

Abstract
The impact of coronary artery stenosis (CAS) to renal injury is unknown. Here we tested whether the existence of CAS, regardless of concurrent atherosclerosis, would induce kidney injury and magnify its susceptibility to damage from co-existing hypertension (HT). Pigs (7 each) were assigned to Sham, left-circumflex CAS, renovascular HT, and CAS plus HT groups. Cardiac and non-stenotic kidney functions, circulating and renal inflammatory and oxidative markers, and renal and microvascular remodeling, were assessed 10 weeks later. Myocardial perfusion declined distal to CAS. Systemic levels of PGF2-α isoprostane, a marker of oxidative stress, increased in CAS and CAS plus HT, while single-kidney blood flow responses to acetylcholine were significantly blunted only in CAS plus HT compared to sham, HT, and CAS, indicating renovascular endothelial dysfunction. Tissue expression of inflammatory and oxidative markers were elevated in the CAS pig kidney, and further magnified in CAS plus HT, whereas angiogenic factor expression was decreased. Bendavia, a mitochondria-targeted peptide, decreased oxidative stress and improved renal function and structure in CAS. Furthermore, CAS and HT synergistically amplified glomerulosclerosis and renal fibrosis. Thus, mild myocardial ischemia, independent of systemic atherosclerosis, induced renal injury, possibly mediated by increased oxidative stress. Superimposed HT aggravates renal inflammation and endothelial dysfunction caused by CAS, and synergistically promotes kidney fibrosis, providing impetus to preserve cardiac integrity in order to protect the kidney.
Keywords: coronary artery stenosis, renovascular hypertension, renal fibrosis
Atherosclerotic coronary artery disease is the most common cause of death in the US,(1) and often accompanied by diffuse vascular disease in other organs. An increase in systemic inflammation and oxidative stress may mediate some of the systemic manifestations of atherosclerosis,(2) yet myocardial ischemia may impose adverse effects on remote organs and vascular territories.(3) For example, myocardial infarction and stroke augment inflammation, and in turn atherosclerosis,(4) and can trigger morphological and functional changes in the kidney.(5) However, the isolated effects on the kidney of myocardial ischemia dissociated from infarction and systemic atherosclerosis have been difficult to discern.
Patients with coronary artery disease often also have hypertension (HT), an important cause of chronic kidney disease (CKD).(6) Previous studies have shown increased prevalence of renovascular disease in patients with coronary artery disease,(7-9) in whom renal insufficiency is the main clinical predictor of renal artery stenosis. Renal artery stenosis is an important cause of secondary HT in the elderly population, which may ultimately lead to end-stage renal disease and represents a sizeable fraction of new patients entering dialysis. Importantly, hypertensive patients at risk for renovascular disease often present with severe coronary artery disease, and renal function correlates with its presence.(10) We have also recently shown that coexistence of coronary and renal artery diseases in human subjects magnifies renal injury.(11,12)
The preglomerular vasculature exposed to the HT develops progressive vascular pathology,(13) magnified by loss of autoregulation with glomerular hypertrophy, hyperfiltration and focal segmental glomerulosclerosis. Peritubular capillary loss and consequent hypoxia lead to tubular atrophy and interstitial fibrosis.(14) We have also shown that renovascular HT blunts anti-oxidant defense mechanisms in the non-stenotic kidney.(15) These functional and structural alterations may render kidneys exposed to HT susceptible to other comorbidities, and amplify renal damage.
However, whether non-atherosclerotic coronary artery stenosis (CAS) alone induces kidney injury or increases its vulnerability to adverse effects of coexisting HT remains unclear. The present study was therefore designed to test the hypothesis that isolated CAS elicits kidney inflammation and oxidative stress, which are exacerbated by HT and impair renal function.
Results
Renal-artery-stenosis increased mean arterial pressure (MAP) in both HT and CAS+HT compared with Sham (Table 1, P=0.03 for both). Body weight and plasma renin activity (PRA) were not significantly different among the groups, but serum creatinine (Scr) was elevated in CAS, HT and CAS+HT (Table 1, P<0.04 each), affected by both CAS and HT. Urinary protein, affected by HT, increased significantly only in CAS+HT compared to Sham and CAS (Table 1, P=0.006 and P=0.045, respectively). Circulating PGF2-α isoprostane levels increased in CAS and CAS+HT compared to Sham and HT (Figure 1A, P<0.05 each), while systemic transforming growth-factor (TGF)-β1 levels increased by HT in HT and CAS+HT (Figure 1B, P=0.02 and P=0.03 vs. Sham, respectively). Norepinephrine levels were slightly reduced in CAS only compared to HT. Systemic levels of granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-1α, IL-1β, IL-1ra, IL-2, IL-4, IL-6, IL-8, IL10, IL12, IL18 and tumor necrosis factor (TNF)-α were not significantly different among the four groups (Table 1s, P>0.05 for all).
Table 1.
Systemic characteristics and cardiac Function in the 4 groups (mean±SEM, n=7 each).
| Sham | HT | CAS | CAS+HT | P Value for Two-way ANOVA | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| HT | CAS | HT×CAS | |||||
| Body weight, kg | 48.0±2.8 | 48.3±5.2 | 51.6±4.6 | 46.1±5.0 | |||
| MAP, mmHg | 96.7±2.8 | 116.5±7.8* | 101.3±5.6 | 112.5±7.0* | 0.028 | 0.961 | 0.519 |
| PRA, pg·ml-1·h-1 | 0.17±0.10 | 0.25±0.16 | 0.22±0.07 | 0.24±0.15 | 0.505 | 0.786 | 0.648 |
| Creatinine, mg·dL-1 | 1.17±0.31 | 1.58±0.17* | 1.62±0.32* | 1.80±0.16* | 0.016 | 0.008 | 0.326 |
| Urine Protein,μg·ml-1 | 16.9±2.5 | 22.1±5.9 | 18.4±6.1 | 33.4±4.7*& | 0.047 | 0.199 | 0.319 |
| Norepinephrine,ng·ml-1 | 0.08±0.02 | 0.10±0.02 | 0.04±0.01# | 0.05±0.01 | 0.290 | 0.01 | 0.583 |
| Degree of CAS, % | 0 | 0 | 77.3±8.9*# | 74.0±10.3*# | |||
| Stoke volume, ml | 48±2 | 50±3 | 48±5 | 37±3*# | 0.262 | 0.045 | 0.078 |
| Ejection fraction, % | 50±3 | 57±4 | 45±5 | 45±3 | 0.421 | 0.046 | 0.426 |
| E/A | 1.07±0.06 | 1.02±0.15 | 1.20±0.15 | 1.14±0.12 | 0.652 | 0.313 | 0.946 |
HT, hypertension; CAS, coronary artery stenosis; MAP, mean arterial pressure; PRA, plasma renin activity; E/A, early-to-late left ventricular filling velocities.
