

Abstract
The macrocyclic depsipeptide Largazole is a potent inhibitor of metal-dependent histone deacetylases (HDACs), some of which are drug targets for cancer chemotherapy. Indeed, Largazole partially resembles Romidepsin (FK228), a macrocyclic depsipeptide already approved for clinical use. Each inhibitor contains a pendant side chain thiol that coordinates to the active site Zn2+ ion, as observed in the X-ray crystal structure of the HDAC8–Largazole complex [Cole, K. E.; Dowling, D. P.; Boone, M. A.; Phillips, A. J.; Christianson, D. W. J. Am. Chem. Soc. 2011, 133, 12474]. Here, we report the X-ray crystal structures of HDAC8 complexed with three synthetic analogues of Largazole in which the depsipeptide ester is replaced with a rigid amide linkage. In two of these analogues, a 6-membered pyridine ring is also substituted (with two different orientations) for the 5-membered thiazole ring in the macrocycle skeleton. The side chain thiol group of each analogue coordinates to the active site Zn2+ ion with nearly ideal geometry, thereby preserving the hallmark structural feature of inhibition by Largazole. Surprisingly, in comparison with the binding of Largazole, these analogues trigger alternative conformational changes in the L1 and L2 loops flanking the active site. However, despite these structural differences, inhibitory potency is generally comparable to, or just moderately less than, the inhibitory potency of Largazole. Thus, this study reveals important new structure-affinity relationships for the binding of macrocyclic inhibitors to HDAC8.
INTRODUCTION
Class I, II, and IV histone deacetylases (HDACs)1,2 are metal-dependent enzymes that utilize a single transition metal ion, either Zn2+ and/or Fe2+ in vivo,3 to catalyze the hydrolysis of trans-acetyl-L-lysine residues4 in histone and non-histone proteins to regulate their function.5,6 These enzymes, also known more broadly as lysine deacetylases, are critical targets for inhibitor design because they are implicated in a variety of biological processes such as gene expression, cellular differentiation, proliferation, and apoptosis.7–11 Upregulated HDAC activity is associated with tumorigenesis, and the HDAC inhibitors suberoylanilide hydroxamic acid (Zolinza)12,13 and Romidepsin (Istodax)14,15 are currently in clinical use for cancer chemotherapy.
Romidepsin (Figure 1) is particularly notable among HDAC inhibitors in that it is a macrocyclic depsipeptide that undergoes disulfide reduction in vivo to liberate a ~7 Å-long thiol side chain that coordinates to the active site metal ion. Although no X-ray crystal structure of an HDAC-Romidepsin complex has been reported to date, the crystal structure of HDAC8 complexed with the active form of a structurally-related marine natural product, Largazole thiol (henceforth simply referred to as “Largazole”; Figure 1),16,17 reveals that the thiol side chain is readily accommodated in the narrow tunnel that otherwise accommodates the substrate trans-acetyl-L-lysine.18 The thiol moiety of Largazole is presumably ionized as the negatively charged thiolate to coordinate to the active site Zn2+ ion, and ideal metal coordination geometry contributes to exceptionally high affinity.18 Indeed, Largazole is one of the most potent HDAC inhibitors known and it exhibits low nanomolar affinity against several HDAC isozymes.17,19 A key difference between Largazole and Romidepsin is that the 16-membered depsipeptide ring of Largazole contains an unusual thiazoline-thiazole moiety, suggesting potential opportunities for the design of diverse macrocyclic analogues.
Figure 1.
