

Abstract
Defects in autophagy have been linked to a wide range of medical illnesses, including cancer as well as infectious, neurodegenerative, inflammatory, and metabolic diseases. These observations have led to the hypothesis that autophagy inducers may prevent or treat certain clinical conditions. Lifestyle and nutritional factors, such as exercise and caloric restriction, may exert their known health benefits through the autophagy pathway. Several currently available FDA-approved drugs have been shown to enhance autophagy, and this autophagy-enhancing action may be repurposed for use in novel clinical indications. The development of new drugs that are designed to be more selective inducers of autophagy function in target organs is expected to maximize clinical benefits while minimizing toxicity. This Review summarizes the rationale and current approaches for developing autophagy inducers in medicine, the factors to be considered in defining disease targets for such therapy, and the potential benefits of such treatment for human health.
The past two decades have witnessed an explosion of research on the molecular mechanisms of autophagy and its roles in physiology and disease. Numerous gene products essential for the induction of autophagy, the formation of autophagosomes, the lysosomal clearance of autophagosomes, and the targeting of specific cargo to the autophagosomes have been identified (1–3). The biochemical and structural mechanisms by which these gene products act in an orchestrated manner to execute autophagy are being defined (4). Through loss-of-function studies in mice and other model organisms, we have learned that autophagy plays crucial roles in differentiation and development (5, 6), cellular and tissue homeostasis (7), protein and organelle quality control (8), metabolism (9), immunity (10), and protection against aging (11) and diverse diseases (refs. 12, 13, and Table 1; supplemental references available online with this article; doi:10.1172/JCI73938DS1). Moreover, an increasing number of human diseases are being linked to polymorphisms or mutations in autophagy genes (Table 2) or deficiencies in autophagy function (14, 15). Based on these advances, considerable interest has emerged in developing new (or exploiting old) strategies to induce autophagy — through either pharmacologic or non-pharmacologic approaches. In this Review we provide an overview of the rationale, potential disease targets, and current efforts and future challenges for the development of autophagy inducers in clinical medicine. Other Reviews in this issue discuss the potential use of autophagy inhibitors in clinical medicine (16–18).
Table 1.
Diseases in mice with mutations in autophagy genes
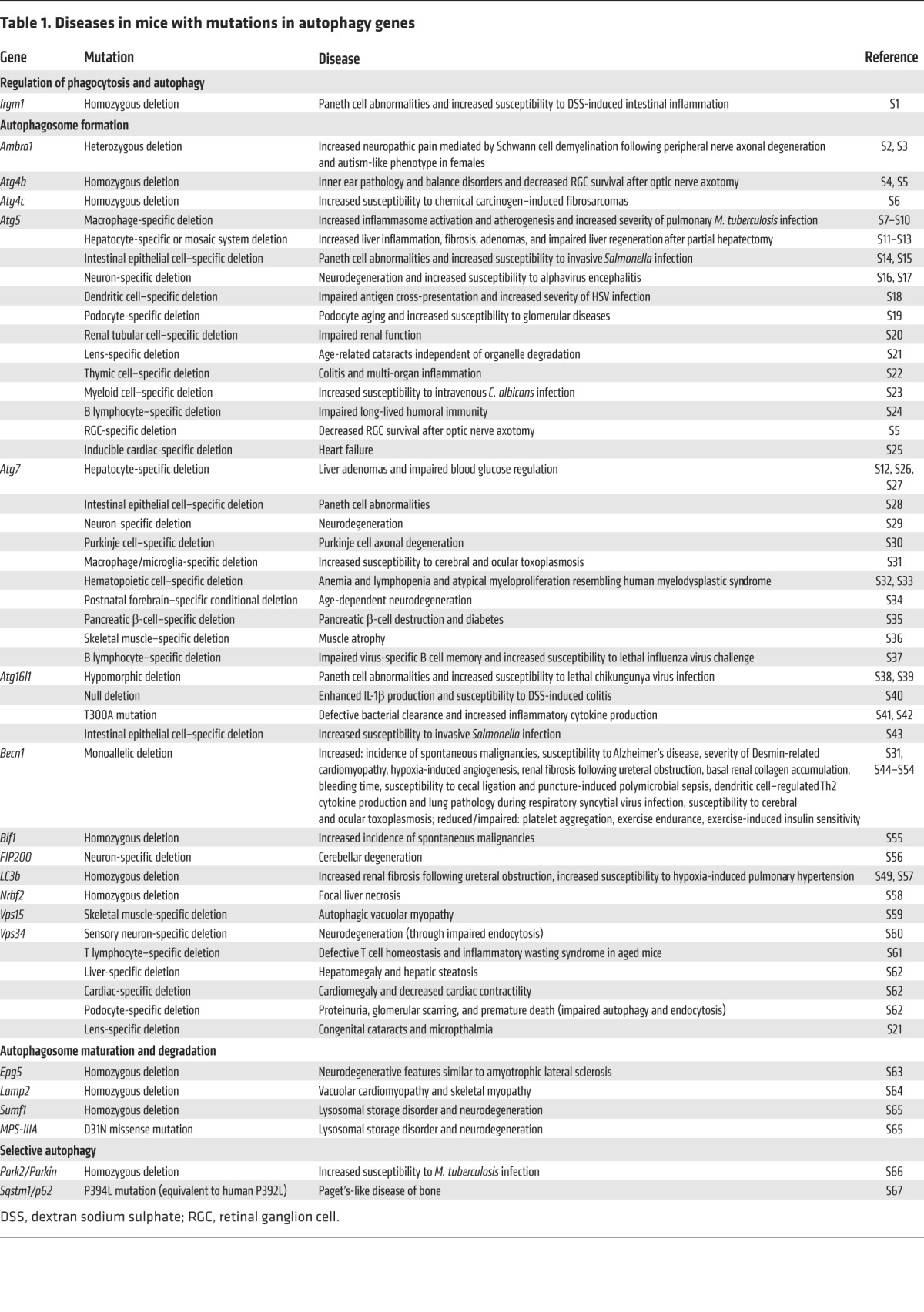
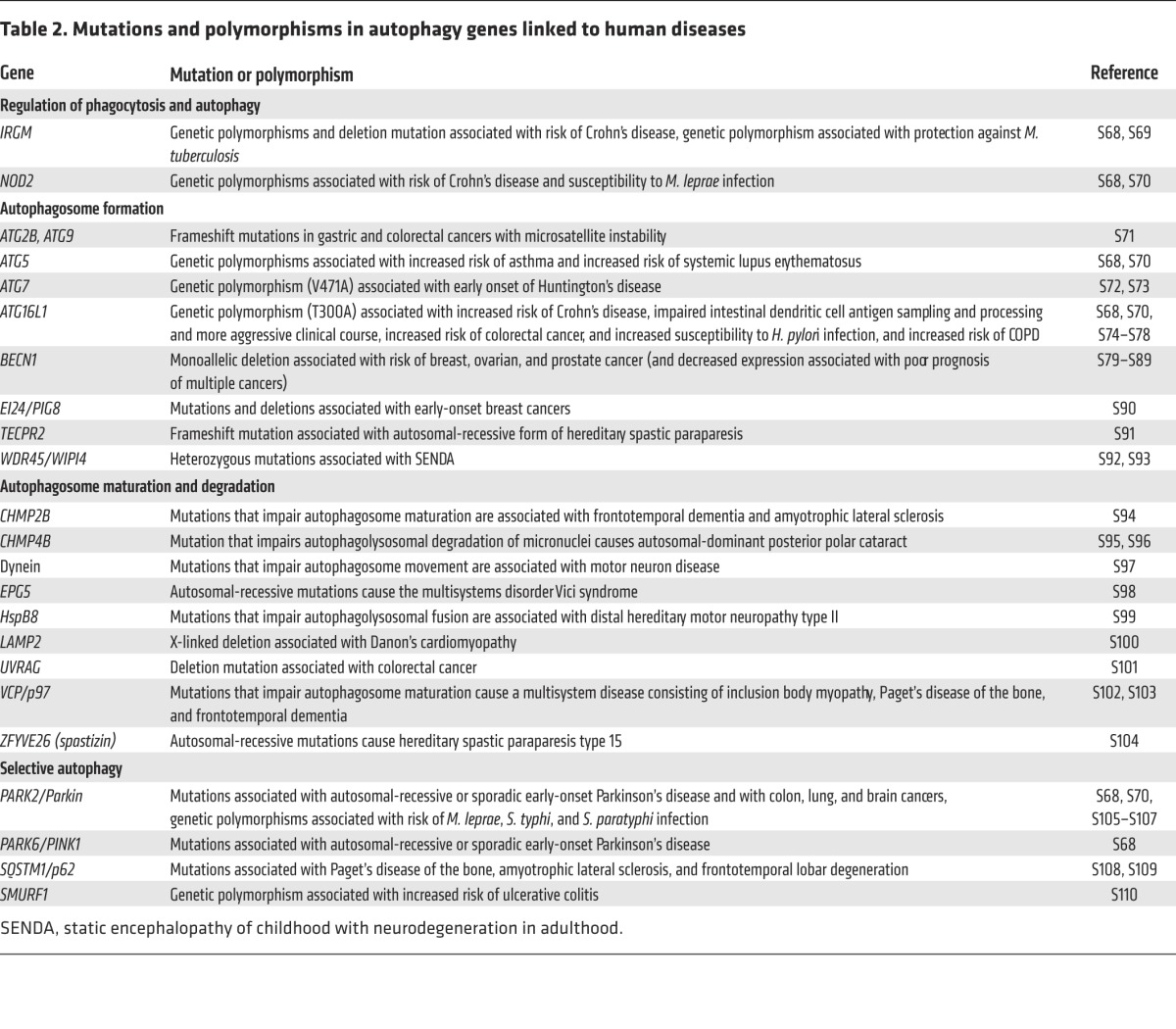
Table 2.
Mutations and polymorphisms in autophagy genes linked to human diseases
Rationale for the development of autophagy inducers
The macroautophagy form of autophagy (herein referred to as autophagy) is an evolutionarily conserved lysosomal degradation pathway that controls cellular bioenergetics (by recycling cytoplasmic constituents) and cytoplasmic quality (by eliminating protein aggregates, damaged organelles, lipid droplets, and intracellular pathogens) (8). In addition, independently of lysosomal degradation, the autophagic machinery can be deployed in the process of phagocytosis, apoptotic corpse clearance, entosis, secretion, exocytosis, antigen presentation, and regulation of inflammatory signaling (7). As a result of the broad range of cellular functions, the autophagy pathway plays a key role in protection against aging and certain cancers, infections, neurodegenerative disorders, metabolic diseases, inflammatory diseases, and muscle diseases (refs. 12, 13, 19–21, and Figure 1).