P<0.05 vs. Sham;
P<0.05 vs. HT;
P<0.05 vs. CAS.
Figure 1.
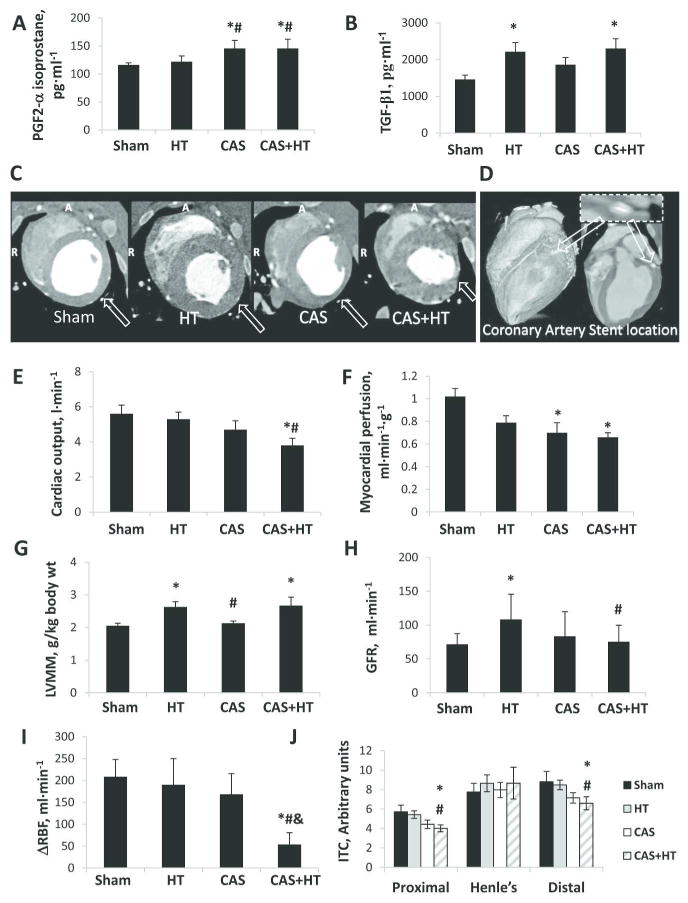
Systemic, cardiac, and renal characteristics in Sham, hypertension (HT), coronary artery stenosis (CAS), and CAS+HT pigs. A. Circulating PGF2-α isoprostane. B. Systemic transforming growth-factor (TGF)-β1. C-D. Representative cardiac CT images obtained at the middle left-ventricle (LV). In hypertensive animals (HT and CAS+HT), the LV wall is thickened. The arrow in C shows myocardial lateral wall thinning distal to a high-grade CAS (illustrated in D). E, Cardiac output. F, Myocardial perfusion. G, LV muscle mass (LVMM). H, Glomerular filtration rate (GFR). I, Change in renal blood flow (ΔRBF) in response to acetylcholine (Ach). J, Intratubular concentration (ITC) in the different nephron segments. *P<0.05 vs. Sham; #P<0.05 vs. HT; &P<0.05 vs. CAS.
Cardiac Function
Ten weeks after induction, the pigs developed significant and similar degree of CAS (Table 1, Figure 1D), but stroke volume and cardiac output fell due to CAS only in CAS+HT compared to Sham and HT (Table 1, Figure 1E, P≤0.03 for each). The ejection fraction (albeit slightly affected by CAS) and early and late LV filling velocities (E/A) were not significantly different among the four groups (Table 1). Myocardial perfusion distal to CAS and CAS+HT was lower than Sham (Figure 1F, P=0.02 and P=0.01, respectively), while LV muscle mass (LVMM) was increased in HT and CAS+HT (Figure 1C, G, P≤0.03 for both).
Renal Hemodynamics and Function
The degree of renal artery stenosis was not different between HT and CAS+HT, but renal blood flow (RBF) and glomerular filtration rate (GFR) of the non-stenotic kidney were elevated only in HT; RBF was increased by HT, suppressed by CAS, and showed a significant interaction between the two (Table 2). GFR also tended to be blunted by the interaction HT×CAS (Figure 1H). RBF response to Ach was attenuated by CAS only in CAS+HT (P=0.08 vs. baseline, Figure 1-I), and became lower than Sham (P=0.004), by a significant interaction CAS×HT (Table 2).
Table 2.
Non-stenotic kidney hemodynamics and function in Sham, coronary artery stenosis (CAS), hypertension (HT) and CAS+HT pigs (mean±SEM, n=7 each).
| Sham | HT | CAS | CAS+HT | P Value for Two-way ANOVA | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| HT | CAS | HT×CAS | |||||
| Degree of renal artery stenosis, % | 0 | 81.7±7.5*& | 0 | 80.7±5.7*& | |||
| RBF, ml·min-1 | |||||||
| Basal | 469.0±63.1 | 779.4±81.1* | 422.3±82.5# | 428.5±51.0# | 0.031 | 0.008 | 0.037 |
| Ach | 677.4±32.5† | 969.4±83.3*† | 590.6±111.0#† | 482.0±48.5*# | 0.192 | 0.0003 | 0.007 |
| Perfusion, ml·min-1·ml-1 | |||||||
| Cortex: baseline | 4.10±0.47 | 5.03±0.75 | 3.80±0.46# | 4.03±0.30# | 0.141 | 0.103 | 0.371 |
| Ach | 6.21±0.29† | 6.30±0.59† | 4.94±0.60*#† | 5.12±0.40*#† | 0.781 | 0.017 | 0.920 |
| Medulla: baseline | 2.84±0.44 | 2.19±0.10 | 1.98±0.35 | 2.00±0.35 | 0.370 | 0.146 | 0.342 |
| Ach | 3.77±0.55† | 3.47±0.43† | 3.82±0.47† | 2.48±0.39*& | 0.095 | 0.328 | 0.281 |
| RVR, mmHg·min-1·ml-1 | 0.18±0.02 | 0.16±0.02 | 0.27±0.04*# | 0.29±0.04*# | 0.511 | 0.012 | 0.174 |
RBF, renal blood flow; Ach, acetylcholine; ITC, intratubular concentration; RVR, renal vascular resistance.