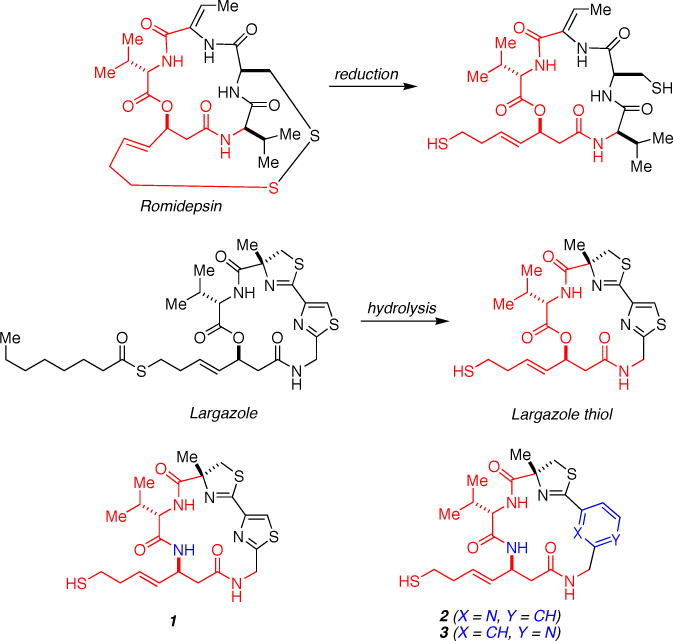
The macrocyclic depsipeptide Romidepsin is an approved drug used for cancer chemotherapy; reduction of the disulfide linkage liberates the active form of the inhibitor. In contrast, the macrocyclic depsipeptide Largazole is activated by hydrolysis of the thioester group to yield the thiol moiety that targets Zn2+ coordination (Largazole thiol is simply referred to as “Largazole” throughout this paper). Both Romidepsin and Largazole share a core structure (red) that is responsible for potent inhibition of HDAC8. In the current study, the Largazole macrocycle has been rigidified by substitution of an amide linkage for the ester linkage in 1. Further elaboration of this macrocyclic peptide platform by two alternative substitution patterns of a pyridine ring in place of the thiazole ring yields 2 and 3. Substituted atoms relative to Largazole are highlighted in blue. IC50 values are reported for HDAC8.26
Among the total syntheses of Largazole reported soon after its discovery,17,20–22 Williams and colleagues reported a modular and scalable total synthesis that facilitated the generation of Largazole analogues containing modified thiol side chains,19 thiazole-pyridine and -oxazole substitutions,19,23 and a macrocyclic ring in which the depsipeptide ester linkage is rigidified by the substitution of an amide linkage.24 The thiazole-pyridine substitution is particularly intriguing because this modification confers 3–4-fold enhancement of inhibitory potency (IC50) against several HDAC isozymes.19 Conversely, the conversion of the macrocyclic depsipeptide into a macrocyclic peptide generally results in a 3–4-fold loss of inhibitory potency.24
In order to understand structure-affinity relationships for Largazole analogues, we now report the high resolution X-ray crystal structures of three macrocyclic peptide analogues, 1, 2, and 3 (Figure 1), complexed with HDAC8. These structures reveal how the enzyme active site accommodates derivatization of the macrocyclic skeleton and may suggest potential new avenues for modifications that could further modulate biological activity. Among the greater family of HDAC isozymes, HDAC8 is the most amenable to the X-ray crystal structure determination of enzyme-inhibitor complexes, and the crystal structure of the HDAC8-Largazole complex18 provides an important reference point for the structures described herein. Accordingly, HDAC8 serves as a paradigm for inhibitor design experiments targeting the class I isozymes (HDACs 1, 2, 3, and 8) that serve as targets for cancer chemotherapy,7–11 and HDAC8 itself has been recently identified as a target for neuroblastoma chemotherapy.25
MATERIALS AND METHODS
Reagents
Most chemicals used for buffers or crystallization were purchased from Fisher or Sigma and used without further purification. The synthesis of compound 1 has been previously described,24 and the preparation of 2 and 3 are being reported elsewhere.26
Crystallization and Structure Determination of HDAC8-Inhibitor Complexes
To facilitate crystallization, the S39D mutation was introduced into the previously described HDAC8-6His-pET20b construct27 according to QuickChange site-directed mutagenesis protocols using forward 5′- CTA AAA TCC CGA AAC GTG CAG ACA TGG TGC ATT CTT TGA TTG AAG -3′ and reverse 5′- CTT CAA TCA AAG AAT GCA CCA TGT CTG CAC GTT TCG GGA TTT TAG -3′ primers. DNA sequencing at the University of Pennsylvania Perelman School of Medicine confirmed the incorporation of the desired mutation. Recombinant S39D HDAC8 was expressed and purified as previously described.28 As noted by Vannini and colleagues,29 this mutant is properly folded and catalytically active, but it can also yield higher quality crystals of enzyme-inhibitor complexes in comparison with the wild-type enzyme. Since the structures of wild-type HDAC8 and S39D HDAC8 are essentially identical, we henceforth refer to S39D HDAC8 simply as “HDAC8”.