Figure 1. Schematic illustration of potential mechanisms by which autophagy-inducing agents may exert beneficial effects in clinical disease.

The precise mechanisms underlying the beneficial effects of autophagy upregulation in preventing or treating different diseases are not fully understood, and multiple different functions of the autophagy pathway or of specific autophagy proteins (acting independently of autophagy) are likely to be contributory. The mechanisms and target diseases shown are representative examples based on animal studies and human genetic data (see Tables 1–3). Other functions of autophagy and autophagy proteins not depicted here may be important, and other diseases not listed here may be targets for autophagy induction.
The recognition that autophagy may prevent the occurrence, delay the progression, and/or decrease the severity of certain diseases provides the primary rationale for the development of pharmacologic agents that induce or enhance autophagy. Several lines of evidence support this approach. First, genetic mutations in autophagy genes in mice (either systemic homozygous or heterozygous deletion, tissue-specific deletion, or knock-in mutations of mutant alleles that are found in human diseases) results in a wide spectrum of disorders (see Table 1) including increased susceptibility to neurodegeneration, cancer, atherosclerosis, diabetes, bone disease, intracellular bacterial infections (e.g., Mycobacterium tuberculosis, Salmonella), and Paneth cell abnormalities associated with Crohn’s disease. Second, mutations or polymorphisms in autophagy genes are associated with susceptibility to human diseases (see Table 2), including Parkinson’s disease, inflammatory bowel disease, breast and other malignancies, mycobacterial infections, asthma, chronic obstructive pulmonary disease (COPD), systemic lupus erythematosus, and hereditary neurologic disorders. Third, autophagy gene therapy (via lentiviral or adenovirus-associated viral delivery) in specific target organs results in clinical improvement in rodent models of obesity, α1-antitrypsin deficiency, Parkinson’s disease, Alzheimer’s disease, Pompe disease, muscular dystrophy, cystic fibrosis, and KRAS-driven lung carcinomas, and systemic transgenic expression of an autophagy gene in mice extends lifespan and improves metabolism (Table 3). Fourth, several commonly used drugs and nutritional supplements induce autophagy (Table 4). Although it is generally unknown whether these agents exert their clinical benefits through autophagy or other pathways, there is considerable overlap between diseases that occur in the setting of autophagy deficiency and diseases that respond to drugs that can induce autophagy. Moreover, some of these agents fail to exert beneficial effects in model organisms lacking autophagy genes. For example, spermidine and resveratrol extend life span in wild-type but not autophagy gene–deficient nematodes (22). Similarly, tyrosine kinase inhibitors do not improve amyloid clearance in the brains of mice with Alzheimer’s-like disease when the essential autophagy gene, beclin 1 (Becn1), is depleted by shRNA-mediated knockdown (23).
