

Abstract
In addition to alleviating depression, trophic responses produced by antidepressants may regulate neural plasticity in the diseased brain, providing not only symptomatic benefit but potentially slowing the rate of disease progression in Parkinson’s disease (PD). Recent in vitro and in vivo data provide evidence that neurotrophic factors such as brain derived-neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) may be key mediators of the therapeutic response to antidepressants. As such, we conducted a cross-sectional time-course study to determine whether antidepressant-mediated changes in neurotrophic factors occur in relevant brain regions in response to amitriptyline (AMI) treatment before and after intrastriatal 6-hydroxydopamine (6OHDA). Adult male Wistar rats were divided into seven cohorts and given daily injections (i.p.) of AMI (5mg/kg) or saline throughout the duration of the study. In parallel, various cohorts of intact or parkinsonian animals were sacrificed at specific time points to determine the impact of AMI treatment on trophic factor levels in the intact and degenerating nigrostriatal system. The left and right hemispheres of the substantia nigra, striatum, frontal cortex, piriform cortex, hippocampus and anterior cingulate cortex were dissected and BDNF and GDNF levels were measured with ELISA. Results show that chronic AMI treatment elicits effects in multiple brain regions and differentially regulates levels of BDNF and GDNF depending on the region. Additionally, AMI halts the progressive degeneration of dopamine (DA) neurons elicited by an intrastriatal 6-OHDA lesion. Taken together, these results suggest that AMI treatment elicits significant trophic changes important to DA neuron survival within both the intact and degenerating nigrostriatal system.
Keywords: Neuroprotection, antidepressants, amitriptyline, trophic factors, Parkinson’s disease
Introduction
Depression is a common co-morbid disorder that affects almost half of all patients with Parkinson’s disease (PD), many of which take antidepressants on a daily basis [1]. Although a number of studies have investigated the trophic response to various antidepressant drugs [2-11], most focus on changes within mesolimbic brain structures. Therefore, little is known about whether antidepressant therapy impacts trophic factors within the nigrostriatal dopamine (DA) system. The trophic factors brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) are of particular importance for nigrostriatal DA neurons vulnerable to degeneration in PD [12]. While both factors have shown promise for PD pre-clinically [13-24], only GDNF has been investigated for utility in human trials. Multiple open label clinical trials demonstrated significant symptomatic improvement in PD patients that received intraputamenal GDNF infusion [25-27]. However, a randomized, controlled trial of intraputamenal GDNF infusion yielded negative results [28], thereby dampening the enthusiasm for exogenous delivery of this protein in the clinic. Though unexpected, this finding does not negate the positive data supporting the neuroprotective potential of trophic factors; it merely indicates that exogenous delivery of trophic support may not be an optimal strategy, especially in end stage PD. Therefore, treatments that induce the synthesis or release of endogenous factors in the brain, particularly earlier in the disease process, may provide a more effective strategy for treating PD patients [29, 30].
Psychopharmacological agents such as antidepressants have been shown to modulate important signaling pathways involved in cell survival and plasticity [3-5], thereby suggesting they may have more pleiotrophic ability than previously anticipated. Such studies provide evidence that antidepressant treatment could evoke changes that induce neuroprotection in neurodegenerative diseases, including PD. The trophic response elicited by chronic antidepressant treatment has been well documented within mesolimbic regions involved in depression and mood disorders; however, little is known about whether similar changes occur in the nigrostriatal pathway and hence may provide dual therapeutic value for PD patients. Additionally, there are conflicting data concerning the onset and duration of antidepressant-mediated trophic responses [31, 32]. Therefore, we performed a cross-sectional time-course study to determine the effects of chronic antidepressant treatment on BDNF and GDNF levels in brain regions relevant to PD. As tricyclic antidepressants (TCAs) have been shown to be more efficacious in treating depression in PD patients [33, 34], we opted to examine the specific TCA amitriptyline (AMI), which is often prescribed to patients with PD [35]. Furthermore, we recently reported that AMI, but not other antidepressants, were associated with a delay in the need to start dopaminergic therapy in an early cohort of patients with PD [36], suggesting it may have disease-modifying properties. More recently, our laboratory demonstrated that the low dose of AMI used in this study afforded significant neuroprotection to the nigrostriatal system in a rat toxin model of parkinsonism [37]. We report here that chronic AMI treatment differentially regulates BDNF and GDNF in the intact and degenerating nigrostriatal system, and that AMI significantly retards the progression of nigral dopamine neuron loss.
Methods
Animals
Animals were housed and treated following the National Institutes of Health guidelines[38]. Each experiment utilized adult (4-6 month old) male Wistar rats (200-225g) as subjects. Rats were obtained from Harlan Animal Research Laboratory (Indianapolis, IN). The animal facility at the University of Cincinnati is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care and complied with all Federal animal care and use guidelines. The Institutional Animal Care and Use Committee (IACUC) at the University of Cincinnati, where these studies were performed, approved all protocols pertaining to these experiments.
Experimental Design
Rats were acclimated to their new environment for 24 hours before beginning experiments then randomized and divided into four groups (saline-no lesion, amitriptyline (AMI)-no lesion, saline +lesion, AMI +lesion) and given daily intraperitoneal (i.p.) injections of sterile saline (Henry Schein) or 5mg/kg of amitriptyline hydrochloride (AMI, Sigma) for 14 days (Fig. 1A). This 14-day pretreatment regimen and low dosage was based on previous studies indicating that this timing is important to achieve antidepressant efficacy [3, 5, 11]. On day 14, rats in the appropriate groups (+lesion) received unilateral intrastriatal injection of the DA neurotoxin 6-hydroxydopamine (6-OHDA), after which all rats continued to receive daily i.p. AMI or saline injections for the remainder of the experiment (four weeks). Lesioned and non-lesioned cohorts of rats from both saline and AMI groups were sacrificed on days 14, 17, 24 and 42, which corresponds to the following days post-lesion: day 0 (D0), day 3 (D3), day 10 (D10) and day 30 (D30). All rats were sacrificed at prescribed time-points and brains extracted and processed for either immunohistochemical or ELISA analyses.
Figure 1. Experimental design and nigrostriatal interconnectivity.
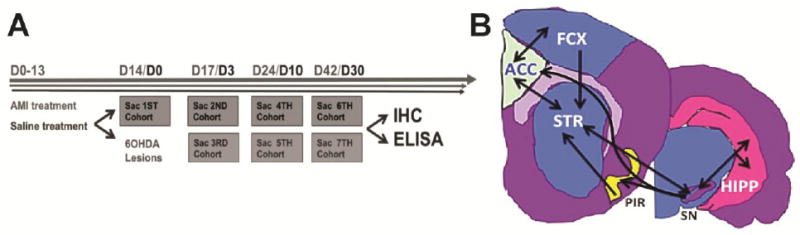
The experimental timeline depicts the cross-sectional time course design highlighting specific time-points at which select cohorts are treated and sacrificed (A). Time course in gray (Days 0-42) shows sacrifice times for non-lesioned animals treated with saline or AMI. Structures dissected at these times are indicative of the trophic response elicited by saline or AMI treatment in intact animals. Time course in black (Days 0-30) depicts sacrifice times post-lesion for animals treated with saline or AMI. Structures dissected at these times are indicative of the trophic response elicited by saline or AMI in the degenerating nigrostriatal system. Representative map detailing the interconnectivity between dissected structures of interest (B). Abbreviations: AMI=amitriptyline hydrochloride, D=day, Sac=sacrifice, 6OHDA=6-hydroxydopamine, IHC=immunohistochemistry, ELISA=enzyme-linked immunosorbent assay, STR=striatum, HIPP=hippocampus, SN=substantia nigra, FCX=frontal cortex, ACC=anterior cingulate cortex, PIR=piriform cortex
To assess trophic factor levels within the nigrostriatal system, we examined BDNF and GDNF protein in the SN and striatal structures. However, BDNF is not produced in the striatum [39, 40], yet receives BDNF protein via anterograde/retrograde transport from various interconnected regions including the SN [41], hippocampus [42, 43], frontal [39, 40, 44], piriform [45, 46] and anterior cingulate cortices [47-50] (Fig. 1B). Therefore, we extended the scope of the project to include these structures, and each hemisphere was microdissected and processed as an independent sample as described previously [51].