Crystals of HDAC8-inhibitor complexes were prepared by cocrystallization at 4°C (except for the complex with 3, which was cocrystallized at 21°C) in sitting drops using the vapor diffusion method. In general, a 500 nL drop containing 5 mg/mL HDAC8, 50 mM Tris (pH 8.0), 150 mM KCl, 5% v/v glycerol, 1 mM DTT, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) (except for 1, which did not require TCEP for cocrystallization), 2 mM inhibitor, 5% v/v DMSO, and 0.03 M glycylglycylglycine was added to a 500 nL drop of precipitant solution and equilibrated against a 100 μL reservoir of precipitant solution. The precipitation solution for the cocrystallization of the HDAC8–1 complex consisted of 100 mM imidazole (pH = 7.0), 8% PEG 8,000 (Hampton Research), and 4 mM TCEP. The precipitation solution for the cocrystallization of the HDAC8–2 complex consisted of 100 mM imidazole (pH = 7.0), 13% PEG 3,350 (Hampton Research), and 4 mM TCEP. Finally, the precipitation solution for the cocrystallization of the HDAC8–3 complex consisted of 100 mM BisTris (pH = 6.5), 8% PEG 3,350, and 4 mM TCEP.
Crystals of each complex appeared within 1–2 days and were flash-cooled in liquid nitrogen after transfer to a cryoprotectant solution consisting of precipitant solution supplemented with 25% glycerol for the HDAC8–1 complex, 30% glycerol for the HDAC8–2 complex, and 30% of a 1:1 (v/v) mixture of PEG 400 (Hampton Research) and 2-methyl-2,4-pentanediol for the HDAC8–3 complex. X-ray diffraction data were collected on beamline X29 at the National Synchrotron Light Source (NSLS, Brookhaven National Laboratory, New York). Data collection statistics are presented in Table 1. Data were indexed, integrated and scaled using HKL2000.30
Table 1.
Data collection and refinement statistics
|
|
|||
|---|---|---|---|
| HDAC8–1 Complex | HDAC8–2 Complex | HDAC8–3 Complex | |
| Unit cell | |||
| space group symmetry | P21 | P21 | P21 |
| a, b, c (Å) | 53.9, 85.0, 94.7 | 54.1, 85.1, 95.4 | 53.8, 84.7, 94.1 |
| α, β, γ (deg) | 90, 100.3, 90 | 90, 100.2, 90 | 90, 100.5, 90 |
| Data collection | |||
| wavelength (Å) | 1.075 | 1.075 | 1.075 |
| resolution limits (Å) | 50.0 – 1.76 | 50.0 – 2.18 | 50.0 – 2.39 |
| total/unique reflections | 621648/82963 | 273766/44473 | 195462/32705 |
| Rmergea,b | 0.073 (0.317) | 0.124 (0.811) | 0.144 (0.736) |
| I/σ(I)a | 21.3 (7.6) | 13.0 (2.8) | 12.9 (2.2) |
| redundancya | 7.5 (7.3) | 6.2 (6.1) | 6.0 (4.5) |
| completeness (%)a | 99.9 (100) | 100 (100) | 98.7 (97.5) |
| Refinement | |||
| reflections used in refinement/test set | 82933/4145 | 44415/2238 | 32682/2526 |
| Rcrystc | 0.137 | 0.182 | 0.187 |
| Rfreed | 0.162 | 0.213 | 0.224 |
| protein atomse | 5715 | 5608 | 5481 |
| water moleculese | 750 | 349 | 182 |
| ligand moleculese | 2 | 2 | 2 |
| Zn2+ ionse | 2 | 2 | 2 |
| K+ ionse | 4 | 4 | 4 |
| glycerol moleculese | 1 | 1 | – |
| imidazole moleculese | 1 | 1 | – |
| R.m.s. deviations from ideal geometry | |||
| bonds (Å) | 0.010 | 0.002 | 0.002 |
| angles (°) | 1.3 | 0.7 | 0.6 |
| dihedral angles (°) | 15 | 11 | 11 |
| Ramachandran plot (%)f | |||
| allowed | 91.2 | 91.2 | 90.7 |
| additionally allowed | 8.8 | 8.8 | 9.3 |
| PDB accession code | 4RN0 | 4RN1 | 4RN2 |
Values in parentheses refer to the highest shell of data.