P<0.05 vs. Sham;
P<0.05 vs. HT;
P<0.05 vs. CAS.
P<0.05 vs. baseline.
Basal cortical perfusion in CAS and CAS+HT was lower than in HT, and after Ach infusion was also lower than in Sham (Table 2, P<0.05 for each). Basal medullary perfusion was not significantly different among the four groups, but its response to Ach was blunted only in CAS+HT. Renal vascular resistance (RVR) was elevated in CAS and CAS+HT compared to Sham and HT (Table 2) due to CAS. CAS also decreased proximal and distal tubular Intratubular concentration (ITC) in CAS+HT (Figure 1J), suggesting impaired tubular fluid reabsorption.
Renal Histology
Trichrome staining showed increased renal fibrosis in HT and CAS compared with Sham pigs (P=0.003 and P=0.001, respectively). Compared with other groups, renal fibrosis and glomerular score in CAS+HT increased significantly (Figure 2A-C, P<0.01 each), showing interactions between CAS and HT on both fibrosis (Figure 2B, P=0.0008) and glomerulosclerosis (Figure 2C, P=0.04). Dihydroethidium (DHE) staining was elevated in CAS (P=0.03 vs. Sham), and further exacerbated in CAS+HT (Figure 2A-DHE, D, P=0.006 vs. Sham and P=0.03 vs. HT).
Figure 2.
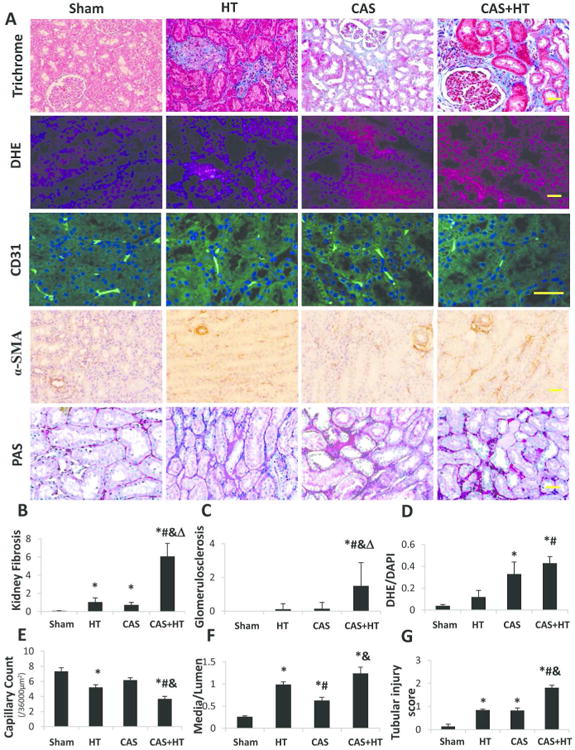
Renal tissue remodeling. A. Representative renal trichrome (×20), dihydroethidium (DHE) (×20), CD31 immunofluorescence (×40), α-SMA (×20) and PAS staining (×20). Kidney fibrosis increased in hypertension (HT) and coronary artery stenosis (CAS) compared with Sham; kidney fibrosis and glomerular score increased synergistically in CAS+HT compared with the other groups (B, C). DHE staining (normalized to DAPI-positive nuclei) increased in CAS compared with Sham, and in CAS+HT compared with Sham and HT (D). Capillary density (CD31 immunofluorescence) in HT decreased compared with Sham, but in CAS +HT fell lower than all other groups (E). Renal microvascular media-to-lumen ratio (α-SMA) increased in CAS compared with Sham, and further in HT and CAS+HT compared with Sham and CAS (F). Tubular injury score (PAS staining) increased in HT and CAS, and further aggravated in CAS+HT (G). *P<0.05 vs. Sham; #P<0.05 vs. HT; &P<0.05 vs. CAS; Δ P<0.05 for synergistic interaction CAS×HT. Scale bar = 50μm.
Compared with Sham, renal capillary density fell in HT (Figure 2, P=0.02), yet in CAS+HT decreased further compared to all groups (P<0.04 each). Microvascular wall thickening (media-to-lumen ratio) increased in CAS (P=0.0003 vs. Sham), but further in HT and CAS+HT (P<0.001 each). Tubular injury observed in HT and CAS was further aggravated in CAS+HT (P<0.001). None showed CAS×HT interactions.
Compared with Sham, renal protein expression of TNFα increased in CAS (Figures 3A and 1s, P=0.03), and further in CAS+HT (P=0.004 and P=0.002, respectively). Monocyte chemoattractant protein (MCP)-1 and GP91-phox expressions were all higher in CAS and CAS+HT than in Sham and HT (Figures 3A and 1s), affected by CAS alone (P<0.003 each), whereas nitrotyrosine was not. Hypoxia-inducible factor (HIF)-1α expression increased by CAS only in CAS+HT kidneys compared to Sham and HT (Figures 3B and 2s), suggesting tissue hypoxia. Vascular endothelial growth factor (VEGF) expression decreased in HT and further fell in CAS+HT, by both CAS and HT. Renal TGF-β1 levels increased in HT compared with Sham, but markedly rose in CAS and CAS+HT (Figures 3B, D and 2s). Tissue inhibitor of metalloproteinases (TIMP)-1 was elevated by HT in CAS+HT compared to other groups (P<0.04 for each). Protein expression of endothelial nitric-oxide synthase (eNOS) was downregulated in HT, and further in CAS and CAS+HT (P≤0.01 each), showing a significant interaction CAS×HT (P=0.03).
Figure 3.