6-OHDA Lesions
Prior to surgery, all rats were anesthetized (30 mg/kg, pentobarbital, i.p.) and placed into a stereotaxic device. The 6-OHDA (dissolved in a 0.2% ascorbic acid-physiological saline) was made immediately prior to surgery and kept on ice. All rats received 2μl of 6-OHDA (5μg/μl) into the ipsilateral left striatum (AP +0.7, ML +3.0, DV -5.5 from skull) at a rate of 0.5μl per minute. After each injection, the needle was left in place for 2 minutes and then slowly withdrawn. Lesions were verified by stereological assessment of tyrosine hydroxylase immunoreactive (THir) neurons within the substantia nigra pars compacta (SNpc).
Immunohistochemistry
Animals were euthanized via pentobarbital overdose (60mg/kg) and intracardially perfused with 0.9% saline followed by ice cold 4% paraformaldehyde (PFA) in 0.1 M PO4 buffer. Brains were extracted and post-fixed in 4% PFA for 24 hours and sunk in 30% sucrose. Brains were cut into 40μm thick sections on a freezing microtome. A 1:6 series of coronal sections was double labeled for tyrosine hydroxylase (TH) (marker of dopamine neurons) and NeuN (general neuronal marker) using the free-floating method. A sequential approach was utilized to visualize these proteins using antibodies from the same host (mouse)[52]. Tissue was incubated in 0.3% H202 for 45 minutes, rinsed and blocked in 10% normal goat serum (1 hour) then incubated in primary mouse anti-NeuN antibody (1:500; Chemicon) overnight at 4° C. Following primary incubation, sections were incubated in biotinylated secondary antisera against mouse IgG (1:400, Chemicon) followed by the Vector ABC detection kit employing horseradishperoxidase (Vector Labs). Antibody labeling was visualized by exposure to 0.5 mg/ml 3,3’ diaminobenzidine (DAB), 2.5mg/ml nickel ammonium sulfate and 0.03% H2O2 in Tris buffer. Tissue was rinsed and blocked again overnight at 4°C. The following day, tissue was incubated in mouse anti-TH antibody (1:4000, Chemicon) for four hours, followed by biotinylated secondary antisera against mouse IgG (same as above) and the Vector ABC detection kit. Labeling was visualized using the NovaRed kit (Vector Labs) and sections were mounted on subbed slides, dehydrated to xylene and coverslipped with Cytoseal (Richard-Allan Scientific). Since TH is localized to the cytoplasm, any cross reactivity to the biotinylated secondary antisera against mouse IgG labeling did not interfere with nuclear NeuN visualization.
ELISA
Animals were anesthetized (60mg/kg, pentobarbital, i.p.) and perfused with heparinized saline (0.9%). Brains were flash frozen in 3-methyl butane (Fisher) then stored at -80°C. 1-2 mm coronal slabs were blocked utilizing a brain blocker (Zivic, Pittsburg, PA) and both hemispheres of the SN, striatum, hippocampus, frontal cortex, anterior cingulate cortex (ACC) and the piriform cortex were microdissected at a constant - 12°C on a cold plate (Teca, Chicago,IL) and stored at − 80°C until analysis. Samples were homogenized on ice in 200ul of lysis buffer (M-PER; Pierce) with protease inhibitors (Sigma-Aldrich). The Pierce BCA Protein Kit (Rockford, IL) was utilized for protein determination. The homogenate was centrifuged at 4°C for 20 minutes at 14,000g and the supernatant was collected. ELISAs were run using the Immunolon 4 HBX 96 well plate according to manufacturer’s instructions (GDNF/BDNF Promega Emax ImmunoAssay Kit). Samples were read (<30 minutes) on a spectrophotometer (Multiskan Spectrum, Thermolab Systems) with unknown values interpolated against standard curves.
Stereology for Cell Counts
MicroBrightfield stereological software (MBF Bioscience, Inc.) was used for stereological quantitation of TH and NeuN cells. Outline of substantia nigra pars compacta (SNpc) region of interest (ROI) was performed under low magnification (1.25X) and 20% of the designated area was sampled via a random series of counting frames systematically distributed throughout the contour. Cell counts were performed using the optical fractionator probe under high magnification (60X with oil) by an investigator blind to the animal experimental status. For the ROI, a z-stack of images was acquired at each sampling site within the stereological probe. Cells were counted by focusing through the tissue at 1-2 μm intervals (dissector height was 25μm with top/bottom guard zones of 2.5μm), and then a marker was placed over each NeuN/THir or NeuNir/TH negative neurons within the counting frame (50x50μm). Between 50 and 300 objects were counted to generate the stereological estimates. The total number of stained neurons (N) in the regions of interest was calculated using the following formula: N = NV · VROI where NV is the numerical density and VROI is the volume of the region of interest. The volume was calculated according to the procedure of Cavalieri and variability within groups was assessed via the Coefficient of Error (CE). The Gunderson method was used for calculating the CE value (< 0.1) for the population estimate [53].
Statistical Analysis
AMI-mediated differences in trophic factor levels over time in intact animals were analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison post-hoc analysis comparing all time points to the baseline control value. Differences between groups in 6-OHDA lesioned animals over time were analyzed by two-way ANOVA. Comparisons in THir and NeuNir/TH negative neurons between groups were analyzed using a one-way ANOVA. When appropriate, post-hoc comparisons were made between groups using the Student-Newman-Keuls or Bonferonni method. The level of significance was set at P ≤ 0.05.
Results
Regional Distribution of BDNF and GDNF in Rat Brain
Baseline values of BDNF and GDNF measured in different brain structures are depicted in Table 1. BDNF and GDNF levels varied significantly across regions, but were in line with previous reports of BDNF and GDNF expression in brain [54, [54-56].
Table 1.
Baseline BDNF and GDNF levels in saline-treated rats
| Regional Levels of BDNF and GDNF in Rat Brain | ||
|---|---|---|
| BDNF | ||
| Region | N | Levels pg/mg protein |
| HIPP | 13 | 37.99 ± 3.95 |
| SN | 8 | 46.28 ± 1.95 |
| STR | 8 | 30.20 ± 3.15 |
| FCX | 11 | 53.42 ± 6.73 |
| ACC | 13 | 30.95 ± 5.68 |
| PIR | 13 | 58.96 ± 3.28 |
| GDNF | ||
| Region | N | Levels pg/mg protein |
| HIPP | 8 | 79.54 ± 24.58 |
| SN | 12 | 245.3 ± 53.09 |
| STR | 14 | 176.3 ± 32.98 |
| FCX | 12 | 220.3 ± 27.81 |
| ACC | 6 | 183.3 ± 87.08 |
| PIR | 14 | 139.0 ± 35.79 |
Values represent baseline BDNF or GDNF levels (mean ± SEM) obtained from intact rats treated with saline for two weeks. Data are combined from two different experimental control groups. Each hemisphere of the region of interest (ROI) was processed independently and represents one sample. Variability in sample size is due to loss or insufficient recovery of protein.
Effects of Amitriptyline (AMI) on BDNF and GDNF Levels in the Intact Brain
BDNF
ELISA results demonstrated that hippocampal BDNF was significantly increased compared to baseline levels and remained elevated after 24 days of AMI treatment (F(4, 62)=5.217, p<0.001; Fig. 2, A). Previous studies indicate that hippocampal BDNF is increased in response to antidepressant treatment [54]; therefore, this structure served as a positive control for AMI efficacy. In the SN, BDNF was significantly increased after 24 days of AMI treatment and remained elevated for the duration of the study (F(4, 59)=3.764, p=0.0009; Fig. 2B). In the striatum, BDNF was significantly decreased after 24 days of AMI treatment (F(4, 52)=8.472, p<0.001; Fig. 2C). In the frontal cortex, AMI did not elicit any significant changes in BDNF (p > 0.05; Fig. 2D). In the ACC, BDNF was significantly decreased after 24 days of AMI treatment (F(4, 58)=4.627, p=0.003; Fig. 2E). In the piriform cortex, there was a transient but significant increase in BDNF after 14 days of AMI treatment, but then levels significantly decreased after 24 days (F(4, 60)=16.22, p<0.0001; Fig. 2F).
Figure 2. Temporal regulation of BDNF in response to AMI.