Rmerge = Σ|Ih − ⟨I⟩h|/ΣIh, where ⟨I⟩h is the average intensity calculated from replicate reflections.
Rcryst = Σ||Fo| − |Fc||/Σ|Fo| for reflections contained in the working set. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
Rfree = Σ||Fo| − |Fc||/Σ|Fo| for reflections contained in the test set held aside during refinement.
Per asymmetric unit.
Calculated with PROCHECK version 3.4.4.
Analysis of X-ray intensity data collected from crystals of the HDAC8–3 complex revealed deviations from ideal intensity statistics suggestive of twinning, which is somewhat unusual for crystallographic space group P21. Intensity statistics can deviate from ideality due to other crystallographic complications, as observed in the structure of the amino acid ester hydrolase from Acetobacter turbidans, where merohedral twinning in space group P21 was complicated by pseudotranslational noncrystallographic symmetry.31 For the HDAC8–3 complex, we reindexed intensity data in space group P1 and used Zanuda (contained in the CCP4 suite of programs32) to validate the P21 space group and origin assignments. We proceeded to solve the structure by molecular replacement using PHENIX33 with the atomic coordinates of the H143A HDAC8–tetrapeptide substrate complex (PDB accession code 3EWF)27 less ligand, ion, and solvent used as a search probe for rotation and translation function calculations. Refinement and manual model adjustment were performed with PHENIX and COOT, respectively.33,34 While the origin of the deviation from ideal intensity statistics remains unknown for these crystals, it is clear that the minor crystallographic defect that might be present in these crystals did not hinder satisfactory refinement based on the refinement statistics recorded in Table 1.
X-ray data collected from crystals of the HDAC8 complexes with 1 and 2 exhibited ideal intensity distributions and did not require further analysis. Structures were solved by molecular replacement using PHENIX,33 and refined and rebuilt with PHENIX and COOT, respectively.33,34 Refinement converged smoothly and detailed refinement statistics are recorded in Table 1.
Certain segments in each structure (the N-terminus, the C-terminus, and a portion of the L2 loop) appeared to be disordered and were accordingly excluded from each final model as follows: HDAC8–1 complex: M1-S13 (monomers A and B), D87-D92 (monomer A), E85-D89 (monomer B), G99-Y100 (monomer B), E379-H389 (monomer A), S383-H389 (monomer B); HDAC8–2 complex: M1-S13 (monomer A), M1-Q12 (monomer B), D88-P91 (monomer A), E85-D88 (monomer B), Y100-D101 (monomer B), E379-H389 (monomer A), I378-H389 (monomer B); and HDAC8–3 complex: M1-L14 (monomers A and B), E85-D92 (monomer A), G99-A104 (monomer A), D87-D92 (monomer B), I378-H389 (monomers A and B). Ambiguous electron density was observed for L2 loop segments E85-D89 and G99-Y100, and Y100-D101 in monomer B of the HDAC8–1 and HDAC8–2 complexes, respectively. Such ambiguous electron density might arise from molecular disorder or multiple conformations, but it was not sufficiently clear to enable clear interpretation. Accordingly, these short segments were not modeled. Likewise, side chains of residues that were completely disordered were excluded from the model as follows: HDAC8–1 complex, monomer A: K60, S93, Q253, and K374; monomer B: L14, K60, K81, V82, Q84, H90, D92, S93, I94, E95, D101, C102, K132, Q253, E379; HDAC8–2 complex, monomer A: D87, D92, S93, Q253, and E358; monomer B: S13, E23, K58, K60, K81, Q84, D89, D92, C102, K221, K249, and K374; HDAC8–3 complex, monomer A: K33, K60, D73, K81, I94, E95, T105, E106, and Q253; monomer B: K33, S93, I94, E95, K325, and E358.