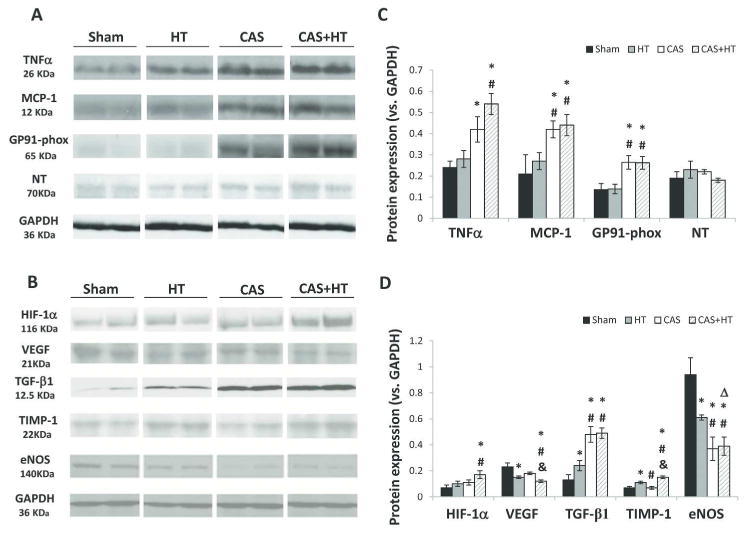
Renal expression of inflammatory, oxidative, and growth factors. Representative (2 bands shown per group) immunoblotting of (A): tumor necrosis-factor (TNF)-α, monocyte chemoattractant protein (MCP)-1, GP91-phox, nitrotyrosine (NT); (B): hypoxia-inducible factor (HIF)-1α, vascular endothelial growth-factor (VEGF), transforming growth-factor (TGF)-β1, tissue inhibitor of metalloproteinase (TIMP)-1, endothelial nitric-oxide synthase (eNOS). Inflammatory and oxidative markers were elevated in coronary artery stenosis (CAS), and further augmented in CAS+Hypertension (HT) (C). Hypoxia and fibrotic markers increased in CAS+HT, whereas eNOS decreased; VEGF expression decreased in HT and further fell in CAS+HT (D). * P<0.05 vs. Sham; # P<0.05 vs. HT; &P<0.05 vs. CAS; Δ P<0.05 for synergistic interaction CAS*HT.
Additional mechanistic exploration
Induction of femoral artery stenosis (FAS) instead of CAS in additional pigs, using the same local-irritant coil, had no effect on renal function or endothelial function (Table 2s), oxidative stress, or fibrosis (Figure 3s) (P>0.05 vs. Sham for all).
In additional animals chronically treated with the mitochondrial-targeted antioxidant bendavia, myocardial perfusion was not different from that in untreated CAS, yet Scr that was elevated in CAS fell in CAS+Bendavia, and MAP and renal hemodynamics were unchanged compared to CAS (Table 3s). Kidney fibrosis, DHE, microvascular media/lumen ratio, and tubular injury in CAS+Bendavia all decreased compared to untreated CAS (Figure 4s), and TNF-α, P67, GP91 and TGF-β1 were downregulated, whereas eNOS expression remained unchanged (Figure 5s).
Discussion
This study demonstrates that a hemodynamically significant but non-atherosclerotic CAS leads to renal dysfunction, oxidative stress, inflammation, endothelial dysfunction, and fibrosis, which are associated with systemic oxidative stress. Furthermore, coexisting experimental HT synergistically accelerates CAS-induced kidney fibrosis and glomerulosclerosis. These were accompanied by renovascular endothelial dysfunction and pronounced systemic and kidney tissue oxidative stress, microvascular injury, and hypoxia, which might contribute to progression of renal injury that characterizes coexisting CAS and HT. The current studies therefore demonstrate an interaction between cardiac and renal function beyond systemic atherosclerosis.
Coronary artery disease remains the main cause of death in the Western society. Its chief etiology is atherosclerosis, in which cytokines instigate release of inflammatory and cytotoxic molecules.(2) Nevertheless, myocardial infarction per-se can also trigger systemic inflammation that can accelerate atherosclerosis(4) or renal inflammation in mice.(16) However, the contribution of chronic low-level myocardial ischemia to renal disease is unknown.
In recent years, deleterious interactions along the cardiac-renal axis have been increasingly recognized(17), particularly in association with heart failure.(18) However, it remained unclear whether myocardial ischemia due to CAS alone, without either atherosclerosis or myocardial infarction, aggravates renal injury. In our study, the slight but significant decrease in myocardial perfusion distal to the CAS indicates the hemodynamic significance of the stenoses. Our chronic CAS model was non-atherosclerotic, non-infarcted, and exhibited preserved cardiac function. Nevertheless, CAS increased kidney oxidative stress (DHE, GP91), inflammation (TNFα, MCP-1), fibrosis, tubular injury, and microvascular remodeling. Moreover, RVR was elevated and cortical perfusion responses to Ach blunted, indicating endothelial dysfunction and decreased nitric oxide bioavailability in CAS,(19) supported by downregulation of eNOS. This impact on the kidney might have been mediated by factors released from the heart, eliciting an increase in systemic oxidative stress (PGF2-α isoprostanes, Figure 6s) observed in CAS, while systemic levels of inflammatory markers remained unaltered. Indeed, the drop in serum creatinine, renal fibrosis, and remodeling after chronic antioxidant treatment of CAS pigs implicates oxidative stress in the mechanisms by which CAS adversely affected the kidney. These observations position CAS as a risk factor for kidney damage independent of, but likely aggravated by, atherosclerosis or infarction.
HT is an important cause for renal disease.(6,20) Importantly, when HT coexisted with CAS, cardiac function and renal hemodynamics further deteriorated. Basal RBF and cortical perfusion and responses to Ach impaired in CAS+HT, indicating exacerbated endothelial dysfunction. Mild capillary loss observed in HT was exacerbated in CAS+HT, evoking tissue hypoxia, suggested by upregulation of HIF-1α, accompanied by microvascular remodeling.(21) Interestingly, ITC, an index of tubular integrity and function, decreased in the CAS+HT proximal and distal tubules, implying a defect in urinary concentration mechanisms. Capillary injury may reduce blood supply to the vulnerable renal tubules(22). Thus, coexisting experimental CAS and HT exacerbate impairment in cardiac and renal function, and induce renal capillary injury, thereby magnifying tubular injury, interstitial fibrosis, and ultimately glomerulosclerosis (Fig. 4s).