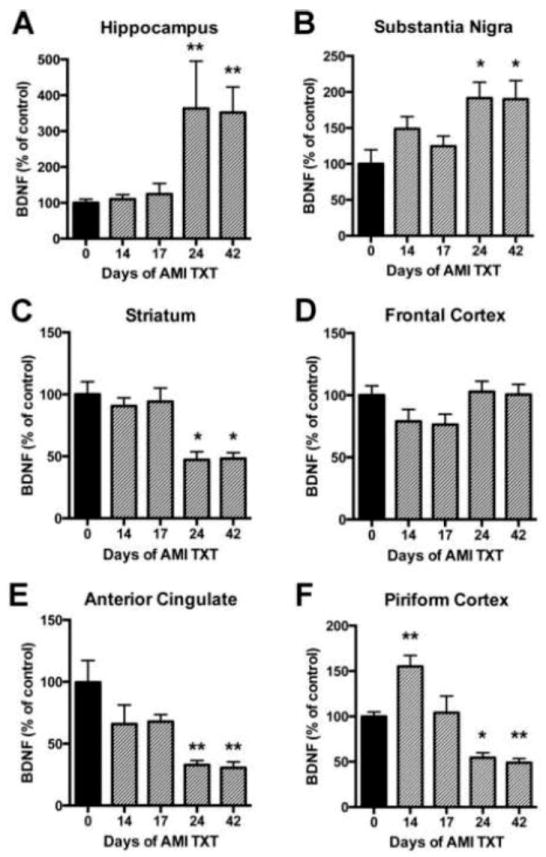
Adult rats received saline or AMI (5mg/kg, i.p.) for 14, 17, 24 or 42 days. Each hemisphere of the region of interest was dissected on ice and analyzed independently with ELISA for BDNF. Levels of BDNF are depicted for each of the following regions: Hippocampus (A), substantia nigra (B), striatum (C), frontal cortex (D), anterior cingulate (E) and piriform cortex (F). Data from AMI-treated rats are depicted in grey and data from saline-treated animals are depicted in black. Values are expressed as a percentage of absolute values in the saline control group. Levels of significance determined by Dunnett’s multiple comparison post-hoc analysis comparing all time points to the baseline control value. Data represent mean +/-SEM and are combined from two separate experiments. Abbreviations: AMI: amitriptyline, TXT: treatment, BDNF: brain derived neurotrophic factor *p<0.05, **p<0.01
GDNF
A transient, but significant increase in hippocampal GDNF was observed after 14 days of AMI treatment (F(4, 49)=5.257, p=0.002); Fig. 3A). In line with BDNF results, SN GDNF levels were significantly increased after 24 days of treatment with AMI and remained elevated (F(4, 52)=3.740, p=0.0095; Fig. 3B). Striatal GDNF did not change in response to chronic AMI treatment (p>0.05; Fig. 3, C). Similarly, AMI did not elicit any significant changes in GDNF in the frontal cortex (Fig. 3D). In contrast to BDNF results, ACC GDNF was significantly increased at 14 days (compared to baseline levels) as a result of AMI treatment (F(4, 50)=10.38, p<0.001; Fig. 3, E), then returned to baseline over time. GDNF levels were not altered in the piriform cortex in response to chronic AMI treatment (p>0.05; Fig. 3, F). Combined, these results suggest AMI mediates substantial trophic changes over time within the intact nigrostriatal system that may significantly impact DA neuron survival.
Figure 3. Temporal regulation of GDNF in response to AMI.
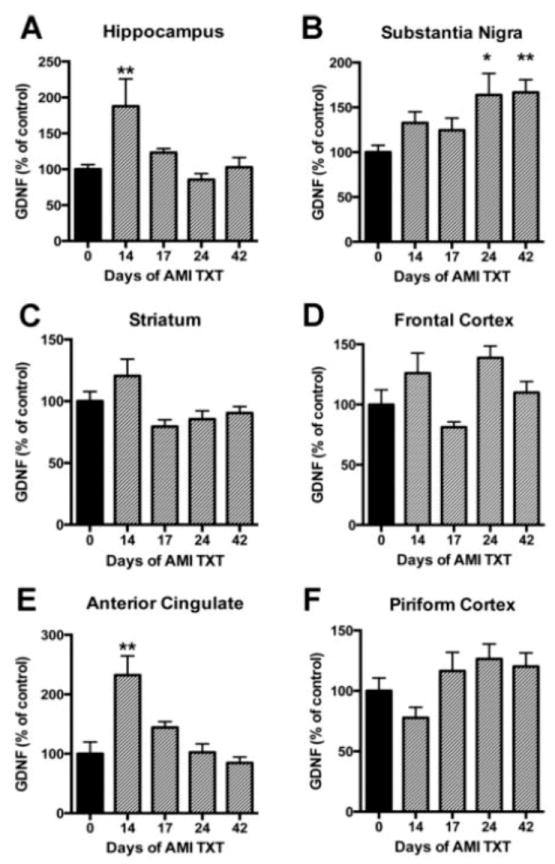
Adult rats received saline or AMI (5mg/kg, i.p.) for 14, 17, 24 or 42 days. Each hemisphere of the region of interest was dissected on ice and analyzed independently with ELISA for GDNF. Levels of GDNF are depicted for each of the following regions: Hippocampus (A), substantia nigra (B), striatum (C), frontal cortex (D), anterior cingulate (E) and piriform cortex (F). Data from AMI-treated rats are depicted in grey and data from saline-treated animals are depicted in black. Levels of significance determined by Dunnett’s multiple comparison post-hoc analysis comparing all time points to the baseline control value. Values are expressed as a percentage of absolute values in the saline control group. Data represent mean+/-SEM and are combined from two separate experiments. Abbreviations: AMI: amitriptyline, TXT: treatment, GDNF: glialderived neurotrophic factor *p<0.05, **p<0.01
DA Degeneration Post 6-OHDA Lesion
Population estimates were derived for both TH immunoreactive (ir) and NeuNir neurons throughout the lesioned substantia nigra in saline- and AMI-treated rats. There were no significant differences in the total number of THir cells in the intact hemisphere between groups (F(1, 31) = 2.78; p>0.05) or over time (F(3, 31) = 0.69; p>0.05) and the combined average population estimate was 15,683.00+/-819.89. Additionally, there were no significant differences between groups (F(1, 32) = 1.90; p>0.05) or over time (F(3, 32) = 0.14; p>0.05) for total NeuNir counts on the intact side and the combined average population estimate was 30,603.00+/-1403.30. Stereological assessment showed that in the SNpc ipsilateral to intrastriatal 6- OHDA injection THir neurons decreased by approximately 25% in both treatment groups 10 days postlesion. Neurodegeneration continued in the saline-treated group (-60%), whereas it was halted in the AMI-treated group (-32%) 30 days post-lesion. A two-way ANOVA revealed a significant overall effect for time (F(3, 33)=11.89, p<0.0001). Post hoc analyses indicate significant differences between groups at the 30-day time point with AMI treatment associated with significantly higher numbers of THir neurons (Fig. 4A; p<0.05). To confirm that the extent of THir neuron loss was the result of neuron death and not phenotype down-regulation, THir neuron counts were compared to total NeuNir neuron counts. Results show an overall effect for time demonstrating that the number of neurons declined significantly over time (F(3, 55)= 8.48, p<0.0001; Fig. 4B). In saline-treated rats, the amount of THir neuron loss was greater at day 3 (-2224.51± 1982.95) and day 10 (-4163.29 ± 2163.38) compared to the total NeuNir neuron loss at day 3 (-587.19±2712) and day 10 (-1623.79 ± 1584.43) post-lesion. However, by day 30 post-lesion, THir neuron loss (-9815.89 ± 1724.57) was comparable to total NeuNir neuron loss (-8961.97 ± 1482.58). Similarly, in AMI-treated rats, the amount of THir neuron loss was greater at the early time points (day 3: -1171.65 ± 952.84 and day 10: -5706.63 ± 1819.03) relative to the total NeuNir neuron loss at the early time points (day 3: -981.95 ± 933.19 and day 10: -2766.99 ± 1884.75) post-lesion. Yet, at the 30-day time point post-lesion, THir neuron loss (-4138.34 ± 1040.97) was equivalent to total NeuNir neuron loss (-4346.23 ± 1264.04). Post hoc analysis revealed a significant difference between the number of THir neurons in AMI- and saline-treated rats 30 days post-lesion (-4138.34 ± 1040.97 vs. -9815.89 ± 1724.57; p<0.05). Combined, these data show that for both AMI- and saline-treated rats, loss of THir (DA phenotype) precedes overt neuron loss at the early time points post-lesion. However, after 30 days post-lesion, both populations decline in comparable numbers indicating that a loss of TH phenotype reflects actual neuron loss. Additionally, AMI treatment significantly attenuated the magnitude of nigral DA neuron loss 30 days post-lesion.