RESULTS
Initial inhibitor cocrystallization experiments with wild-type HDAC8 were unsuccessful. Therefore, the site-specific mutant S39D HDAC8 was utilized for cocrystallization experiments with Largazole analogues 1, 2, and 3. As noted by Vannini and colleagues,29 this mutant is properly folded and catalytically active, but it can also yield higher quality crystals of enzyme-inhibitor complexes in comparison with the wild-type enzyme due to additional crystal contacts. The side chain of S39 is ~20 Å from the active site and undergoes phosphorylation by cyclic AMP-dependent protein kinase A.35 However, the structures of wild-type HDAC8 and S39D HDAC8 are essentially identical, so inhibitor binding modes are expected to be similarly identical in each enzyme active site. Accordingly, we henceforth refer to S39D HDAC8 simply as “HDAC8”.
In the structure of each enzyme-inhibitor complex, the Largazole analogue binds to monomers A and B in the asymmetric unit with full occupancy and thermal B factors comparable to those of surrounding residues. The thiol side chain of each inhibitor extends into the narrow active site tunnel and coordinates to the Zn2+ ion, presumably as the negatively charged thiolate. This is the hallmark of HDAC inhibition by Largazole, and these structures reveal that ideal thiolate-zinc coordination geometry is conserved in the binding of Largazole analogues. As previously measured by Williams and colleagues,26 IC50 values for Largazole and analogues 1, 2, and 3 are summarized in Table 2. The specific details of each enzyme-inhibitor complex are outlined below.
Table 2.
Inhibitory Activity of Largazole Analogues against Class I HDACs (IC50, nM)a
|
|
||||
|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | |
| Largazole | 2.5 | 4.2 | 2.8 | 228 |
| 1 | 2.0 | 3.4 | 2.6 | 255 |
| 2 | 42 | 70 | 42 | ~10,000 |
| 3 | 13 | 21 | 15 | 1500 |
Reported in reference 26.
HDAC8–1 Complex
Largazole analogue 1 is essentially isosteric with Largazole except for the substitution of the cyclic depsipeptide ester with an amide group. The overall structure of the HDAC8–1 complex determined at 1.76 Å resolution is similar to that of the wild-type HDAC8–Largazole complex (r.m.s. deviations of 0.58 Å for 350 Cα atoms and 0.29 Å for 345 Cα atoms in monomers A and B, respectively). An electron density map of the enzyme-inhibitor complex (Figure 2a) reveals that the geometry of thiolate-Zn2+ coordination is nearly ideal36 with an average S–Zn2+ distance of 2.3 Å, a C–S–Zn2+ angle of 101°, and a C–C–S–Zn2+ dihedral angle of 95° (values averaged over monomers A and B). Additionally, the phenolic side chain of Y306 donates a hydrogen bond to the Zn2+-bound thiolate anion. The Zn2+ ion is coordinated with distorted tetrahedral geometry (average ligand–Zn2+–ligand angles range 98°–119°).
Figure 2.