Table 3.
Rodent studies demonstrating beneficial clinical outcomes with autophagy gene transgenic expression, autophagy gene therapy, or autophagy-inducing peptides

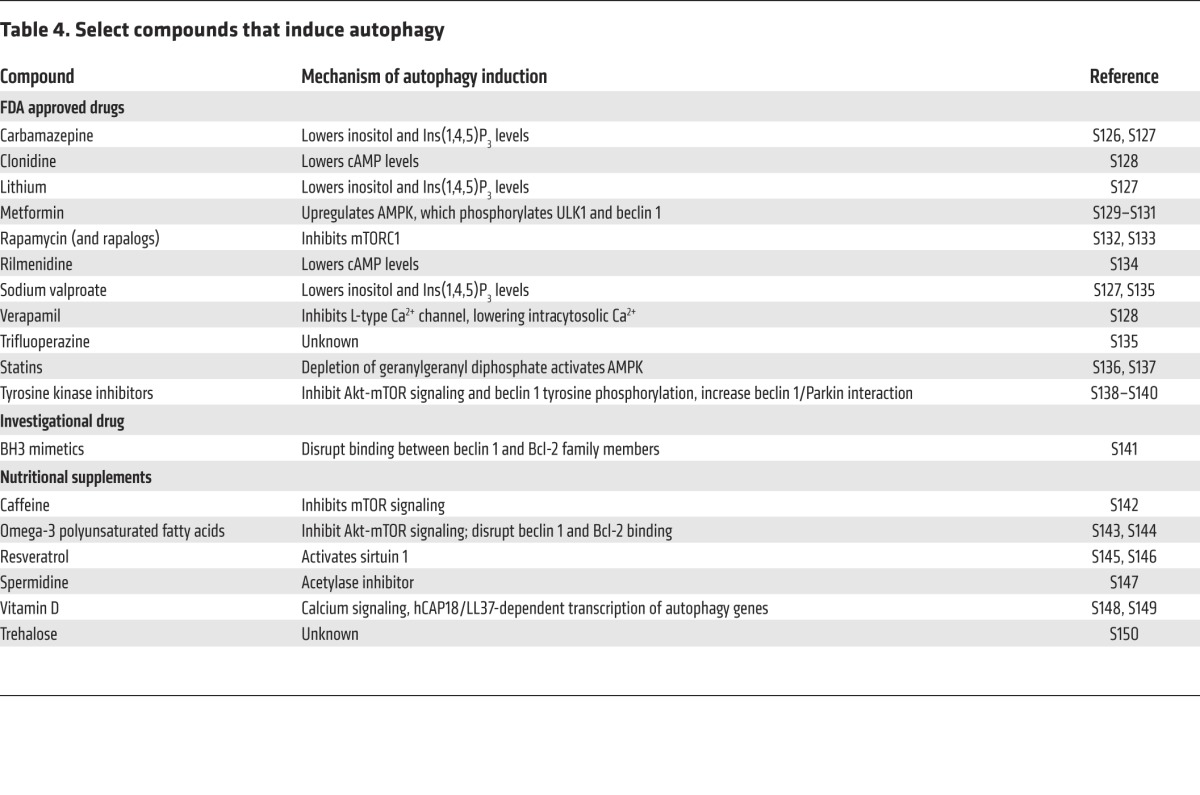
Table 4.
Select compounds that induce autophagy
Factors to consider in defining disease targets for autophagy induction
Considerable enthusiasm has emerged for the development of autophagy-inducing agents for the prevention or treatment of diseases in which the upregulation of autophagy is thought to be clinically beneficial. The spectrum of potential disease targets is broad and has been reviewed extensively elsewhere (9, 11, 24, 25); the major focus has been on neurodegenerative disorders, infectious diseases, aging, and metabolic diseases. A common underlying pathophysiologic event in these diseases is the accumulation of harmful contents inside the cell — damaged organelles, protein aggregates, lipid droplets, or pathogens. In these circumstances, the pharmacologic (or non-pharmacologic) enhancement of autophagy-mediated delivery of deleterious structures for lysosomal destruction may be beneficial (Figure 1).
For such manipulations to have therapeutic value, the machinery involved in autophagosome formation, cargo recognition and targeting, or autophagic delivery to lysosomes should not be rate limiting. If polymorphisms in autophagy genes associated with autophagosomal formation were to cause disease via loss of autophagy (which has not yet been determined for the mutations listed in Table 2), it is possible that such mutations could also block the successful upregulation of autophagy by agents that would otherwise induce autophagy in wild-type cells with an intact autophagy machinery. Similarly, it is not known whether patients with mutations in factors involved in autophagic cargo recognition and targeting (Table 2) (such as PARK2 and PINK1 mutations, which occur in Parkinson’s disease, or SQSTM1/p62 mutation, which occurs in Paget’s disease of the bone) will respond to autophagy inducers. Furthermore, several diseases involve an impairment of the delivery of autophagosomes to lysosomes, including human motor neuron disease associated with mutations in the dynein apparatus (26), lysosomal storage diseases (27), and familial Alzheimer’s disease caused by presenilin 1 mutations (28). In these cases, increasing autophagosomal membrane formation will not necessarily enhance autophagic substrate degradation and may result in a toxic buildup of cellular membranes, polyubiquitinated aggregates, and dysfunctional mitochondria (29). One way to enhance successful autophagic substrate degradation in the setting of lysosomal dysfunction may be to upregulate transcription factor EB (TFEB), a master regulator of both autophagy gene expression and lysosomal biogenesis (30). As a proof of principle, Tfeb gene therapy decreases glycogen storage and excess accumulation of autophagosomes in a murine model of the lysosomal storage disease, Pompe disease (31). However, if the defects in autophagy (at the stage of induction, autophagosome formation, cargo targeting, or autophagolysosomal maturation) are only partial, diseases may still benefit from pharmacologic upregulation of autophagosome formation. Indeed, autophagy-enhancing agents show beneficial effects in induced pluripotent stem cells from patients with the lysosomal storage disorder Niemann-Pick type C disease (32) and in NPC1 mutant mouse cells (33). Further studies are warranted to examine the effects of autophagy inducers in other lysosomal storage disorders and diseases associated with impaired cargo delivery to the autophagosome.