Figure 4. Amitriptyline slows the progressive loss of dopamine (DA) neurons in the 6- OHDA rat model of PD.
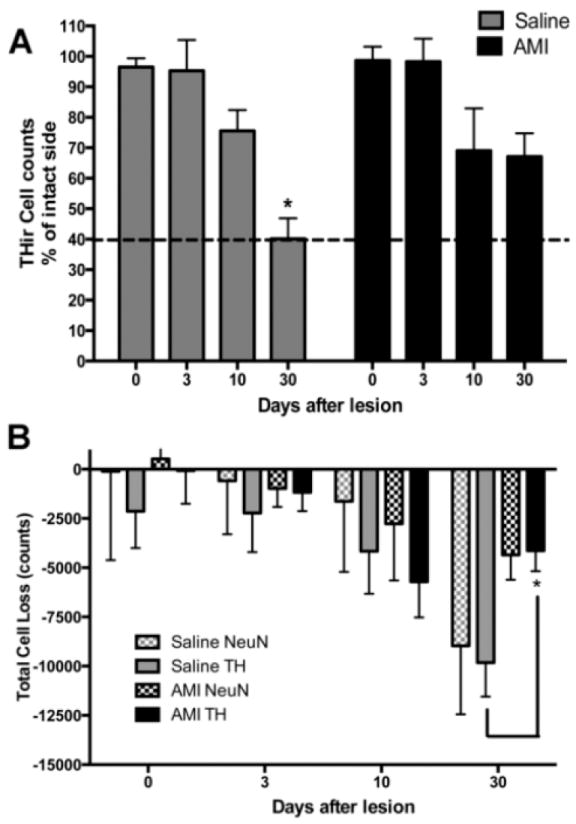
Stereological population estimates show DA neuron loss is significantly attenuated in the AMItreated animals compared to the saline-treated animals 30 days post-lesion (A). To confirm that the extent of DA (THir) neuron loss was the result of cell death and not phenotype downregulation, THir cell counts were compared to NeuNir counts. Results indicate both populations of cells decline in comparable numbers by the 30-day time point (B); however, there is significantly less THir cell loss in the AMI group compared to the saline group. For comparison, dashed line illustrates percent lesion for control animals at 30-day time point. Data represent mean+/-SEM and are combined from two separate experiments. Abbreviations: AMI: amitriptyline, TH: Tyrosine hydroxylase, THir: tyrosine hydroxylase immunoreactive, NeuN: neuronal nuclei, NeuNir: NeuN immunoreactive. Panel A: *p<0.05 compared to saline D0, $p<0.05 compared to AMI D0, ##p<0.01compared to saline D3, ˆˆp<0.01 compared to AMI D3; Panel B: *p<0.05 compared to Saline TH at D30
Effects of Amitriptyline (AMI) on BDNF and GDNF Levels in Brain During Nigrostriatal Degeneration
BDNF
In the 6-OHDA lesioned brain, there was a significant interaction between treatment and time for hippocampal BDNF (F(2, 36)=11.37, p<0.0001), suggesting that AMI impacted BDNF levels over time. Post hoc analysis revealed that hippocampal BDNF was significantly increased in AMI treated animals at the 30-day time point compared to either treatment at other time points (Fig 5A). Furthermore, this increase was significantly elevated compared to saline treated animals at the 30-day time point (~2.5 fold; p<0.0001), suggesting there is a temporal increase in hippocampal BDNF mediated by AMI that occurs independent of the lesion. In the SN, there was a significant effect of treatment (F(1, 25)=5.550, p<0.05) suggesting that AMI impacted BDNF levels over time. At the 3 day post-lesion time point, post hoc analysis indicates SN BDNF was significantly elevated in response to AMI (~2.6 fold; p<0.05) compared to saline (Fig 5B); but that levels normalized by the 30 day time-point. In the lesioned striatum, there was no significant effect overall; however, there was a substantial (-56%), albeit non-significant, AMImediated decrease in BDNF levels over time (Fig. 5C). In the frontal cortex (Fig. 5D), ACC (Fig. 5E) and piriform (Fig. 5F) cortices, there was no effect of time or treatment (p>0.05); however, there was a trend suggesting AMI-mediated a decrease in ACC BDNF over time (p=0.06).
Figure 5. BDNF response to amitriptyline (AMI) post-lesion.
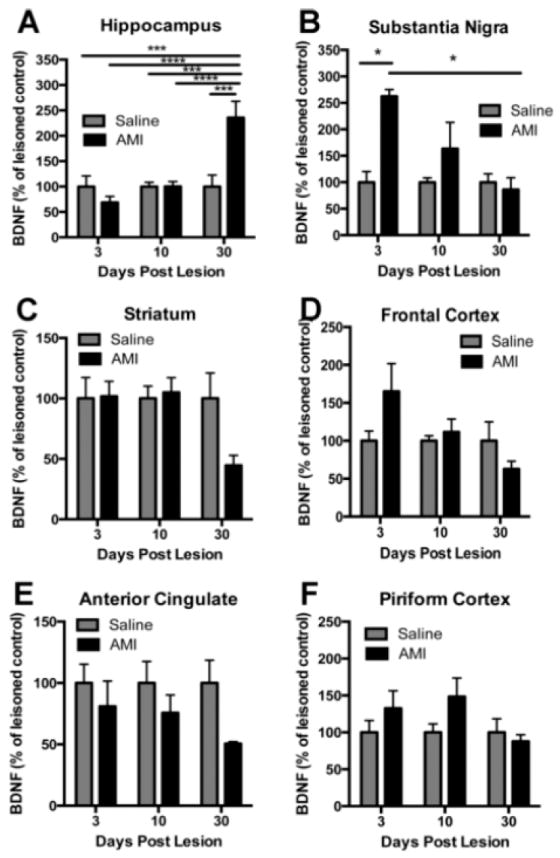
BDNF is differentially regulated in response to chronic antidepressant treatment and/or lesion. The region of interest was dissected and each hemisphere was analyzed independently with ELISA for BDNF. Adult rats were treated with saline or AMI (5mg/kg, i.p.) for 2 weeks prior to receiving 6-OHDA lesions on day 14. Subsets of these rats were sacrificed at 3, 10 or 30 days post-lesion. Levels of BDNF are depicted for each of the following regions: Hippocampus (A), substantia nigra (B), striatum (C), frontal cortex (D), anterior cingulate (E) and piriform cortex (F). Values are expressed as a percentage of saline lesioned controls. Data represent the mean+/- SEM and are combined from two separate experiments. **p<0.01, ****p<0.0001, $$p<0.001
GDNF
In contrast to BDNF, there was no significant effect of time or treatment for GDNF in the hippocampus (p>0.05; Fig. 6A). However, there was a significant effect of treatment on GDNF levels in the SN (F(1, 29)=4.37, p<0.05; Fig 6B). Post hoc analysis reveals a significant increase in SN GDNF at the early time point immediately post lesion compared to saline control (p<0.05), which returns to control levels by the 10-day time-point. There were no significant effects for striatal (p>0.05, Fig. 6C) or frontal cortex (p>0.05; Fig. 6D) GDNF levels. There was an overall effect of treatment for ACC GDNF levels (F(1, 24)=5.32, p<0.05). Post hoc analyses indicate that AMI-mediated a significant increase in ACC GDNF at the early time points compared to saline treated animals post-lesion, and these levels returned to normal by the 10-day time-point (Fig. 6E). There were no significant effects of time or treatment for GDNF levels in the piriform cortex (p>0.05; Fig 6F). Taken together, results illustrate a divergent pattern of trophic changes elicited by antidepressant treatment within the degenerating brain.
Figure 6. GDNF response to amitriptyline (AMI) post-lesion.
