mTORC2 signaling in cancer cells. Intriguingly, one report suggests that mTORC2 activity is essential for development and survival of prostate cancer cells, but not for normal prostate epithelium (102). Furthermore, rapamycin has differential effects on S6K and eukaryotic initiation factor 4E binding protein 1 (4E-BP1), two major mTORC1 substrates that regulate protein synthesis (103). Additionally, a recent study showed that the sequence composition of substrates determines the substrate quality of mTORC1, thereby dictating the substrate’s sensitivity to rapamycin (104). Phosphorylation of poor mTORC1 substrates is more sensitive to inhibition by rapamycin than is phosphorylation of good mTORC1 substrates. Interestingly, the autophagy-related mTORC1 substrates TFEB Ser142 and ULK1 Ser758 are less sensitive to rapamycin than the mTORC1 substrate S6K (104), which highlights the need for other types of mTORC1 inhibitors to efficiently regulate autophagy. For these reasons, several groups have independently developed mTOR inhibitors that directly target the kinase activity of both mTORC1 and mTORC2 (83–85, 105). These inhibitors (listed in Table 1) are ATP analogs, collectively called ATP-competitive mTOR inhibitors or mTOR kinase inhibitors (mTOR-KIs). Some of these inhibitors have dual inhibitory effects on mTOR and PI3K due to the similarity of their kinase domain structures (105).
As anticipated, treatment with mTOR-KIs induces autophagy and cytotoxicity in various cell types. The mTOR-KI torin 1 blocks the phosphorylation of all mTORC1 substrates more efficiently than does rapamycin, and as a result torin 1 elicits stronger autophagy induction in both mouse and human cell lines (106). Consistently, the mTOR-KIs Ku-0063794 and WYE-354 induce autophagy in rapamycin-resistant cancer cells, and a subefficacious dose of mTOR-KIs in combination with rapamycin can synergistically inhibit mTORC1 to induce autophagy (107). Additionally, another mTOR-KI, AZD8055, potently induces autophagy and inhibits proliferation in a broad range of cancer cell lines (108). Interestingly, it has been shown that AZD8055 treatment can be cytoprotective via autophagy induction in a cytotoxic chemotherapy setting (109). Some of these mTOR-KIs are undergoing or recently completed phase I or II clinical trials as anticancer agents (ClinicalTrials.gov identifiers NCT01316809, NCT00973076, NCT00999882, NCT00731263, NCT01545947, and NCT01177397.
Metformin.
Metformin is a biguanide that is widely used for the treatment of type II diabetes. Metformin activates AMPK indirectly by inhibiting the mitochondrial respiratory chain complex I, thus increasing the cellular AMP/ATP ratio (110). As discussed above, AMPK is an upstream negative regulator of mTORC1. Thus, metformin treatment inhibits mTORC1-mediated protein synthesis in breast cancer cells (111). It has also been shown that metformin can inhibit mTORC1 in an AMPK-independent manner, possibly via Rag GTPases or REDD1 (112, 113). Therefore, since active AMPK promotes autophagy by phosphorylating key autophagy regulatory proteins such as ULK1, VPS34, and Beclin 1 (70, 72), metformin can induce autophagy by simultaneously activating AMPK and inhibiting mTORC1. Consequently, metformin-induced autophagy and its beneficial effects have been demonstrated in various cancer cells in vitro as well as in in vivo models (114–118).
Conclusion
mTOR promotes anabolic metabolism and inhibits autophagy induction. Therefore, the regulation of autophagy with mTOR inhibitors provides a new therapeutic strategy for a variety of diseases, including neurodegenerative diseases, diabetes, and cancer. Most available mTOR inhibitors that have been rigorously tested for clinical uses are rapamycin derivatives, and the majority of these tests have been focused on their anti-proliferation effects for cancer treatment (84). These compounds must be further evaluated in autophagy-related diseases such as neurodegeneration and cardiac myopathy, which are often associated with lysosomal and autophagy defects (82). One critical factor that must be considered is the potential side effects of mTOR inhibitors. mTOR-KIs are cytotoxic, likely due to the inhibition of mTORC2, whereas rapamycin is generally cytostatic with less toxicity (83). It might be advantageous to use mTOR-KIs for cancer treatment but not for chronic diseases such as neurodegeneration. Therefore, in the treatment of neurodegenerative or metabolic diseases, rapalogs are probably more desirable, as they have fewer side effects. Both rapalogs and mTOR-KIs have immunosuppressive effects that could also limit their potential application. Further pharmacokinetic studies are needed to determine the effective doses of mTOR inhibitors for inducing autophagy with minimal side effects.