The mechanism by which HT aggravates the effects of CAS on the kidney may be multifactorial. Impaired systolic function may partly account for the lack of an increase in RBF and GFR, observed in HT, despite a similar increase in renal perfusion pressure. Furthermore, systemic oxidative stress was elevated similarly in CAS and CAS+HT, likely originating from the ischemic myocardium. The fall in serum creatinine after antioxidant treatment implicates oxidative-stress in renal dysfunction. Notably, CAS+HT additionally showed elevated circulating TGF-β1, possibly derived from the stenotic kidney.(23) The elevated renal TGFβ1 expression in HT, amplified in CAS and CAS+HT, was likely produced locally in response to injury. TIMP-1 also increased further in CAS+HT, which likely exacerbated renal fibrosis. Hence, several parallel injury pathways might converge to amplify kidney injury when CAS+HT coexist. Yet, unaltered circulating levels of multiple inflammatory mediators argue against systemic inflammation underpinning the effects of CAS or its interaction with HT.
In addition, prolonged and severe tissue inflammation and oxidative stress interfere with upregulation of angiogenic factors,(24,25) and HT is characterized by capillary loss.(14) In the present study, CAS+HT showed a small additive effect on renal inflammation (e.g., TNFα) and oxidative stress, which downregulated protein expression of VEGF and eNOS compared with HT, suppressing capillary density. Downregulation of eNOS may also underlie endothelial dysfunction and unaltered nitrotyrosine expression. Thus, co-existence of CAS and HT may promote microvascular regression through amplified inflammation and oxidative stress.
Limitations: In our models CAS and HT developed over 10 weeks, which is shorter than the usual disease durations in humans, who often also have other comorbidities. This might limit the clinical translation power of our observations. Nevertheless, this exposure sufficed to illustrate that CAS+HT has significant pathophysiological implications for evolution of kidney fibrosis. The unchanged renal function and structure during FAS underscore the specific effects of CAS and mild myocardial ischemia on the kidney. As typical for the renovascular HT, systemic PRA levels were not increased in chronic HT and CAS+HT, yet local angiotensin-II activity cannot be excluded.
In conclusion, this study demonstrates that non-atherosclerotic CAS alone augments renal inflammation, increases systemic and renal oxidative stress, and elicits renal injury and dysfunction. Coexistence of CAS and HT aggravates renal microvascular injury and consequently tissue hypoxia, synergistically magnifies kidney fibrosis, and may thereby contribute to increased incidence of renal failure seen when CAS and HT coexist. These observations underscore the cross talk between the myocardium and the kidney and the need for careful screening in order to assess the relative risk and ensure adequacy of management in patients with concurrent CAS and HT, regardless of the atherosclerosis burden. Further studies are also needed to examine the ability of percutaneous coronary intervention to preserve renal function.
Methods
Experimental Design
Twenty-eight female domestic pigs (initially weighing 25-35kg) were studied after approval of the Institutional Animal Care and Use Committee and randomized to four groups (n=7 each): control, CAS only, HT only, and CAS with HT.
Induction of HT and CAS
Animals were anesthetized with intramuscular Telazol® (Fort Dodge Animal Health, New York, NY) (5mg·kg-1) and xylazine (2mg·kg-1), intubated, and anesthesia maintained with intravenous ketamine (0.2mg·kg-1·min-1) and xylazine (0.03 mg·kg-1·min-1). (26) The femoral and carotid arteries were catheterized, followed by a heparin bolus (5000U). Under fluoroscopic guidance, local-irritant coils were implanted in the left circumflex coronary artery and/or the proximal-middle right renal artery, as previously described.(21,27,28) MAP was subsequently measured by a PhysioTel® telemetry system (Data Sciences International, St. Paul, MN) implanted at baseline in the left femoral artery. Controls underwent Sham renal and/or coronary angiography.
Measurement of Kidney and Cardiac Function
Ten weeks later, all pigs underwent renal and cardiac angiography, for which they were similarly anesthetized, intubated, and mechanically ventilated with room air.(24,29) After angiography, non-stenotic regional kidney perfusion, RBF, and GFR were evaluated using multidetector CT (MDCT, SOMATOM Definition-64; Siemens, Forchheim, Germany), and again after a 10-minute suprarenal arterial infusion of acetylcholine (Ach, 5μg·kg-1·min-1). Thirty minutes later, CT studies were performed to assess cardiac structure, systolic and diastolic function in-vivo.(21,27,30)
All images were analyzed with the ANALYZE™ software package (Biomedical Imaging Resource, Mayo Clinic, Rochester, MN).
For renal function, regions of interest were selected in the cross-sectional images from the aorta, renal cortex, and medulla, and time-attenuation curves generated.(30) The parameters obtained from the vascular curve in each region of the kidney were used to calculate cortical and medullary perfusion. ITC was calculated for each nephron segment as the ratio of the area under each tubular curve to that of the cortical vascular curve, divided by normalized GFR. Renal volume was measured using planimetry, and RBF calculated as the sum of cortical and medullary blood flows (product of cortical and medullary perfusion and volumes), and GFR from the cortical proximal-tubular curve.(30) RVR was calculated as MAP/RBF.
For cardiac function, left ventricular (LV) stroke volume, cardiac output, ejection fraction, E/A, and LVMM, were calculated as previously described.(30) For myocardial perfusion, regions of interest were traced at the lateral LV wall distal to the CAS, and time-attenuation curves analyzed as we have shown before.(21,27,30)
Angiography
A guide catheter was positioned under fluoroscopic guidance in the right renal artery and left main coronary artery for selective injections of contrast media. The degree of stenosis was measured by quantitative angiography, as described previously,(24) and assessed as the decrease in arterial luminal diameter and area at the most stenotic point compared with a stenosis-free segment.
Sample Collection
Blood samples were collected from the inferior vena cava for measurement of SCr (Arbor Assays, Ann Arbor, MI), PGF2-α isoprostane (ELISA, Cayman, Ann Arbor, MI), PRA (radioimmunoassay, DiaSorin, Stillwater, MN), norepinephrine (Abnova GmbH, Germany) and TGF-β1 (R&D, Minneapolis, MN). GM-CSF, IL-1α, IL-1β, IL-1ra, IL-2, IL-4, IL-6, IL-10, IL-12, IL-18 and TNFα were measured by Luminex (Millipore, Billerica, MA). Urine samples were collected for urinary protein (Coomassie Plus assay, Thermo Scientific, Wlatham, MA). After completion of all studies, the pigs were euthanized with a lethal intravenous dose of sodium pentobarbital (100mg/kg; Sleepaway®, Fort-Dodge Laboratories, Fort-Dodge, Iowa). Kidneys were removed and fresh frozen or preserved in formalin.