An interesting question is whether autophagy induction is warranted in clinical conditions without an apparent defect in the autophagic machinery. Drugs that induce autophagy in autophagy-competent animals have favorable effects in diseases characterized by abnormal accumulation of protein substrates (such as Huntingtin’s disease treated with rapamycin [ref. 34] or rilmenidine [ref. 35], and α1-antitryspin deficiency treated with carbamazepine [ref. 36]) or pathogens (such as arboviral infections treated with the autophagy-inducing peptide Tat–beclin 1 [ref. 37] and pulmonary M. tuberculosis infection treated with statins [ref. 38]), suggesting that enhancement of autophagy may be beneficial in the absence of an overt autophagy deficiency. Unfortunately, given our current limitations in measuring autophagic flux in patients, we do not know what constitutes a “normal” range of autophagic activity. However, autophagy function declines with aging in humans and other species, and such a decline likely contributes to aging itself as well as age-related increases in susceptibility to neurodegenerative disorders, infectious diseases, and cancer (11). Preliminary studies also indicate that critically ill patients have an autophagy-deficient phenotype, at least in skeletal muscle and liver (39). Thus, many patients, by virtue of advanced age or severe illness, may have a deficiency in autophagy function in the absence of specific mutations in the autophagy pathway.
In defining disease targets appropriate for autophagy induction therapy, it is important to consider whether autophagy acts in a cytoprotective or cytotoxic manner in the specific disease context, and whether such cytoprotective or cytotoxic actions contribute to disease progression. Although autophagy is classically regarded as a means of promoting cell survival during nutrient-limited conditions (5, 40), autophagy can also contribute to cell death (41).
The pro-survival role of autophagy is commonly believed to promote the progression of cancers driven by RAS mutations (42, 43), which has led to intense efforts to inhibit autophagy in this context (discussed in other Reviews in this issue and in refs. 44, 45). However, this postulated pro-survival action is not necessarily sufficient to override the tumor suppressor effects of autophagy in all cancers. For example, enhanced suppression of autophagy in EGFR-driven non–small cell lung adenocarcinoma xenografts increases tumor cell death but also promotes enhanced proliferation, increased tumor growth and tumor dedifferentiation, as well as resistance to EGFR tyrosine kinase inhibitor therapy (46). Even in RAS-driven tumor cells, autophagy inhibition does not have predictable antitumor effects. Specifically, in RAS-driven oncogenesis, autophagy gene knockdown enhances clonogenic survival in human ovarian epithelial cells (47); the presence of a homozygous p53 mutation transforms the actions of autophagy from a pro-tumorigenic to anti-tumorigenic effect in pancreatic carcinoma (48); autophagy suppresses early oncogenesis in lung adenocarcinoma through effects on regulatory T cells (49); and beclin 1 gene transfer prevents the progression from lung adenomas to adenocarcinomas and enhances tumor cell death (50).
Moreover, autophagy genes are often required for the cytotoxic effects of chemotherapy (51–53), and the combination of antimetabolite and receptor tyrosine kinase inhibitor therapy can increase autophagy-dependent tumor cell death (54). Autophagy also contributes to radiosensitivity in vivo (through immune-dependent responses) even if it contributes to radioresistance in vitro (55). In addition, multiple myeloma cells utilize caspase-10 to restrain autophagy and undergo autophagic cell death following caspase-10 inhibition (56). Interestingly, glucose-starved yeast and mammalian cells do not engage in prosurvival autophagy (56, 57), challenging the notion that glucose deprivation (one of the most common forms of metabolic stress in the tumor microenvironment) induces prosurvival autophagy in vivo. Thus, because the role of autophagy in cancer progression and response to therapy is complex and context dependent, it is possible that — despite the prosurvival effects of autophagy in some tumor cells — the induction of autophagy may still be useful in certain cancers through autophagy-dependent antitumor immunity, autophagy-dependent cytotoxic effects, or other tumor-suppressor effects.
While cytotoxic effects of autophagy or autophagy-dependent anticancer immune responses may be beneficial in certain malignancies, the cytotoxic effects of autophagy may be pathogenic in other diseases. These include mouse models of cigarette smoke–induced COPD (58), acute lung injury caused by avian influenza A H5N1 infection (59), diabetes-induced and pressure overload–induced cardiomyopathies (60–62), pancreatic β-cell death in the setting of Pdx1 deficiency (63), ischemic brain damage in diabetes (64), and traumatic, ischemic, ischemic/reperfusion, and/or hypoxic injury in the brain, heart, or kidney of non-diabetic subjects (65–69). Such “pro-death” effects of autophagy (whether they are direct through autophagic cell death, indirect through enhanced apoptosis as postulated in lung epithelial cells exposed to cigarette smoke, or a combination of both) have yet to be confirmed in patients. However, if cigarette smoking or diabetes (or other comorbid conditions) increase susceptibility to autophagy-dependent enhancement of organ pathology in the clinical setting, the coexistence of these common comorbid conditions might affect the safety of utilizing autophagy inducers, particularly if they are not organ specific. Even if present, these unwanted effects could occur at doses higher than or durations longer than those needed to enhance the autophagy-mediated delivery of deleterious structures for lysosomal destruction. This would allow for the identification of a useful therapeutic window and the safe development of low doses or short-term or intermittent treatment strategies.
Besides potential unwanted cytoprotective effects or cytotoxic effects, another possible concern regarding autophagy induction relates to the complex roles of autophagy proteins in infectious diseases, immunity, and inflammation. Autophagy plays important roles in protection against several medically important intracellular pathogens through different mechanisms including xenophagy (the selective autophagic degradation of microbes), enhanced adaptive immunity, and the prevention of excessive inflammatory responses (Figure 1), leading to significant optimism that autophagy inducers may represent an important new class of host-direct anti-infective therapy (10, 70). However, the autophagy pathway and/or autophagy pathway–independent functions of autophagy proteins may also enhance the replication of certain viruses and intracellular bacteria. For example, pancreatic cell–specific knockout of Atg5 dramatically reduces Coxsackie virus replication and virus-induced pathology (71); similarly, liver-specific knockout of Atg5 reduces HBV DNA replication in mice that transgenically express HBV (72). It is not known whether these phenotypes reflect a role for the autophagy pathway or for autophagy pathway–independent effects of Atg5, which can also function in the negative regulation of inflammasomes, recruitment of immunity-related GTPases, secretion, exocytosis, and formation of membranes that serve as scaffolds for viral replication (10, 70). If the mechanism involves autophagy, autophagy inducers might be contraindicated in the setting of certain infections such as HBV. However, if the mechanism involves autophagy-independent functions of Atg5, it is unknown whether the upstream upregulation of autophagic flux will also enhance autophagy-independent pro-pathogen functions of individual autophagy proteins. Suppression of inflammatory responses by autophagy induction might also impair the host capacity to clear infectious organisms.