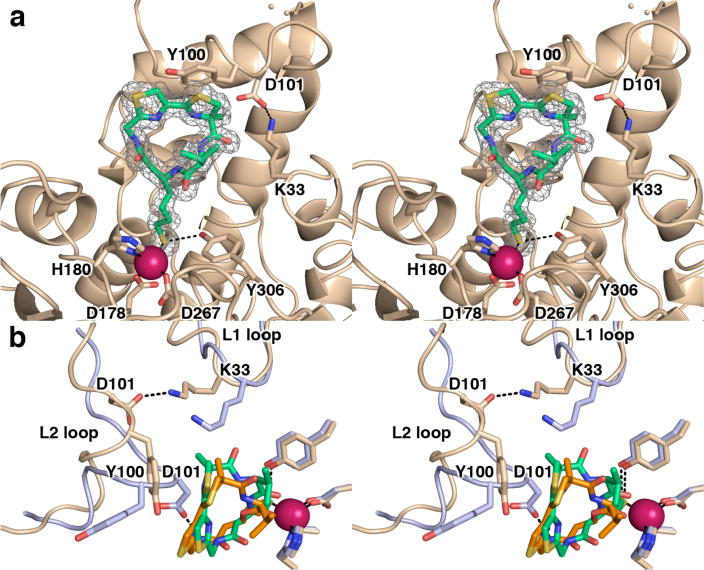
(a) Simulated annealing omit map of 1 bound in the active site of HDAC8 (monomer A, contoured at 3.0σ). Atomic color codes are as follows: C = wheat (protein) or green (inhibitor), N = blue, O = red, S = gold, Zn2+ = magenta. Metal coordination and selected hydrogen bond interactions are shown as solid black or dashed black lines, respectively. Wheat-colored dots indicate the disordered D87-D92 segment in the L2 loop. (b) Superposition of the HDAC8–Largazole and HDAC8–1 complexes (monomer A of each). Significant conformational changes of the L2 loop (especially for Y100 and D101) accommodate a slightly different orientation of the macrocycle in the active site, such that the macrocycle pivots by ~25° around an axis roughly defined by the Zn2+-bound thiolate and the thiazole-thiazoline bond. The HDAC8–1 complex is color coded as in (a); for the HDAC8-Largazole complex, C = light blue (protein) or orange (Largazole).
Notably, the ester-to-amide substitution in the macrocyclic peptide moiety of 1 results in a slight conformational difference, in that the average dihedral angle of the ester linkage of Largazole is 171° for the ester and 180° for the amide. The higher energetic barrier for the distortion of an amide compared with an ester serves to rigidify this linkage with a planar conformation. However, comparison of the HDAC8–1 complex with the wild-type HDAC8-Largazole complex reveals that this conformational difference is readily tolerated and does not perturb the overall binding mode of the macrocycle. Even so, while the macrocycle conformations of Largazole and 1 are essentially identical (r.m.s. deviation = 0.32 Å for the 32 nonhydrogen atoms of each inhibitor), the macrocycle pivots by ~25° around an axis roughly defined by the Zn2+-bound thiolate and the thiazole-thiazoline bond as the structures of the two enzyme-inhibitor complexes are superimposed (Figure 2b).
Surprisingly, major structural differences are observed in the L1 and L2 loops that accommodate the slightly-pivoted binding orientation of 1. Whereas D101 in the L2 loop hydrogen bonds with Largazole, the L2 loop in the HDAC8–1 complex adopts an alternative conformation such that D101 is more than 7 Å away from 1. Instead, adjacent residue Y100 (L2 loop) tends toward a van der Waals interaction with 1, and D101 accepts hydrogen bonds from K33 (L1 loop) and Y154 (Figure 2b). Additionally, K33 of monomer B hydrogen bonds with the carbonyl group close to the thiazoline group of 1 in both monomers A and B. This interaction can only be relevant in monomer B in solution because HDAC8 is a monomer.