Renal Histology and Immunohistochemistry
Trichrome stainings were performed in 5-μm paraffin midhilar renal cross-sections to assess fibrosis by a computer-aided image analysis program (AxioVision 4.8.2, Carl Zeiss Microscopy, Thornwood, NY). In each slide, trichrome staining was semiautomatically quantified in 6-10 fields, expressed as fraction of kidney surface area, and the results from all fields averaged. Glomerular score (% of sclerotic out of 100 glomeruli) was assessed as described.(24) CD31 (1:50; AbD Serotec, UK) immunofluorescence was used to investigate capillary density.(31) Microvascular remodeling was assessed by wall-to-lumen ratio using α-smooth muscle actin (α-SMA) staining (1:50; Dako, Glostrup, Denmark).(32) Tubular injury was scored as described previously.(33) Oxidative stress indicated by in-situ production of superoxide anion was quantified in 30 μm DHE stained slides.(34)
Western blotting
Standard Western blotting protocols were followed as described (35,36), using specific antibodies against TGF-β1 (1:200, Santa-Cruz, Dallas, TX), MCP-1, HIF-1α (both 1:1000, Abcam, Cambridge, England), TNFα (1:100, Santa-Cruz), GP91-phox, and p67-phox (both 1:200, Santa-Cruz), nitrotyrosine (1:200, Cayman), TIMP-1, VEGF (both 1:200, Santa-Cruz) and eNOS (1:1000, BD BioSciences, San Jose, CA). Protein expression was determined in one Sham, CAS, or non-stenotic HT and CAS+HT kidney in each animal, and the intensities of the protein bands quantified and normalized for a GAPDH (1:5000, Abcam) loading control.
Mechanistic exploration
To exclude the possible non-specific effects of an intravascular local-irritant coil on the remote kidney, coils were implanted endovascularly in the femoral arteries of 2 additional pigs, and studies performed 10 weeks later (n=4 kidneys).
To further explore the role of oxidative stress in mediating the effect of CAS on the kidney, additional animals with CAS received chronic daily mitochondria-targeted antioxidant Bendavia (0.1mg·kg-1·d-1, subcutaneous) for 10 weeks.(37) After 10 weeks of diet and intervention, studies were performed in-vivo using MDCT to measure myocardial and renal perfusion, and ex-vivo for representative tissue studies (n=4 kidneys).
Statistical Analysis
Results are expressed as mean±SEM. 2-way ANOVA was used to test for the effect of CAS and HT; their interaction was analyzed with Tukey's multiple-comparison adjustment for the least-squares means, and post-hoc comparisons among groups were performed using unpaired Student's t-test. Paired Student's t-test was performed for comparisons within groups (baseline vs. Ach). For data not normally distributed, comparisons were done using nonparametric tests. All analyses were performed in JMP10 and significance accepted for P≤0.05.
Supplementary Material
Figure 1s. Full blots showing renal expression of inflammatory and oxidative factors. Tumor necrosis-factor (TNF)-α, monocyte chemoattractant protein (MCP)-1, GP91-phox and nitrotyrosine (NT). Inflammatory and oxidative markers were elevated in coronary artery stenosis (CAS), and further augmented in CAS+Hypertension (HT).
Figure 2s. Full blots showing renal expression of growth factors. Hypoxia-inducible factor (HIF)-1α, vascular endothelial growth-factor (VEGF), transforming growth-factor (TGF)-β1, tissue inhibitor of metalloproteinase (TIMP)-1 and endothelial nitric-oxide synthase (eNOS). Hypoxia and fibrotic markers increased in CAS+HT, whereas eNOS decreased; VEGF expression decreased in HT and further fell in CAS+HT (D).
Figure 3s. Evaluation of pigs with femoral artery stenosis (FAS). Representative femoral artery angiography shows the femoral artery stent location (A). Kidney fibrosis (Trichrome, ×20), DHE staining (normalized to DAPI-positive nuclei, ×20), renal microvascular media-to-lumen ratio (α-SMA, ×20) and tubular injury score (PAS staining, ×20) (B-F) did not increase in FAS pigs compared with Sham.
Figure 4s. Renal tissue remodeling in pigs with coronary artery stenosis (CAS) treated by the mitochondria-targeted peptide Bendavia. A. Representative renal trichrome (×20), dihydroethidium (DHE) (×20), α-SMA (×20) and PAS staining (×20). Kidney fibrosis increased in CAS compared with Sham, and decreased after treatment by Bendavia (B). DHE staining (normalized to DAPI-positive nuclei) increased in CAS compared with Sham, and decreased after treatment by Bendavia (C). Renal microvascular media-to-lumen ratio (α-SMA) increased in CAS compared with Sham, and decreased after treatment by Bendavia (D). Tubular injury score (PAS staining) increased in CAS, and decreased after treatment by Bendavia (E). *P<0.05 vs. Sham.
Figure 5s. Renal expression of inflammatory, oxidative, and growth factors in CAS+Bendavia pigs. Representative (2 bands shown per group) immunoblotting of (A): tumor necrosis-factor (TNF)-α, p67-phox, GP91-phox, transforming growth factor (TGF)-β1, endothelial nitric-oxide synthase (eNOS). Inflammatory and oxidative markers were elevated in coronary artery stenosis (CAS), and decreased after treatment by Bendavia (B-D). TGF-β1 increased in CAS, and eNOS decreased in CAS. After Bendavia treatment, TGF-β1 decreased in CAS, whereas eNOS did not increase. *P<0.05 vs. Sham.
Figure 6s. Proposed mechanisms of the cardio-renal interaction in the study. Experimental coronary artery stenosis increases systemic oxidative stress, which leads to renal inflammation, oxidative stress, and endothelial dysfunction. Hypertension induces renal microvascular loss and glomerular hyperfiltration, which also lead to renal damage, and their interaction is synergistic.
Supplemental Table 1. Circulating inflammatory mediators in the experimental groups (mean±SEM, n=7 each).
Supplemental Table 2. Systemic characteristics, kidney hemodynamics and function in Sham and femoral artery stenosis (FAS) pigs (mean±SEM).
Supplemental Table 3. Systemic characteristics, kidney hemodynamics and function in Sham, coronary artery stenosis (CAS) and CAS+Bendavia pigs (mean±SEM).
Acknowledgments
This study was partly supported by NIH Grant numbers DK73608, HL77131, HL121561, DK100081, C06-RR018898, HL-92954, and AG-31750.