Studies of the loss of autophagy gene function in B cells and dendritic cells suggests a crucial role for autophagy in antigen-specific immune responses. For example, B cell–specific deletion of Atg7 impairs virus-specific B cell memory in mice, leading to lethal influenza virus infection (73). Dendritic cell–specific deletion of Atg5 results in impaired antigen presentation and increased susceptibility to lethal herpes simplex virus (HSV) infection (74). Moreover, autophagy-competent but not autophagy-deficient tumor cells attract dendritic cells and T cells into tumor beds, leading to enhanced antitumor immunity (75). However, it is unknown whether enhancement of autophagy will augment these antigen-specific immune responses; if so, autophagy inducers may have an important clinical role in enhancing vaccine and antitumor immunity. Of note, rapamycin increases the generation of memory CD8+ T cells in mice following lymphocytic choriomeningitis virus infection or vaccination with a modified vaccinia virus (76), although it is not yet known whether this effect of mTOR inhibition is mediated through autophagy.
Polymorphisms linked to certain genes involved in autophagy regulation or autophagosome formation have been identified in human autoimmune diseases (such as ATG5 and systemic lupus erythematosus) and inflammatory disorders (such as IRGM, NOD2, and ATG16L1 and inflammatory bowel disease) (Table 2), raising the possibility that these disorders may be targets for autophagy induction therapy. However, the effects of the polymorphisms on gene function have not been sufficiently well characterized to allow a prediction of the effects of autophagy induction. For example, the ATG16L1T300A risk allele for Crohn’s disease impairs intestinal Paneth cell function, regulation of proinflammatory IL-1β production, and likely, bacterial autophagy (77–79). However, this region of ATG16L1 is not conserved in yeast Atg16 and may not be required for general autophagy (80); thus, it is unknown whether pharmacologic enhancement of autophagy in Crohn’s disease will correct the pathophysiologic defects imposed by the ATG16L1T300A mutation. Further studies are required to dissect the molecular function of autophagy risk alleles and their specific roles in the pathogenesis of diseases to determine effective strategies for correcting molecular defects imposed by such genetic variations.
Current approaches to developing autophagy inducers
Non-pharmacologic interventions such as caloric restriction and regular exercise induce autophagy and may improve overall health. Exercise-induced autophagy may be required for exercise-mediated protection against high fat diet–induced diabetes in mice (81); this raises the possibility that autophagy enhancement may also underlie other beneficial health effects of exercise, such as delaying the onset or progression of human cancers and neurodegenerative diseases (82). Similarly, autophagy is required for caloric restriction–induced lifespan extension in C. elegans (83) and may underlie the health benefits of caloric restriction in mammals. Intriguingly, the EPaNIC trial found that delayed onset of parenteral nutrition (and hence macronutrient deficiency) in patients in intensive care units led to enhanced autophagy (as measured by light chain 3 class II/light chain 3 class I [LC3-II/LC3-I] ratios and decreased ubiquitin staining) in muscle biopsies and reduced muscle weakness (84). In mice, restriction of food intake decreased the severity of post-infarction heart failure by increasing autophagy in surviving cardiomyocytes (85).
In addition to caloric restriction, other nutritional factors such as the consumption of coffee and vitamin D, may also influence health through autophagy induction. Caffeine-induced autophagy reduces hepatic steatosis in mice with nonalcoholic fatty liver disease (86) and protects against human prion protein–mediated neurotoxicity in cultured cells (87). It is thus tempting to speculate that caffeine-induced autophagy may explain why coffee consumption is associated with dose-dependent decreases in total and cause-specific mortality in middle-aged people (88). Vitamin D is a potent inducer of autophagy (89), and through an autophagy-dependent mechanism it inhibits HIV and M. tuberculosis replication in human macrophages and kills human breast tumor cells (90–92). Defects in vitamin D–induced autophagy might therefore underlie the epidemiologic associations between vitamin D deficiency and adverse health outcomes, including susceptibility to certain cancers and infectious diseases (89). Further clinical studies are needed to determine the optimal regimens for caloric restriction, exercise, caffeine consumption, and vitamin D intake that will yield safe and effective autophagy induction and improved clinical outcomes.
Several drugs currently approved by the FDA induce autophagy (Table 4) but generally have pleiotropic actions, making it difficult to parse out the role of autophagy induction in their therapeutic actions in patients. Nonetheless, preclinical studies demonstrate that certain autophagy-inducing agents fail to induce their beneficial effects in host organisms that are deficient in autophagy genes. FDA-approved compounds have been “repurposed” for use in preclinical models of diseases that are believed to respond favorably to autophagy enhancement, e.g., mTOR inhibitors in neurodegenerative diseases (93), EGFR and other tyrosine kinase inhibitors in diabetic nephropathy (94) and neurodegenerative diseases (20, 95–97), carbamazepine in α1-antitrypsin deficiency (36), trifluoperazine in Salmonella infection (98), and statins in M. tuberculosis infection (38). It is unknown whether autophagy upregulation contributes to the therapeutic effects of these agents for their currently approved clinical indications, but if this proves to be the case it would indicate a broader role than previously appreciated for autophagy in physiology and pathophysiology.
The lack of specificity of known autophagy-inducing agents is not necessarily problematic, provided that the non–autophagy-inducing effects are tolerable for the duration of intended use. For short-term indications such as acute infectious diseases, the non–autophagy-inducing (off-target) actions may lead to unwanted effects that may be tolerable if present for only a few days or weeks. For long-term use, such as the prevention of aging and the treatment of neurodegenerative disorders and metabolic diseases, the adverse effects related to non–autophagy-inducing actions may also be acceptable if they are mild or asymptomatic or are apparent only at doses substantially higher than those needed to enhance autophagy. Nevertheless, the development of highly specific autophagy inducers is strongly desirable (Figure 2), since such agents would be expected to provide the most favorable ratio of benefit to risk and would also provide direct proof-of-principle evidence to support (or refute) a role for autophagy upregulation in a specific disease context.