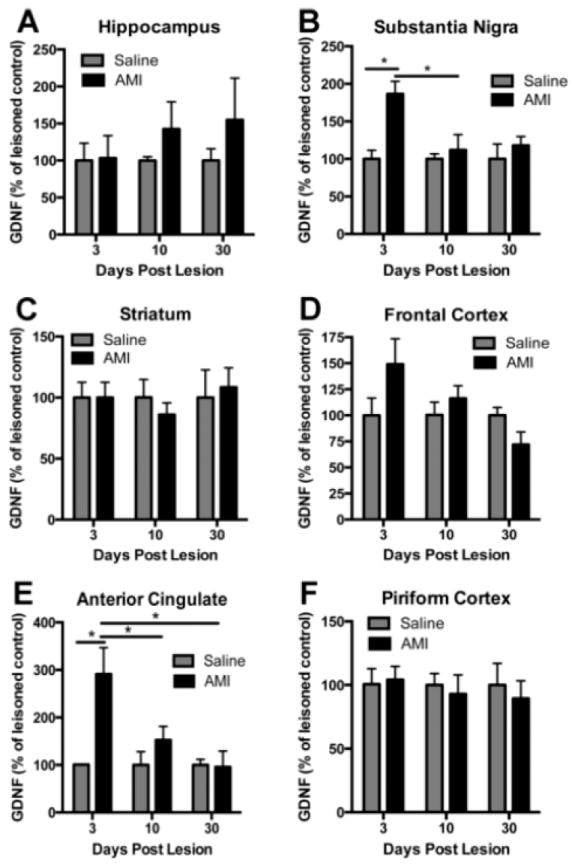
GDNF is differentially regulated in response to chronic antidepressant treatment and/or lesion. The region of interest was dissected and each hemisphere was analyzed independently with ELISA for GDNF. Adult rats were treated with saline or AMI (5mg/kg, i.p.) for 2 weeks prior to receiving 6-OHDA lesions on day 14. Subsets of these rats were sacrificed at 3, 10 or 30 days post-lesion. Levels of GDNF are depicted for each of the following regions: Hippocampus (A), substantia nigra (B), striatum (C), frontal cortex (D), anterior cingulate (E) and piriform cortex (F). Values are expressed as a percentage of saline lesioned controls. Data represent the mean+/-SEM and are combined from two separate experiments. *p<0.05, **p<0.001
Discussion
The findings of this study are highly relevant to people afflicted with PD because these individuals often experience depression and take antidepressants daily. In our evaluation of the temporal response of trophic support in the nigrostriatal system, the current study: 1) extends the scope of our previous finding that AMI treatment is indeed neuroprotective [37], and 2) shows that chronic AMI treatment elicits a trophic response that is both dynamic and divergent depending on the region examined. Our data demonstrate that within the intact nigrostriatal system, AMI-mediates a more pronounced effect on BDNF than GDNF. Pertinent to the notion that AMI may provide ongoing neuroprotection for PD patients, we observed a temporal increase in BDNF levels within both the SN and HIPP, which was maintained with chronic drug administration. Conversely, levels of BDNF were reduced by half over the course of treatment in the striatum and interconnected regions (ie., ACC and piriform cortices). Of potential added trophic benefit, AMI also increased GDNF in the SN in a time-dependent manner, which again was maintained through the extent of drug treatment. The return of GDNF to basal levels over time in the HIPP and ACC contrasts with that seen in the SN, and its significance for antidepressant activity is uncertain. While it is reasonable to suggest that the increased trophic support elicited by AMI is a likely contributor to the nigral DA neuron protection observed in this and our previous study, it is unclear why BDNF levels are decreased in the striatum, anterior cingulate and piriform cortices, and what impact this might have on the dopamine circuitry or the survival of neuronal sub-types that reside therein.
Although it is important to understand how antidepressant treatment impacts the intact nigrostriatal system, it is essential to detail these effects in the degenerating system. To determine the effect of AMI on nigral DA degeneration, the extent of DA neuron loss was measured at select time points after 6-OHDA lesion. While AMI binds primarily to norepinephrine and serotonin transporters and its affinity for the dopamine transporter is negligible [57], animals did not receive an injection of AMI/saline on the day of surgery to ensure AMI did not impede the uptake of 6-OHDA. Since AMI is rapidly cleared from plasma and brain (<12 hours) [58], we feel confident that AMI was not present to interfere with the uptake of 6-OHDA. Furthermore, we’ve previously shown that chronic AMI pre-treatment does not alter DAT levels [37], suggesting 6-OHDA could be readily taken up by dopamine neurons to induce a lesion. Results indicate both AMI- and saline-treated rats lost equivalent numbers of SN DA neurons at early post-lesion time points (ie., 3-10 days post-lesion); however, neurodegeneration continued in the saline treated animals, whereas it was halted in the AMI-treated group. We also report a dynamic trophic response mediated by AMI in the SN immediately post-lesion. This robust increase in BDNF and GDNF soon after the insult importantly suggests that AMI-mediated trophic support of DA neurons, may be a means to preserve these cells during the axonopathic “die back” that has been shown to occur early in the disease [59]. This confirms our previous result highlighting the neuroprotective ability of AMI [37] and is in line with previous reports showing antidepressants from multiple classes elicit neuroprotective effects in neurodegenerative diseases [33, 60, 61]. For example, nortriptyline (the metabolite of AMI) has been shown to prevent the death of motor neurons in an amyotrophic lateral sclerosis (ALS) mouse model and prevent cell death in a Huntington’s disease (HD) cellular model. These effects were attributed to nortriptyline’s anti-apoptotic effects (i.e., inhibition of release of cytochrome c and activation of caspase 3). Additionally, the SSRI sertraline has been shown to prolong survival, improve motor performance and decrease brain atrophy in the R6/2 Huntington’s disease (HD) mouse model [61]. The beneficial effects of sertraline were attributed to its ability to increase hippocampal BDNF and enhance neurogenesis. Similarly, treatment with the SSRI, paroxetine, prevented degeneration of nigrostriatal DA neurons, increased striatal DA levels, and improved motor function in a mouse model of PD [33]. These authors attribute paroxetine’s neuroprotective effects to anti-inflammatory mechanisms and reduction of oxidative stress. Finally, a more recent study reports that the SSRI fluoxetine ameliorates motor deficits in a transgenic model of multiple systems atrophy (MSA), with a concomitant decrease in neurodegenerative pathology in the basal ganglia, neocortex and hippocampus [62]. These investigators [62] attribute the neuroprotective effects mediated by antidepressant treatment to increased levels of hippocampal BDNF and GDNF, as well as ERK activation. Taken together, existing data suggests that antidepressants may provide neuroprotection to DA neurons via a number of different mechanisms and may have dual therapeutic value in treating both the motor and non-motor features of PD.
As previously reported, some of the therapeutic effects elicited by antidepressants may be due to increased trophic factor levels in the brain. Of all the neurotrophic factors, BDNF [13-17, 63] and GDNF [18-24, 64-72] have shown the most promising results in animal models of PD. These factors exert their effects by binding to tyrosine kinase receptors (TrkB and GDNFα/RET, respectively) that activate tyrosine kinases through receptor dimerization, followed by autophosphorylation and resultant intracellular signaling. The intracellular pathways involved in cell survival include the extracellular signal-regulated-protein kinase/ mitogen-activated protein kinase (ERK/MAPK) [7, 73] and phosphatidylinositol-3-kinase (PI3K) signaling pathways [74-76]. Moreover, the expression of these factors is activity dependent and is involved in neuronal adaptation and plasticity. Although antidepressant-mediated changes have been demonstrated in brain regions involved in depression, little is known about the morphological and biochemical changes induced by antidepressants within the degenerating nigrostriatal system. Therefore, we examined levels of BDNF and GDNF at various time points after lesion in AMI- and saline-treated rats. This cross-sectional time course study allowed us to visualize the amount of DA neuron loss relative to the changes in BDNF or GDNF at specific time-points after lesion.
The present study demonstrates AMI treatment was associated with a significant increase in SN BDNF early post-lesion, suggesting it may participate in the nigrostriatal neuroprotection afforded by AMI treatment. Additionally, levels of GDNF were increased immediately post-lesion in both the SN and ACC, which may further contribute to the AMI-mediated preservation of the DA system. Hippocampal BDNF was significantly elevated over time by AMI treatment, suggesting antidepressant efficacy despite ongoing degeneration. In fact, we previously demonstrated that the low dose and time-course of AMI administration utilized in this study exhibited antidepressant activity and attenuated motor deficits induced by the 6-OHDA lesion [37]. Although not specifically investigated, the AMI-mediated increase in hippocampal BDNF may also preserve the loss of hippocampal neurogenesis associated with PD-related depression [77]. Taken together, the present findings indicate the AMI-mediated trophic response elicited here could potentially restore diminished trophic factor levels in the aged or injured brain, thereby providing dual therapeutic value by attenuating both depression and neurodegeneration.