Autophagy induction could be beneficial or detrimental depending on the type or stage of disease (82). For example, autophagy may promote survival during tumor initiation and development by providing nutrients to tumor cells when nutrients are limited. Thus inhibition of autophagy may sensitize cancer cells to metabolic stress conditions, leading to cell death. Autophagy can have growth-suppressive functions, and defects in autophagy may enhance genomic instability and promote cancer development. Given the potential dual functions of autophagy in tumor suppression and promotion, more studies are needed to elucidate the precise function of autophagy in individual cancer types before a therapeutic approach can be considered. In-depth discussions regarding the role of autophagy in tumor development and its implications in tumor therapy may be found in other articles in this Review series (119–121). For degenerative diseases, a mild induction of autophagy should protect cells from damaged proteins and organelles; thus, partial mTORC1 inhibition rather than a complete inhibition may be a possible therapeutic strategy.
Acknowledgments
We thank all members of the Guan lab for their discussion, and especially thank Steven Plouffe for critical reading of this manuscript. This work is supported by grants from the NIH (GM51586 and CA18941).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(1):25–32. doi:10.1172/JCI73939.
References
- 1.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim DH, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11(4):895–904. doi: 10.1016/S1097-2765(03)00114-X. [DOI] [PubMed] [Google Scholar]
- 3.Jacinto E, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 4.Peterson TR, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaizuka T, et al. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem. 2010;285(26):20109–20116. doi: 10.1074/jbc.M110.121699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hara K, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–189. doi: 10.1016/S0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 7.Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 8.Sancak Y, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25(6):903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 10.Thedieck K, et al. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One. 2007;2(11):e1217. doi: 10.1371/journal.pone.0001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Harris TE, Roth RA, Lawrence JC., Jr PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007;282(27):20036–20044. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 12.Sarbassov DD, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 13.Jacinto E, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127(1):125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 14.Frias MA, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16(18):1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Pearce LR, et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405(3):513–522. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20(20):2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15(3):155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- 18.Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126(pt 8):1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 20.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–162. doi: 10.1016/S1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 21.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4(9):658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 22.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13(15):1259–1268. doi: 10.1016/S0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 24.Menon S, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156(4):771–785. doi: 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5(6):566–571. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- 26.Stocker H, et al. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5(6):559–565. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- 27.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004;101(37):13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121(2):179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 29.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 30.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brugarolas J, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18(23):2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22(2):239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14(3):133–139. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24(7):400–406. doi: 10.1016/j.tcb.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10(8):935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J Biol Chem. 2001;276(10):7246–7257. doi: 10.1074/jbc.M004389200. [DOI] [PubMed] [Google Scholar]
- 38.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thedieck K, et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell. 2013;154(4):859–874. doi: 10.1016/j.cell.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat Cell Biol. 2013;15(10):1186–1196. doi: 10.1038/ncb2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150(6):1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bar-Peled L, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340(6136):1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsun ZY, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013;52(4):495–505. doi: 10.1016/j.molcel.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334(6056):678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han JM, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149(2):410–424. doi: 10.1016/j.cell.2012.02.044. [DOI] [PubMed] [Google Scholar]
- 46.Efeyan A, et al. RagA, but not RagB, is essential for embryonic development and adult mice. Dev Cell. 2014;29(3):321–329. doi: 10.1016/j.devcel.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim YC, et al. Rag GTPases are cardioprotective by regulating lysosomal function. Nat Commun. 2014;5:4241. doi: 10.1038/ncomms5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144(5):757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 49.Cybulski N, Polak P, Auwerx J, Ruegg MA, Hall MN. mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proc Natl Acad Sci U S A. 2009;106(24):9902–9907. doi: 10.1073/pnas.0811321106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 51.Harrington LS, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Um SH, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431(7005):200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 53.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30(4):908–921. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol. 2006;26(1):63–76. doi: 10.1128/MCB.26.1.63-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsu PP, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332(6035):1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu Y, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332(6035):1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 58.Clark SL., Jr Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J Biophys Biochem Cytol. 1957;3(3):349–362. doi: 10.1083/jcb.3.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 60.Mitchener JS, Shelburne JD, Bradford WD, Hawkins HK. Cellular autophagocytosis induced by deprivation of serum and amino acids in HeLa cells. Am J Pathol. 1976;83(3):485–492. [PMC free article] [PubMed] [Google Scholar]