Figure 2. Schematic illustration of different approaches to developing autophagy inducers for the treatment of human diseases.

IND, investigational new drug.
Beyond optimizing the clinical use of current autophagy-inducing pharmacologic and non-pharmacologic approaches, a key challenge will be to identify agents that can specifically induce autophagy with fewer unwanted side effects than those that accompany currently available drugs. To identify novel autophagy-inducing compounds, chemical screens have been performed using fluorescent measurement of autophagosomes (GFP-LC3–positive puncta) or FACS-based measurement of total levels of LC3 as readouts of autophagy (17, 99–101). For newly identified autophagy-inducing compounds, it is important to (a) determine whether the compound alters autophagy activity at physiologic or clinically meaningful concentrations; (b) demonstrate that the compound exerts its biologic effects through autophagy by showing that it is inactive when autophagy genes are silenced; and (c) identify and confirm its targets by showing that target knockdown or overexpression alters the activity of the compound. Established strategies in medicinal chemistry could then be used to optimize specificity and minimize off-target effects. Although screens based on readouts of autophagy activity have identified new compounds that can induce autophagy, it is not clear whether such approaches will be superior to current repurposing strategies in identifying agents that are specific inducers of autophagy.
To achieve the goal of synthesizing highly specific agents, new approaches will be needed (Figure 2). Strategies most likely to be useful include those that target regulatory steps that are unique to the autophagy pathway or that enhance the activity of specific components of the molecular machinery that are rate-limiting in the process. Since presently known upstream signals that activate autophagy also function in multiple downstream pathways, there is an urgent need to identify new autophagy-specific regulatory molecules or post-translational modification events. In this regard, the use of newer, unbiased proteomic mapping methods in living cells, such as spatially restricted enzymatic tagging (102), may be helpful to uncover such autophagy-specific regulatory steps. In addition, some of the phosphorylation, acetylation/deacetylation, and ubiquitination reactions involved in activation of the Unc-51 like autophagy activating kinase 1 (ULK1) complex (involved in autophagy induction) and the beclin 1/class III PI3K complex (involved in initial formation of the autophagosomal membrane) may become viable pharmacologic targets (103). The binding between beclin 1 and Bcl-2 anti-apoptotic family members (which inhibit beclin 1 function) can also be pharmacologically disrupted; investigational drugs such as the BH3 mimetics upregulate autophagy via this mechanism (104), but they are not specific for autophagy, as they also upregulate apoptosis by disrupting interactions between Bcl-2 family members and BH3 domains of pro-apoptotic molecules. Based on differences in structural determinants between the BH3 domain of beclin 1 versus those of pro-apoptotic family members, it may be possible to design BH3 mimetics that selectively enhance autophagy. Structure-based design is currently being used to develop inhibitors of class III PI3K, of the E1-activating enzyme, ATG7, and of the ATG4B cysteine protease that cleaves LC3 at its carboxyl terminus (25); these approaches may possibly be used to develop agonists rather than antagonists of these molecules. However, it is unknown whether increased activity of these molecules will increase autophagic flux in specific clinical settings, although in some mouse studies, overexpression of ATG7 (as well as ATG5 or beclin 1) results in increased levels of autophagy (Table 3).
Approaches that may also more specifically induce autophagy include gene therapy (involving the tissue-specific delivery of vectors that express autophagy genes) or the use of cell-penetrating peptides (or drug-like derivatives thereof) (Table 3). In animal models, tissue-specific gene therapy with core autophagy genes, Atg7 or beclin 1, the selective autophagy factor Parkin, or the transcriptional regulator of autophagy gene expression and lysosomal activity Tfeb exerts beneficial effects in a wide ranges of diseases including obesity/diabetes, neurodegenerative diseases, cystic fibrosis, α1-antitrypsin deficiency, Pompe disease, muscular dystrophy, and KRAS-induced lung cancers. The protective effects of Tfeb gene therapy in Pompe disease are abolished in the absence of Atg7, suggesting the mechanism of action is enhanced autophagy. Similarly, the cell-permeable Tat–beclin 1 peptide, which contains a short amino acid sequence of beclin 1 necessary and sufficient for autophagy induction, reduces the replication of several pathogens in vitro including HIV, enhances the clearance of the exon 1 fragment of mutant huntingtin protein, and decreases mortality in mice infected with West Nile virus or chikungunya virus. These antiviral effects are abolished in mice partially deficient in beclin 1, suggesting that the peptide acts through enhancement of autophagy. Since subcutaneous administration of this peptide induces autophagy in a variety of mouse tissues at doses that lack apparent toxicity, it may exert beneficial effects in other disease models. The possibility of long-term therapy would be enhanced by the development of an orally active peptidomimetic agent with actions similar to those of Tat–beclin 1. One potential advantage of gene therapy approaches and modified peptides that have spatially restricted in vivo activity is the potential to enhance autophagy in an organ-specific manner.
In addition to developing agents that increase autophagosome formation, it may also be possible to increase autophagic substrate recruitment and lysosomal activity. Considerable progress has been made in understanding the molecular machinery of selective autophagy (2, 105), and indeed, the neuroprotective effects of tyrosine kinase inhibitors may be mediated by enhanced functional interaction between the selective autophagy factor Parkin and beclin 1 (23). Innovative new chemical screens are needed to identify compounds that increase autophagic substrate clearance, in parallel with rational drug-based design to increase autophagic targeting through modulating the activity of specific steps in selective autophagy. For example, phosphorylation of the autophagy receptor optineurin promotes the autophagic clearance of ubiquitin-coated bacteria (106) and protein aggregates associated with neurodegenerative disease (107), suggesting that kinases that promote optineurin phosphorylation may enhance certain forms of selective autophagy. With respect to increasing lysosomal activity, modulation of the activity of TFEB, a transcriptional master regulator of lysosomal biogenesis that is regulated by protein phosphorylation (30), is a promising target.
Challenges for translation of preclinical discoveries into human clinical trials
Even if autophagy-inducing agents demonstrate significant activity in animal models of disease, their clinical development into useful drugs will be challenging. Early-phase clinical trials of any new pharmacologic agent are generally designed to identify a dose or range of potentially useful doses based on their ability to produce the expected changes in a biologic or physiologic system in a proof-of-concept study. However, because the magnitude of autophagy induction cannot readily be assessed in humans, no autophagy-specific surrogate endpoint currently exists. Consequently, unless biomarkers capable of assessing the levels of autophagic flux are identified, doses will need to be defined based on disease-specific intermediate markers, but the relationship of such effects to the induction of autophagy will be difficult to determine. Furthermore, for diseases requiring chronic treatment, it is not clear whether the therapeutic effects of autophagy-inducing agents require continuous long-term treatment or can be achieved by only intermittent therapy. The possibility that intermittent enhancement may be sufficient is supported by observations that intermittent fasting or exercise are associated with clinical benefits (82, 108) that may be attributable to autophagy stimulation. Long-term trials will be needed to assess both the effects of autophagy inducers on patient-centered outcomes and the safety and tolerability of these drugs.