In contrast to the upregulation of BDNF in the SN and HIPP, striatal BDNF decreased substantially post-lesion. Although striatal BDNF levels are equivalent in AMI- and saline-treated lesioned rats at the early time-points, over time BDNF levels decline only in AMI-treated rats. Contrary to previous reports utilizing near-complete lesions that show elevated BDNF in the striatum post-lesion [78, 79], the present findings indicate striatal BDNF levels do not change substantially in response to a partial lesion. However, it is important to note that previous reports used different lesion parameters and also compared the lesioned hemisphere to the contralateral (non-lesioned) hemisphere, which we have found is changed post-lesion (data not shown). Accordingly, prior reported changes were not normalized to baseline levels in lesioned animals. Interestingly, after 6 weeks of AMI treatment, striatal BDNF levels fall to half (-50%) of control levels in both the intact and lesioned animals. This provides strong evidence that such an effect is the direct result of chronic AMI treatment, independent of ongoing neurodegeneration. Additionally, it shows that antidepressants can elicit divergent effects across the brain. To this end, BDNF has been shown to have very different effects in animal models of depression depending on the brain region involved. For example, BDNF is best characterized in the hippocampus where its induction by antidepressants is important for alleviating depressive symptoms [5, 80]. Conversely, local depletion of BDNF in the ventral tegmental area (VTA)/nucleus accumbens pathway also elicits an antidepressant-like effect [81]. Furthermore, intact BDNF function in the VTA is required for the development of social aversion in mice [82, 83]. These results are in opposition to previous reports linking increased BDNF to antidepressant efficacy. However, the increase in BDNF relative to antidepressant action has been shown primarily in the hippocampus. It is not clear why striatal BDNF decreases in response to chronic AMI treatment. Furthermore, it is not known whether this effect occurs with other antidepressants and what impact the striatal BDNF deficiency may have on the DA system or the neuronal subtypes that rely on BDNF for survival. This will be of interest in future studies.
In summary, our results show chronic AMI treatment elicits effects in brain regions that extend beyond the mesolimbic system and that AMI differentially regulates levels of BDNF and GDNF depending on the structure, and time of exposure to drug. The current studies also serve to confirm our previous report that AMI has neuroprotective effects and can halt the continuing degeneration of DA neurons elicited by intrastriatal 6-OHDA. Furthermore, our findings suggest that the most pronounced BDNF and GDNF increases are associated with the DA neuron cell bodies in SN, rather than DA terminals in striatum. Thus, based on the correlative relationship documented here between AMI, BDNF/GDNF, and DA neuron survival, it is reasonable to suggest that chronic AMI treatment induces trophic changes that may help retard neurodegeneration in the diseased brain. These data provide rationale for continuing investigations to 1) confirm a causal relationship between AMI effects on these trophic factors and DA neuron survival, 2) determine whether these factors work independently or synergistically, and 3) delve into additional factors which might be activated by AMI for development as targetable therapeutics in PD.
Highlights.
Chronic amitriptyline treatment significantly increases BDNF and GDNF in substantia nigra
Chronic amitriptyline treatment substantially decreases BDNF in striatum
Chronic amitriptyline treatment attenuates dopamine neuron loss
Tricyclic antidepressants may have dual therapeutic value in Parkinson’s disease
Acknowledgments
Funding: This work was supported by the Davis Phinney Foundation for Parkinson’s and the Morris K. Udall Center of Excellence for Parkinson’s Disease Research at Michigan State University and the University of Cincinnati (NS 058830). The authors thank Drs. Alberto Espay and Freddy Revilla at the University of Cincinnati for their endless encouragement toward this pursuit. The authors appreciate the technical expertise and intellectual contributions of Allyson Cole-Strauss, Valerie Thompson and Sara Gombash. Also, special thanks to Nathan Levine for his dedication and help with daily injections.
Footnotes
Author Roles: Katrina L. Paumier contributed to the design and conceptualization of the study, interpretation of data, draft and revision of the manuscript. Caryl Sortwell contributed to the design and conceptualization of the study, interpretation of data, draft and revision of the manuscript. Lalitha Madhavan contributed to the design and conceptualization of study, interpretation of data and revision of the manuscript. Brian Terpstra contributed to the design and conceptualization of study, interpretation of data and revision of the manuscript. Brian Daley contributed to the collection and interpretation of data and revision of the manuscript. Kathy Steece-Collier contributed to the design and conceptualization of the study, collection of and interpretation of data, draft and revision of the manuscript. Timothy J. Collier contributed to the design and conceptualization of study, interpretation of data and revision of the manuscript.
Relevant conflicts of interest/financial disclosures: All funding sources provided unrestricted support and had no role in the oversight or review of the research data or reporting.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ravina B, et al. The impact of depressive symptoms in early Parkinson disease. Neurology. 2007;69(4):342–7. doi: 10.1212/01.wnl.0000268695.63392.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez-Turrillas R, Del Rio J, Frechilla D. Sequential changes in BDNF mRNA expression and synaptic levels of AMPA receptor subunits in rat hippocampus after chronic antidepressant treatment. Neuropharmacology. 2005;49(8):1178–88. doi: 10.1016/j.neuropharm.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Xu H, Steven Richardson J, Li XM. Dose-related effects of chronic antidepressants on neuroprotective proteins BDNF, Bcl-2 and Cu/Zn-SOD in rat hippocampus. Neuropsychopharmacology. 2003;28(1):53–62. doi: 10.1038/sj.npp.1300009. [DOI] [PubMed] [Google Scholar]
- 4.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15(11):7539–47. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16(7):2365–72. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nibuya M, et al. Repeated stress increases catalytic TrkB mRNA in rat hippocampus. Neurosci Lett. 1999;267(2):81–4. doi: 10.1016/s0304-3940(99)00335-3. [DOI] [PubMed] [Google Scholar]
- 7.Hisaoka K, et al. Antidepressants increase glial cell line-derived neurotrophic factor production through monoamine independent activation of protein tyrosine kinase and extracellular signal-regulated kinase in glial cells. J Pharmacol Exp Ther. 2007 doi: 10.1124/jpet.106.116558. [DOI] [PubMed] [Google Scholar]
- 8.Hisaoka K, et al. Antidepressant drug treatments induce glial cell line-derived neurotrophic factor (GDNF) synthesis and release in rat C6 glioblastoma cells. J Neurochem. 2001;79(1):25–34. doi: 10.1046/j.1471-4159.2001.00531.x. [DOI] [PubMed] [Google Scholar]
- 9.Mercier G, et al. MAP kinase activation by fluoxetine and its relation to gene expression in cultured rat astrocytes. J Mol Neurosci. 2004;24(2):207–16. doi: 10.1385/JMN:24:2:207. [DOI] [PubMed] [Google Scholar]
- 10.Rogoz Z, Legutko B. Combined treatment with imipramine and metyrapone induces hippocampal and cortical brain-derived neurotrophic factor gene expression in rats. Pharmacol Rep. 2005;57(6):840–4. [PubMed] [Google Scholar]
- 11.Coppell AL, Pei Q, Zetterstrom TS. Bi-phasic change in BDNF gene expression following antidepressant drug treatment. Neuropharmacology. 2003;44(7):903–10. doi: 10.1016/s0028-3908(03)00077-7. [DOI] [PubMed] [Google Scholar]