- 61.Mortimore GE, Schworer CM. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature. 1977;270(5633):174–176. doi: 10.1038/270174a0. [DOI] [PubMed] [Google Scholar]
- 62.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273(7):3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 63.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7(2):167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 64.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014;24(1):42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jung CH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150(6):1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nazio F, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15(4):406–416. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 72.Kim J, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy. 2013;9(12):1983–1995. doi: 10.4161/auto.26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Settembre C, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Settembre C, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martina JA, Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 2013;200(4):475–491. doi: 10.1083/jcb.201209135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oh WJ, et al. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010;29(23):3939–3951. doi: 10.1038/emboj.2010.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 83.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121(4):1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10(11):868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 85.Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 2013;123(3):980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19(3):373–379. doi: 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 88.Brown EJ, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369(6483):756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 89.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78(1):35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 90.Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci U S A. 1994;91(26):12574–12578. doi: 10.1073/pnas.91.26.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sabers CJ, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270(2):815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- 92.Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2010;29(26):3733–3744. doi: 10.1038/onc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5(8):671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 94.Takeuchi H, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65(8):3336–3346. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 95.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11(9):1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 96.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 97.Nam HY, Han MW, Chang HW, Kim SY, Kim SW. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy. 2013;9(10):1631–1632. doi: 10.4161/auto.25879. [DOI] [PubMed] [Google Scholar]
- 98.Tanemura M, et al. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am J Transplant. 2012;12(1):102–114. doi: 10.1111/j.1600-6143.2011.03771.x. [DOI] [PubMed] [Google Scholar]
- 99.Castillo K, et al. Measurement of autophagy flux in the nervous system in vivo. Cell Death Dis. 2013;4:e917. doi: 10.1038/cddis.2013.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Parkhitko A, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A. 2011;108(30):12455–12460. doi: 10.1073/pnas.1104361108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pazdur R. FDA Approval for Everolimus. National Cancer Institute Web site. http://www.cancer.gov/cancertopics/druginfo/fda-everolimus Updated July 3, 2013. Accessed September 26, 2014
- 102.Guertin DA, et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15(2):148–159. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105(45):17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kang SA, et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013;341(6144):1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2(67):pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 106.Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nyfeler B, et al. Relieving autophagy and 4EBP1 from rapamycin resistance. Mol Cell Biol. 2011;31(14):2867–2876. doi: 10.1128/MCB.05430-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chresta CM, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70(1):288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 109.Huang S, Yang ZJ, Yu C, Sinicrope FA. Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J Biol Chem. 2011;286(46):40002–40012. doi: 10.1074/jbc.M111.297432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hardie DG. Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett. 2008;582(1):81–89. doi: 10.1016/j.febslet.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 111.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 112.Kalender A, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11(5):390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ben Sahra I, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71(13):4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 114.Buzzai M, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67(14):6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 115.Shi WY, et al. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012;3:e275. doi: 10.1038/cddis.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tomic T, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011;2:e199. doi: 10.1038/cddis.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xie Z, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60(6):1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Feng Y, et al. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 2014;5:e1088. doi: 10.1038/cddis.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):5564–5568. doi: 10.1172/JCI73941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125(1):5536–5546. doi: 10.1172/JCI73938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vakifahmetoglu-Norberg H, Xia H, Yuan J. Pharmacologic agents targeting autophagy. J Clin Invest. 2015;125(1):5527–5535. doi: 10.1172/JCI73937. [DOI] [PMC free article] [PubMed] [Google Scholar]