Conclusion
The development of autophagy-inducing drugs heralds the potential for highly effective treatments for a wide range of clinical diseases, many of which have responded poorly to current therapy. Studies of the benefits and toxicity of these novel agents are not only likely to affect clinical care, but they will also inform our understanding of the multifaceted roles of autophagy in normal physiology and pathophysiology.
Supplementary Material
Acknowledgments
We thank Haley Harrington and Cindy Jozefiak for help with manuscript preparation. The work in the authors’ laboratories was supported by Cancer Prevention and Research Institute of Texas grant RP120718-P1 (to B. Levine); NIH grants R01CA84254 (to B. Levine), R01CA109618 (to B. Levine), and U19AI109725 (to B. Levine); INSERM (to P. Codogno); CNRS (to P. Codogno); University Paris Descartes (to P. Codogno); and grants from ANR (to P. Codogno) and INCa (to P. Codogno).
Footnotes
Conflict of interest: Beth Levine has received consulting fees from Novus Biologicals, and Milton Packer has received consulting fees from Acetelion, Amgen, Bard, Boston Scientific, Cardiorentis, CardioMEMS, Janssen, Novartis, Pfizer, Philip Morris, and Piramal.
Reference information:J Clin Invest. 2015;125(1):14–24. doi:10.1172/JCI73938.
References
- 1.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. 2013;20(1):21–30. doi: 10.1038/cdd.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 4.Hurley JH, Schulman BA. Atomistic autophagy: the structures of cellular self-digestion. Cell. 2014;157(2):300–311. doi: 10.1016/j.cell.2014.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–477. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 6.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720. doi: 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Kim KH, Lee MS. Autophagy-a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10(6):322–337. doi: 10.1038/nrendo.2014.35. [DOI] [PubMed] [Google Scholar]
- 10.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–325. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 12.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 15.Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24(1):69–79. doi: 10.1038/cr.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vakifahmetoglu-Norberg H, Xia H, Yuan J. Pharmacologic agents targeting autophagy. J Clin Invest. 2015;125(1):5–13. doi: 10.1172/JCI73937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim YC, Guan K-L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang X, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest. 2015;125(1):47–54. doi: 10.1172/JCI73942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beau I, Mehrpour M, Codogno P. Autophagosomes and human diseases. Int J Biochem Cell Biol. 2011;43(4):460–464. doi: 10.1016/j.biocel.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Sarkar S. Chemical screening platforms for autophagy drug discovery to identify therapeutic candidates for Huntington’s disease and other neurodegenerative disorders. Drug Discov Today Technol. 2013;10(1):e137–e144. doi: 10.1016/j.ddtec.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 2014;26C:16–23. doi: 10.1016/j.gde.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morselli E, et al. Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol. Aging (Albany NY). 2009;1(12):961–970. doi: 10.18632/aging.100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lonskaya I, Hebron ML, Desforges NM, Franjie A, Moussa CE. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol Med. 2013;5(8):1247–1262. doi: 10.1002/emmm.201302771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng Y, Ren X, Hait WN, Yang JM. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev. 2013;65(4):1162–1197. doi: 10.1124/pr.112.007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ravikumar B, et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37(7):771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 27.Settembre C, et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17(1):119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 28.Lee JH, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Settembre C, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011. 33260361429–1433. 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spampanato C, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med. 2013;5(5):691–706. doi: 10.1002/emmm.201202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maetzel D, et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Reports. 2014;2(6):866–880. doi: 10.1016/j.stemcr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarkar S, et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013;5(5):1302–1315. doi: 10.1016/j.celrep.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 35.Rose C, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet. 2010;19(11):2144–2153. doi: 10.1093/hmg/ddq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hidvegi T, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329(5988):229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 37.Shoji-Kawata S, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parihar SP, et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. 2014;209(5):754–763. doi: 10.1093/infdis/jit550. [DOI] [PubMed] [Google Scholar]
- 39.Vanhorebeek I, et al. Insufficient activation of autophagy allows cellular damage to accumulate in critically ill patients. J Clin Endocrinol Metab. 2011;96(4):E633–E645. doi: 10.1210/jc.2010-2563. [DOI] [PubMed] [Google Scholar]
- 40.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6(6):439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 41.Ryter SW, Mizumura K, Choi AM. The impact of autophagy on cell death modalities. Int J Cell Biol. 2014;2014:502676. doi: 10.1155/2014/502676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo JY, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27(13):1447–1461. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang A, et al. Autophagy is critical for pancreatic tumor growth progression in tumors with p53 alterations. Cancer Discov. 2014;4(8):905–913. doi: 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155(6):1216–1219. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White E. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 2013;27(19):2065–2071. doi: 10.1101/gad.228122.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei Y, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154(6):1269–1284. doi: 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42(1):23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 48.Rosenfeldt MT, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 49.Rao S, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 50.Shin JY, Hong SH, Kang B, Minai-Tehrani A, Cho MH. Overexpression of beclin1 induced autophagy and apoptosis in lungs of K-rasLA1 mice. Lung Cancer. 2013;81(3):362–370. doi: 10.1016/j.lungcan.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 51.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6(3):322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma K, Le N, Alotaibi M, Gewirtz DA. Cytotoxic autophagy in cancer therapy. Int J Mol Sci. 2014;15(6):10034–10051. doi: 10.3390/ijms150610034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Macintosh RL, Ryan KM. Autophagy in tumour cell death. Semin Cancer Biol. 2013;23(5):344–351. doi: 10.1016/j.semcancer.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 54.Bareford MD, et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Res. 2011;71(14):4955–4967. doi: 10.1158/0008-5472.CAN-11-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ko A, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014;21(1):92–99. doi: 10.1038/cdd.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramirez-Peinado S, et al. Glucose-starved cells do not engage in prosurvival autophagy. J Biol Chem. 2013;288(42):30387–30398. doi: 10.1074/jbc.M113.490581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lang MJ, et al. Glucose Starvation inhibits autophagy via vacuolar hydrolysis and induces plasma membrane internalization by down-regulating recycling. J Biol Chem. 2014;289(24):16736–16747. doi: 10.1074/jbc.M113.525782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen ZH, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107(44):18880–18885. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun Y, et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci Signal. 2012;5(212):ra16. doi: 10.1126/scisignal.2001931. [DOI] [PubMed] [Google Scholar]