The authors thank Stealth Peptides Incorporated for providing Bendavia for the studies.
Footnotes
Disclosure: Dr. Lilach O. Lerman received research funding from Stealth Peptides Incorporated. Other authors declared no competing interests.
References
- 1.Bitton A, Choudhry NK, Matlin OS, et al. The impact of medication adherence on coronary artery disease costs and outcomes: a systematic review. Am J Med. 2013;126:357 e7–e27. doi: 10.1016/j.amjmed.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.Kim EJ, Kim S, Kang DO, et al. Metabolic Activity of the Spleen and Bone Marrow in Patients With Acute Myocardial Infarction Evaluated by 18F-Fluorodeoxyglucose Positron Emission Tomograpic Imaging. Circ Cardiovasc Imaging. 2014;7:454–60. doi: 10.1161/CIRCIMAGING.113.001093. [DOI] [PubMed] [Google Scholar]
- 4.Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–9. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu J, Wang X, Wang W, et al. Abrogation of lectin-like oxidized LDL receptor-1 attenuates acute myocardial ischemia-induced renal dysfunction by modulating systemic and local inflammation. Kidney Int. 2012;82:436–44. doi: 10.1038/ki.2012.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Appel LJ, Wright JT, Jr, Greene T, et al. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med. 2010;363:918–29. doi: 10.1056/NEJMoa0910975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber-Mzell D, Kotanko P, Schumacher M, et al. Coronary anatomy predicts presence or absence of renal artery stenosis. A prospective study in patients undergoing cardiac catheterization for suspected coronary artery disease. Eur Heart J. 2002;23:1684–91. doi: 10.1053/euhj.2002.3314. [DOI] [PubMed] [Google Scholar]
- 8.Liang F, Hu DY, Wu MY, et al. The incidence of renal artery stenosis in the patients referred for coronary artery bypass grafting. Indian J Nephrol. 2012;22:13–7. doi: 10.4103/0971-4065.91181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ollivier R, Boulmier D, Veillard D, et al. Frequency and predictors of renal artery stenosis in patients with coronary artery disease. Cardiovasc Revasc Med. 2009;10:23–9. doi: 10.1016/j.carrev.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Macedo TA, Pedrosa RP, Costa-Hong V, et al. Renal artery stenosis predicts coronary artery disease in patients with hypertension. PLoS One. 2013;8:e58635. doi: 10.1371/journal.pone.0058635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khangura KK, Eirin A, Kane GC, et al. Cardiac function in renovascular hypertensive patients with and without renal dysfunction. Am J Hypertens. 2014;27:445–53. doi: 10.1093/ajh/hpt203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khangura KK, Eirin A, Kane GC, et al. Extrarenal atherosclerotic disease blunts renal recovery in patients with renovascular hypertension. J Hypertens. 2014;32:1300–6. doi: 10.1097/HJH.0000000000000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bidani AK, Polichnowski AJ, Loutzenhiser R, et al. Renal microvascular dysfunction, hypertension and CKD progression. Curr Opin Nephrol Hypertens. 2013;22:1–9. doi: 10.1097/MNH.0b013e32835b36c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill GS. Hypertensive nephrosclerosis. Curr Opin Nephrol Hypertens. 2008;17:266–70. doi: 10.1097/MNH.0b013e3282f88a1f. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez-Porcel M, Krier JD, Lerman A, et al. Combination of hypercholesterolemia and hypertension augments renal function abnormalities. Hypertension. 2001;37:774–80. doi: 10.1161/01.hyp.37.2.774. [DOI] [PubMed] [Google Scholar]
- 16.Ruparelia N, Digby JE, Jefferson A, et al. Myocardial infarction causes inflammation and leukocyte recruitment at remote sites in the myocardium and in the renal glomerulus. Inflamm Res. 2013;62:515–25. doi: 10.1007/s00011-013-0605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ronco C, McCullough P, Anker SD, et al. Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. 2010;31:703–11. doi: 10.1093/eurheartj/ehp507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heywood JT, Fonarow GC, Costanzo MR, et al. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail. 2007;13:422–30. doi: 10.1016/j.cardfail.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 19.Urbieta-Caceres VH, Lavi R, Zhu XY, et al. Early atherosclerosis aggravates the effect of renal artery stenosis on the swine kidney. Am J Physiol Renal Physiol. 2010;299:F135–40. doi: 10.1152/ajprenal.00159.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension. 2004;44:595–601. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- 21.Zhu XY, Daghini E, Chade AR, et al. Role of oxidative stress in remodeling of the myocardial microcirculation in hypertension. Arterioscler Thromb Vasc Biol. 2006;26:1746–52. doi: 10.1161/01.ATV.0000227469.40826.01. [DOI] [PubMed] [Google Scholar]
- 22.Sun D, Bu L, Liu C, et al. Therapeutic effects of human amniotic fluid-derived stem cells on renal interstitial fibrosis in a murine model of unilateral ureteral obstruction. PLoS One. 2013;8:e65042. doi: 10.1371/journal.pone.0065042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Urbieta-Caceres VH, Zhu XY, Jordan KL, et al. Selective improvement in renal function preserved remote myocardial microvascular integrity and architecture in experimental renovascular disease. Atherosclerosis. 2012;221:350–8. doi: 10.1016/j.atherosclerosis.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chade AR, Zhu X, Lavi R, et al. Endothelial progenitor cells restore renal function in chronic experimental renovascular disease. Circulation. 2009;119:547–57. doi: 10.1161/CIRCULATIONAHA.108.788653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chade AR, Zhu X, Mushin OP, et al. Simvastatin promotes angiogenesis and prevents microvascular remodeling in chronic renal ischemia. FASEB J. 2006;20:1706–8. doi: 10.1096/fj.05-5680fje. [DOI] [PubMed] [Google Scholar]