- 12.Allen SJ, et al. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther. 2013;138(2):155–75. doi: 10.1016/j.pharmthera.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 13.Sauer H, et al. Brain-derived neurotrophic factor enhances striatal neuropeptide expression in both the intact and the dopamine-depleted rat striatum. Neuroreport. 1994;5(5):609–12. doi: 10.1097/00001756-199401000-00019. [DOI] [PubMed] [Google Scholar]
- 14.Zhou J, Bradford HF, Stern GM. Influence of BDNF on the expression of the dopaminergic phenotype of tissue used for brain transplants. Brain Res Dev Brain Res. 1997;100(1):43–51. doi: 10.1016/s0165-3806(97)00019-9. [DOI] [PubMed] [Google Scholar]
- 15.Olson L. Grafts and growth factors in CNS. Basic science with clinical promise. Stereotact Funct Neurosurg. 1990;54-55:250–67. doi: 10.1159/000100220. [DOI] [PubMed] [Google Scholar]
- 16.Yurek DM, et al. BDNF enhances the functional reinnervation of the striatum by grafted fetal dopamine neurons. Exp Neurol. 1996;137(1):105–18. doi: 10.1006/exnr.1996.0011. [DOI] [PubMed] [Google Scholar]
- 17.Altar CA, et al. Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc Natl Acad Sci U S A. 1992;89(23):11347–51. doi: 10.1073/pnas.89.23.11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bjorklund A, et al. Studies on neuroprotective and regenerative effects of GDNF in a partial lesion model of Parkinson’s disease. Neurobiol Dis. 1997;4(3-4):186–200. doi: 10.1006/nbdi.1997.0151. [DOI] [PubMed] [Google Scholar]
- 19.Zawada WM, et al. Growth factors improve immediate survival of embryonic dopamine neurons after transplantation into rats. Brain Res. 1998;786(1-2):96–103. doi: 10.1016/s0006-8993(97)01408-x. [DOI] [PubMed] [Google Scholar]
- 20.Kordower JH, et al. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science. 2000;290(5492):767–73. doi: 10.1126/science.290.5492.767. [DOI] [PubMed] [Google Scholar]
- 21.Mendez I, et al. Enhancement of survival of stored dopaminergic cells and promotion of graft survival by exposure of human fetal nigral tissue to glial cell line--derived neurotrophic factor in patients with Parkinson’s disease. Report of two cases and technical considerations. J Neurosurg. 2000;92(5):863–9. doi: 10.3171/jns.2000.92.5.0863. [DOI] [PubMed] [Google Scholar]
- 22.Hebb AO, et al. Glial cell line-derived neurotrophic factor-supplemented hibernation of fetal ventral mesencephalic neurons for transplantation in Parkinson disease: longterm storage. Neurosurg Focus. 2002;13(5):e4. doi: 10.3171/foc.2002.13.5.5. [DOI] [PubMed] [Google Scholar]
- 23.McBride JL, Kordower JH. Neuroprotection for Parkinson’s disease using viral vector-mediated delivery of GDNF. Prog Brain Res. 2002;138:421–32. doi: 10.1016/S0079-6123(02)38091-9. [DOI] [PubMed] [Google Scholar]
- 24.Kirik D, Georgievska B, Bjorklund A. Localized striatal delivery of GDNF as a treatment for Parkinson disease. Nat Neurosci. 2004;7(2):105–10. doi: 10.1038/nn1175. [DOI] [PubMed] [Google Scholar]
- 25.Patel NK, et al. Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: a two-year outcome study. Ann Neurol. 2005;57(2):298–302. doi: 10.1002/ana.20374. [DOI] [PubMed] [Google Scholar]
- 26.Slevin JT, et al. Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J Neurosurg. 2005;102(2):216–22. doi: 10.3171/jns.2005.102.2.0216. [DOI] [PubMed] [Google Scholar]
- 27.Gill SS, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9(5):589–95. doi: 10.1038/nm850. [DOI] [PubMed] [Google Scholar]
- 28.Lang AE, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59(3):459–66. doi: 10.1002/ana.20737. [DOI] [PubMed] [Google Scholar]
- 29.Castren E. Neurotrophins as mediators of drug effects on mood, addiction, and neuroprotection. Mol Neurobiol. 2004;29(3):289–302. doi: 10.1385/MN:29:3:289. [DOI] [PubMed] [Google Scholar]
- 30.Castren E. Neurotrophic effects of antidepressant drugs. Curr Opin Pharmacol. 2004;4(1):58–64. doi: 10.1016/j.coph.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 31.De Foubert G, et al. Fluoxetine-induced change in rat brain expression of brainderived neurotrophic factor varies depending on length of treatment. Neuroscience. 2004;128(3):597–604. doi: 10.1016/j.neuroscience.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 32.Khundakar AA, Zetterstrom TS. Biphasic change in BDNF gene expression following antidepressant drug treatment explained by differential transcript regulation. Brain Res. 2006;1106(1):12–20. doi: 10.1016/j.brainres.2006.05.063. [DOI] [PubMed] [Google Scholar]
- 33.Chung YC, Kim SR, Jin BK. Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson’s disease. J Immunol. 2010;185(2):1230–7. doi: 10.4049/jimmunol.1000208. [DOI] [PubMed] [Google Scholar]
- 34.Menza M, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology. 2009;72(10):886–92. doi: 10.1212/01.wnl.0000336340.89821.b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen P, et al. Antidepressant treatment of veterans with Parkinson’s disease and depression: analysis of a national sample. J Geriatr Psychiatry Neurol. 2007;20(3):161–5. doi: 10.1177/0891988707301866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paumier KL, et al. Tricyclic antidepressants delay the need for dopaminergic therapy in early Parkinson’s disease. Mov Disord. 2012;27(7):880–7. doi: 10.1002/mds.24978. [DOI] [PubMed] [Google Scholar]
- 37.Paumier KL, et al. Chronic Amitriptyline Treatment Attenuates Nigrostriatal Degeneration and Significantly Alters Trophic Support in a Rat Model of Parkinsonism. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Institute of Laboratory Animal Research, C.o.L.S. Guide for the Care and Use of Laboratory Animals. Washington, D.C: The National Academies Press; 1996. National Research Council; p. 124. [Google Scholar]
- 39.Altar CA, DiStefano PS. Neurotrophin trafficking by anterograde transport. Trends Neurosci. 1998;21(10):433–7. doi: 10.1016/s0166-2236(98)01273-9. [DOI] [PubMed] [Google Scholar]
- 40.Altar CA, et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389(6653):856–60. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 41.Mendez I, Hong M. Reconstruction of the striato-nigro-striatal circuitry by simultaneous double dopaminergic grafts: a tracer study using fluorogold and horseradish peroxidase. Brain Res. 1997;778(1):194–205. doi: 10.1016/s0006-8993(97)01055-x. [DOI] [PubMed] [Google Scholar]
- 42.Gasbarri A, et al. Anterograde and retrograde tracing of projections from the ventral tegmental area to the hippocampal formation in the rat. Brain Res Bull. 1994;33(4):445–52. doi: 10.1016/0361-9230(94)90288-7. [DOI] [PubMed] [Google Scholar]
- 43.Gasbarri A, et al. The projections of the retrorubral field A8 to the hippocampal formation in the rat. Exp Brain Res. 1996;112(2):244–52. doi: 10.1007/BF00227643. [DOI] [PubMed] [Google Scholar]
- 44.Mufson EJ, et al. Distribution and retrograde transport of trophic factors in the central nervous system: functional implications for the treatment of neurodegenerative diseases. Prog Neurobiol. 1999;57(4):451–84. doi: 10.1016/s0301-0082(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 45.Rite I, et al. Divergent regulatory mechanisms governing BDNF mRNA expression in cerebral cortex and substantia nigra in response to striatal target ablation. Exp Neurol. 2005;192(1):142–55. doi: 10.1016/j.expneurol.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 46.Datiche F, Cattarelli M. Catecholamine innervation of the piriform cortex: a tracing and immunohistochemical study in the rat. Brain Res. 1996;710(1-2):69–78. doi: 10.1016/0006-8993(95)01279-6. [DOI] [PubMed] [Google Scholar]
- 47.Shibata H, Naito J. Organization of anterior cingulate and frontal cortical projections to the retrosplenial cortex in the rat. J Comp Neurol. 2008;506(1):30–45. doi: 10.1002/cne.21523. [DOI] [PubMed] [Google Scholar]
- 48.Calzavara R, Mailly P, Haber SN. Relationship between the corticostriatal terminals from areas 9 and 46, and those from area 8A, dorsal and rostral premotor cortex and area 24c: an anatomical substrate for cognition to action. Eur J Neurosci. 2007;26(7):2005–24. doi: 10.1111/j.1460-9568.2007.05825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Domesick VB. Projections from the cingulate cortex in the rat. Brain Res. 1969;12(2):296–320. doi: 10.1016/0006-8993(69)90002-x. [DOI] [PubMed] [Google Scholar]
- 50.Takada M, Hattori T. Collateral projections from the substantia nigra to the cingulate cortex and striatum in the rat. Brain Res. 1986;380(2):331–5. doi: 10.1016/0006-8993(86)90230-1. [DOI] [PubMed] [Google Scholar]