- 60.Xu X, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288(25):18077–18092. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu H, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117(7):1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yin X, et al. miR-30a downregulation aggravates pressure overload-induced cardiomyocyte hypertrophy. Mol Cell Biochem. 2013;379(1–2):1–6. doi: 10.1007/s11010-012-1552-z. [DOI] [PubMed] [Google Scholar]
- 63.Fujimoto K, et al. Autophagy regulates pancreatic beta cell death in response to Pdx1 deficiency and nutrient deprivation. J Biol Chem. 2009;284(40):27664–27673. doi: 10.1074/jbc.M109.041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei N, et al. The involvement of autophagy pathway in exaggerated ischemic brain damage in diabetic mice. CNS Neurosci Ther. 2013;19(10):753–763. doi: 10.1111/cns.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li WL, et al. The regulatory role of NF-κB in autophagy-like cell death after focal cerebral ischemia in mice. Neuroscience. 2013;244:16–30. doi: 10.1016/j.neuroscience.2013.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luo CL, et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011;184:54–63. doi: 10.1016/j.neuroscience.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 67.Liu Y, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110(51):20364–20371. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matsui Y, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 69.Koike M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172(2):454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alirezaei M, Flynn CT, Wood MR, Whitton JL. Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo. Cell Host Microbe. 2012;11(3):298–305. doi: 10.1016/j.chom.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tian Y, Sir D, Kuo CF, Ann DK, Ou JH. Autophagy required for hepatitis B virus replication in transgenic mice. J Virol. 2011;85(24):13453–13456. doi: 10.1128/JVI.06064-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen M, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med. 2014;20(5):503–510. doi: 10.1038/nm.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee HK, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32(2):227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Michaud M, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 76.Araki K, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Murthy A, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506(7489):456–462. doi: 10.1038/nature13044. [DOI] [PubMed] [Google Scholar]
- 78.Lassen KG, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014;111(21):7741–7746. doi: 10.1073/pnas.1407001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456(7219):259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fujita N, Saitoh T, Kageyama S, Akira S, Noda T, Yoshimori T. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: analysis of the organization of the Atg16L1 complex in fibroblasts. J Biol Chem. 2009;284(47):32602–32609. doi: 10.1074/jbc.M109.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.He C, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481(7382):511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Handschin C, Spiegelman BM. The role of exercise and PGC1α in inflammation and chronic disease. Nature. 2008;454(7203):463–469. doi: 10.1038/nature07206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3(6):597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- 84.Hermans G, et al. Effect of tolerating macronutrient deficit on the development of intensive-care unit acquired weakness: a subanalysis of the EPaNIC trial. Lancet Respir Med. 2013;1(8):621–629. doi: 10.1016/S2213-2600(13)70183-8. [DOI] [PubMed] [Google Scholar]
- 85.Watanabe T, et al. Restriction of food intake prevents postinfarction heart failure by enhancing autophagy in the surviving cardiomyocytes. Am J Pathol. 2014;184(5):1384–1394. doi: 10.1016/j.ajpath.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 86.Sinha RA, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59(4):1366–1380. doi: 10.1002/hep.26667. [DOI] [PubMed] [Google Scholar]
- 87.Moon JH, et al. Caffeine prevents human prion protein-mediated neurotoxicity through the induction of autophagy. Int J Mol Med. 2014;34(2):553–558. doi: 10.3892/ijmm.2014.1814. [DOI] [PubMed] [Google Scholar]
- 88.Freedman ND, Park Y, Abnet CC, Hollenbeck AR, Sinha R. Association of coffee drinking with total and cause-specific mortality. N Engl J Med. 2012;366(20):1891–1904. doi: 10.1056/NEJMoa1112010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoyer-Hansen M, Nordbrandt SP, Jaattela M. Autophagy as a basis for the health-promoting effects of vitamin D. Trends Mol Med. 2010;16(7):295–302. doi: 10.1016/j.molmed.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 90.Hoyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jaattela M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005;12(10):1297–1309. doi: 10.1038/sj.cdd.4401651. [DOI] [PubMed] [Google Scholar]
- 91.Campbell GR, Spector SA. Hormonally active vitamin D3 (1α,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J Biol Chem. 2011;286(21):18890–18902. doi: 10.1074/jbc.M110.206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Campbell GR, Spector SA. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012;8(5):e1002689. doi: 10.1371/journal.ppat.1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans. 2013;41(5):1103–1130. doi: 10.1042/BST20130134. [DOI] [PubMed] [Google Scholar]
- 94.Zhang MZ, Wang Y, Paueksakon P, Harris RC. Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes. 2014;63(6):2063–2072. doi: 10.2337/db13-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lonskaya I, Hebron ML, Desforges NM, Schachter JB, Moussa CE. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J Mol Med (Berl). 2014;92(4):373–386. doi: 10.1007/s00109-013-1112-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Le Pichon CE, et al. EGFR inhibitor erlotinib delays disease progression but does not extend survival in the SOD1 mouse model of ALS. PLoS One. 2013;8(4):e62342. doi: 10.1371/journal.pone.0062342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang L, et al. Epidermal growth factor receptor is a preferred target for treating amyloid-β-induced memory loss. Proc Natl Acad Sci U S A. 2012;109(41):16743–16748. doi: 10.1073/pnas.1208011109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Conway KL, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. 2013;145(6):1347–1357. doi: 10.1053/j.gastro.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sarkar S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3(6):331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang L, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104(48):19023–19028. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hundeshagen P, Hamacher-Brady A, Eils R, Brady NR. Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of autophagy. BMC Biol. 2011;9:38. doi: 10.1186/1741-7007-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rhee HW, et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339(6125):1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wirth M, Joachim J, Tooze SA. Autophagosome formation — the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol. 2013;23(5):301–309. doi: 10.1016/j.semcancer.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 104.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4(5):600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. 2014;15(4):403–411. doi: 10.1016/j.chom.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wild P, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Korac J, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126(pt 2):580–592. doi: 10.1242/jcs.114926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Varady KA, Hellerstein MK. Alternate-day fasting and chronic disease prevention: a review of human and animal trials. Am J Clin Nutr. 2007;86(1):7–13. doi: 10.1093/ajcn/86.1.7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.