- 26.Eirin A, Ebrahimi B, Zhang X, et al. Changes in Glomerular Filtration Rate After Renal Revascularization Correlate With Microvascular Hemodynamics and Inflammation in Swine Renal Artery Stenosis. Circulation: Cardiovascular Interventions. 2012;5:720–8. doi: 10.1161/CIRCINTERVENTIONS.112.972596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urbieta Caceres VH, Lin J, Zhu XY, et al. Early experimental hypertension preserves the myocardial microvasculature but aggravates cardiac injury distal to chronic coronary artery obstruction. Am J Physiol Heart Circ Physiol. 2011;300:H693–701. doi: 10.1152/ajpheart.00516.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Favreau F, Zhu XY, Krier JD, et al. Revascularization of swine renal artery stenosis improves renal function but not the changes in vascular structure. Kidney Int. 2010;78:1110–8. doi: 10.1038/ki.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chade AR, Rodriguez-Porcel M, Herrmann J, et al. Antioxidant intervention blunts renal injury in experimental renovascular disease. J Am Soc Nephrol. 2004;15:958–66. doi: 10.1097/01.asn.0000117774.83396.e9. [DOI] [PubMed] [Google Scholar]
- 30.Daghini E, Primak AN, Chade AR, et al. Assessment of renal hemodynamics and function in pigs with 64-section multidetector CT: comparison with electron-beam CT. Radiology. 2007;243:405–12. doi: 10.1148/radiol.2432060655. [DOI] [PubMed] [Google Scholar]
- 31.Muczynski KA, Cotner T, Anderson SK. Unusual expression of human lymphocyte antigen class II in normal renal microvascular endothelium. Kidney Int. 2001;59:488–97. doi: 10.1046/j.1523-1755.2001.059002488.x. [DOI] [PubMed] [Google Scholar]
- 32.Chade AR, Mushin OP, Zhu X, et al. Pathways of renal fibrosis and modulation of matrix turnover in experimental hypercholesterolemia. Hypertension. 2005;46:772–9. doi: 10.1161/01.HYP.0000184250.37607.da. [DOI] [PubMed] [Google Scholar]
- 33.Nangaku M, Alpers CE, Pippin J, et al. CD59 protects glomerular endothelial cells from immune-mediated thrombotic microangiopathy in rats. J Am Soc Nephrol. 1998;9:590–7. doi: 10.1681/ASN.V94590. [DOI] [PubMed] [Google Scholar]
- 34.Ebrahimi B, Eirin A, Li Z, et al. Mesenchymal stem cells improve medullary inflammation and fibrosis after revascularization of Swine atherosclerotic renal artery stenosis. PLoS One. 2013;8:e67474. doi: 10.1371/journal.pone.0067474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eirin A, Zhu XY, Urbieta-Caceres VH, et al. Persistent kidney dysfunction in swine renal artery stenosis correlates with outer cortical microvascular remodeling. Am J Physiol Renal Physiol. 2011;300:F1394–401. doi: 10.1152/ajprenal.00697.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chade AR, Rodriguez-Porcel M, Grande JP, et al. Mechanisms of renal structural alterations in combined hypercholesterolemia and renal artery stenosis. Arterioscler Thromb Vasc Biol. 2003;23:1295–301. doi: 10.1161/01.ATV.0000077477.40824.52. [DOI] [PubMed] [Google Scholar]
- 37.Eirin A, Li Z, Zhang X, et al. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension. 2012;60:1242–9. doi: 10.1161/HYPERTENSIONAHA.112.199919. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1s. Full blots showing renal expression of inflammatory and oxidative factors. Tumor necrosis-factor (TNF)-α, monocyte chemoattractant protein (MCP)-1, GP91-phox and nitrotyrosine (NT). Inflammatory and oxidative markers were elevated in coronary artery stenosis (CAS), and further augmented in CAS+Hypertension (HT).
Figure 2s. Full blots showing renal expression of growth factors. Hypoxia-inducible factor (HIF)-1α, vascular endothelial growth-factor (VEGF), transforming growth-factor (TGF)-β1, tissue inhibitor of metalloproteinase (TIMP)-1 and endothelial nitric-oxide synthase (eNOS). Hypoxia and fibrotic markers increased in CAS+HT, whereas eNOS decreased; VEGF expression decreased in HT and further fell in CAS+HT (D).
Figure 3s. Evaluation of pigs with femoral artery stenosis (FAS). Representative femoral artery angiography shows the femoral artery stent location (A). Kidney fibrosis (Trichrome, ×20), DHE staining (normalized to DAPI-positive nuclei, ×20), renal microvascular media-to-lumen ratio (α-SMA, ×20) and tubular injury score (PAS staining, ×20) (B-F) did not increase in FAS pigs compared with Sham.
Figure 4s. Renal tissue remodeling in pigs with coronary artery stenosis (CAS) treated by the mitochondria-targeted peptide Bendavia. A. Representative renal trichrome (×20), dihydroethidium (DHE) (×20), α-SMA (×20) and PAS staining (×20). Kidney fibrosis increased in CAS compared with Sham, and decreased after treatment by Bendavia (B). DHE staining (normalized to DAPI-positive nuclei) increased in CAS compared with Sham, and decreased after treatment by Bendavia (C). Renal microvascular media-to-lumen ratio (α-SMA) increased in CAS compared with Sham, and decreased after treatment by Bendavia (D). Tubular injury score (PAS staining) increased in CAS, and decreased after treatment by Bendavia (E). *P<0.05 vs. Sham.
Figure 5s. Renal expression of inflammatory, oxidative, and growth factors in CAS+Bendavia pigs. Representative (2 bands shown per group) immunoblotting of (A): tumor necrosis-factor (TNF)-α, p67-phox, GP91-phox, transforming growth factor (TGF)-β1, endothelial nitric-oxide synthase (eNOS). Inflammatory and oxidative markers were elevated in coronary artery stenosis (CAS), and decreased after treatment by Bendavia (B-D). TGF-β1 increased in CAS, and eNOS decreased in CAS. After Bendavia treatment, TGF-β1 decreased in CAS, whereas eNOS did not increase. *P<0.05 vs. Sham.
Figure 6s. Proposed mechanisms of the cardio-renal interaction in the study. Experimental coronary artery stenosis increases systemic oxidative stress, which leads to renal inflammation, oxidative stress, and endothelial dysfunction. Hypertension induces renal microvascular loss and glomerular hyperfiltration, which also lead to renal damage, and their interaction is synergistic.
Supplemental Table 1. Circulating inflammatory mediators in the experimental groups (mean±SEM, n=7 each).
Supplemental Table 2. Systemic characteristics, kidney hemodynamics and function in Sham and femoral artery stenosis (FAS) pigs (mean±SEM).
Supplemental Table 3. Systemic characteristics, kidney hemodynamics and function in Sham, coronary artery stenosis (CAS) and CAS+Bendavia pigs (mean±SEM).