- 51.Paumier KatrinaL, S CE, Madhavan Lalitha, Terpstra Brian, Celano StephanieL, Green JoshuaJ, Imus NastassjaM, Marckini Nathan, Daley Brian, Collier KathySteece, Collier TimothyJ. Chronic amitriptyline treatment significantly attenuates DA neuron loss, preserves striatal fibers and motor function in a rat model of parkinsonism. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.262. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Loos CM. Multiple immunoenzyme staining: methods and visualizations for the observation with spectral imaging. J Histochem Cytochem. 2008;56(4):313–28. doi: 10.1369/jhc.2007.950170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gundersen HJ, et al. The efficiency of systematic sampling in stereology--reconsidered. J Microsc. 1999;193(Pt 3):199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- 54.Okamoto H, et al. Dynamic changes in AP-1 transcription factor DNA binding activity in rat brain following administration of antidepressant amitriptyline and brain-derived neurotrophic factor. Neuropharmacology. 2003;45(2):251–9. doi: 10.1016/s0028-3908(03)00148-5. [DOI] [PubMed] [Google Scholar]
- 55.Lapchak PA, et al. Glial cell line-derived neurotrophic factor: distribution and pharmacology in the rat following a bolus intraventricular injection. Brain Res. 1997;747(1):92–102. doi: 10.1016/s0006-8993(96)01265-6. [DOI] [PubMed] [Google Scholar]
- 56.Pong K, et al. Inhibition of glial cell line-derived neurotrophic factor induced intracellular activity by K-252b on dopaminergic neurons. J Neurochem. 1997;69(3):986–94. doi: 10.1046/j.1471-4159.1997.69030986.x. [DOI] [PubMed] [Google Scholar]
- 57.Tatsumi M, et al. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol. 1997;340(2-3):249–58. doi: 10.1016/s0014-2999(97)01393-9. [DOI] [PubMed] [Google Scholar]
- 58.Coudore F, et al. Plasma and brain pharmacokinetics of amitriptyline and its demethylated and hydroxylated metabolites after one and six half-life repeated administrations to rats. Gen Pharmacol. 1996;27(2):215–9. doi: 10.1016/0306-3623(95)02008-x. [DOI] [PubMed] [Google Scholar]
- 59.Kordower JH, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136(Pt 8):2419–31. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H, et al. Nortriptyline delays disease onset in models of chronic neurodegeneration. Eur J Neurosci. 2007;26(3):633–41. doi: 10.1111/j.1460-9568.2007.05663.x. [DOI] [PubMed] [Google Scholar]
- 61.Peng Q, et al. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington’s disease mouse model. Exp Neurol. 2008;210(1):154–63. doi: 10.1016/j.expneurol.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ubhi K, et al. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of alpha-synucleinopathy. Exp Neurol. 2012;234(2):405–16. doi: 10.1016/j.expneurol.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoglinger GU, et al. Influence of time in culture and BDNF pretreatment on survival and function of grafted embryonic rat ventral mesencephalon in the 6-OHDA rat model of Parkinson’s disease. Exp Neurol. 2001;167(1):148–57. doi: 10.1006/exnr.2000.7546. [DOI] [PubMed] [Google Scholar]
- 64.Johansson M, et al. Effects of glial cell line-derived neurotrophic factor on developing and mature ventral mesencephalic grafts in oculo. Exp Neurol. 1995;134(1):25–34. doi: 10.1006/exnr.1995.1033. [DOI] [PubMed] [Google Scholar]
- 65.Winkler C, et al. Short-term GDNF treatment provides long-term rescue of lesioned nigral dopaminergic neurons in a rat model of Parkinson’s disease. J Neurosci. 1996;16(22):7206–15. doi: 10.1523/JNEUROSCI.16-22-07206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Granholm AC, et al. Glial cell line-derived neurotrophic factor improves survival of ventral mesencephalic grafts to the 6-hydroxydopamine lesioned striatum. Exp Brain Res. 1997;116(1):29–38. doi: 10.1007/pl00005741. [DOI] [PubMed] [Google Scholar]
- 67.Lapchak PA, et al. Adenoviral vector-mediated GDNF gene therapy in a rodent lesion model of late stage Parkinson’s disease. Brain Res. 1997;777(1-2):153–60. doi: 10.1016/s0006-8993(97)01100-1. [DOI] [PubMed] [Google Scholar]
- 68.Bauer M, et al. Nonviral glial cell-derived neurotrophic factor gene transfer enhances survival of cultured dopaminergic neurons and improves their function after transplantation in a rat model of Parkinson’s disease. Hum Gene Ther. 2000;11(11):1529–41. doi: 10.1089/10430340050083261. [DOI] [PubMed] [Google Scholar]
- 69.Espejo M, et al. Increased survival of dopaminergic neurons in striatal grafts of fetal ventral mesencephalic cells exposed to neurotrophin-3 or glial cell line-derived neurotrophic factor. Cell Transplant. 2000;9(1):45–53. doi: 10.1177/096368970000900107. [DOI] [PubMed] [Google Scholar]
- 70.Kirik D, et al. Long-term rAAV-mediated gene transfer of GDNF in the rat Parkinson’s model: intrastriatal but not intranigral transduction promotes functional regeneration in the lesioned nigrostriatal system. J Neurosci. 2000;20(12):4686–700. doi: 10.1523/JNEUROSCI.20-12-04686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akerud P, et al. Neuroprotection through delivery of glial cell line-derived neurotrophic factor by neural stem cells in a mouse model of Parkinson’s disease. J Neurosci. 2001;21(20):8108–18. doi: 10.1523/JNEUROSCI.21-20-08108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kirik D, et al. Delayed infusion of GDNF promotes recovery of motor function in the partial lesion model of Parkinson’s disease. Eur J Neurosci. 2001;13(8):1589–99. doi: 10.1046/j.0953-816x.2001.01534.x. [DOI] [PubMed] [Google Scholar]
- 73.Worby CA, et al. Glial cell line-derived neurotrophic factor signals through the RET receptor and activates mitogen-activated protein kinase. J Biol Chem. 1996;271(39):23619–22. doi: 10.1074/jbc.271.39.23619. [DOI] [PubMed] [Google Scholar]
- 74.Onyango IG, Tuttle JB, Bennett JP., Jr Brain-derived growth factor and glial cell line-derived growth factor use distinct intracellular signaling pathways to protect PD cybrids from H2O2-induced neuronal death. Neurobiol Dis. 2005;20(1):141–54. doi: 10.1016/j.nbd.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 75.Salvatore MF, et al. Striatal GDNF administration increases tyrosine hydroxylase phosphorylation in the rat striatum and substantia nigra. J Neurochem. 2004;90(1):245–54. doi: 10.1111/j.1471-4159.2004.02496.x. [DOI] [PubMed] [Google Scholar]
- 76.Soler RM, et al. Receptors of the glial cell line-derived neurotrophic factor family of neurotrophic factors signal cell survival through the phosphatidylinositol 3-kinase pathway in spinal cord motoneurons. J Neurosci. 1999;19(21):9160–9. doi: 10.1523/JNEUROSCI.19-21-09160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suzuki K, et al. Destruction of dopaminergic neurons in the midbrain by 6- hydroxydopamine decreases hippocampal cell proliferation in rats: reversal by fluoxetine. PLoS One. 5(2):e9260. doi: 10.1371/journal.pone.0009260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yurek DM, Fletcher-Turner A. Differential expression of GDNF, BDNF, and NT-3 in the aging nigrostriatal system following a neurotoxic lesion. Brain Res. 2001;891(1-2):228–35. doi: 10.1016/s0006-8993(00)03217-0. [DOI] [PubMed] [Google Scholar]
- 79.Yurek DM, Fletcher-Turner A. Lesion-induced increase of BDNF is greater in the striatum of young versus old rat brain. Exp Neurol. 2000;161(1):392–6. doi: 10.1006/exnr.1999.7274. [DOI] [PubMed] [Google Scholar]
- 80.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54(7):597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 81.Fanous S, et al. Viral depletion of VTA BDNF in rats modulates social behavior, consequences of intermittent social defeat stress, and long-term weight regulation. Neurosci Lett. 2011;502(3):192–6. doi: 10.1016/j.neulet.2011.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berton O, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311(5762):864–8. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 83.Eisch AJ, et al. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biol Psychiatry. 2003;54(10):994–1005. doi: 10.1016/j.biopsych.2003.08.003. [DOI] [PubMed] [Google Scholar]