Interestingly, the structures of monomers A and B are not exactly identical (r.m.s. deviation = 0.59 Å for 351 Cα atoms); in particular, significant structural differences are observed for the L1 and L2 loops (Figure 3a). Although 1 binds similarly in each monomer, portions of the L2 loop of each monomer adopt different conformations, and the L2 loop in monomer B is more disordered than in monomer A: the E85-H90 and D99-Y100 segments are not modeled due to ambiguous or missing electron density presumably due to disorder. However, certain modeled segments of the L2 loop clearly exhibit alternative conformations. The most significant structural difference is observed for T105, which moves by 9 Å between monomers A and B due to the partial unwinding of helix B4. These structural differences propagate to the L1 loop, where L31 moves by more than 3 Å, and helix B2, where the V82-Q84 segment shifts by ~2.5 Å.
Figure 3.
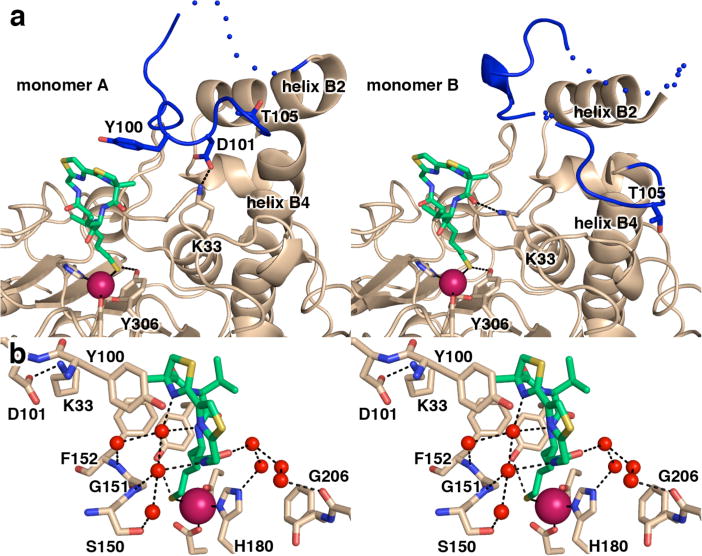
(a) Side-by-side comparison of monomers A and B of the HDAC8–1 complex reveals significant conformational differences for the L2 loop involving the partial unwinding of helix B4, as noted by the position of T105. Atoms are color coded as in Figure 2a, except the L2 loop is blue. Dotted lines indicate the disordered D87-D92 segment and the disordered E85-D89 and G99-Y100 segments in the L2 loops of monomers A and B, respectively. (b) Close-up stereoview of the HDAC8–1 complex showing active site water molecules (red spheres) that make bridging hydrogen bond interactions between 1 and the protein.
Finally, we note that the peptide macrocycle of 1 makes several solvent-mediated hydrogen bond interactions with active site residues that may contribute to inhibitory activity (Figure 3b). These hydrogen bond networks generally involve 1–3 intervening water molecules. Interestingly, an imidazole molecule from the crystallization buffer interacts with a peptide carbonyl group of 1 (monomer A only; not shown). Some, but not all, enzyme-solvent-inhibitor hydrogen bond interactions are similarly observed in the HDAC8-Largazole complex.
HDAC8–2 Complex
The overall structure of the HDAC8–2 complex determined at 2.18 Å resolution is very similar to that of the HDAC8–1 complex (r.m.s. deviations of 0.16 Å for 359 Cα atoms and 0.24 Å for 356 Cα atoms in monomers A and B, respectively). The geometry of thiolate-Zn2+ coordination is comparable to that observed for the binding of 1, with an average S–Zn2+ distance of 2.4 Å, a C–S–Zn2+ angle of 95°, and a C–C–S–Zn2+ dihedral angle of 113°; Y306 donates a hydrogen bond to the thiolate group (Figure 4a). The Zn2+ ion is coordinated in distorted tetrahedral fashion with ligand–Zn2+–ligand angles ranging 101°–119°. The ester-to-amide substitution similarly rigidifies the macrocycle with a nearly planar linkage (amide dihedral angle = 179°). Solvent-mediated hydrogen bond contacts with active site residues are similar to those observed in the HDAC–1 complex.
Figure 4.
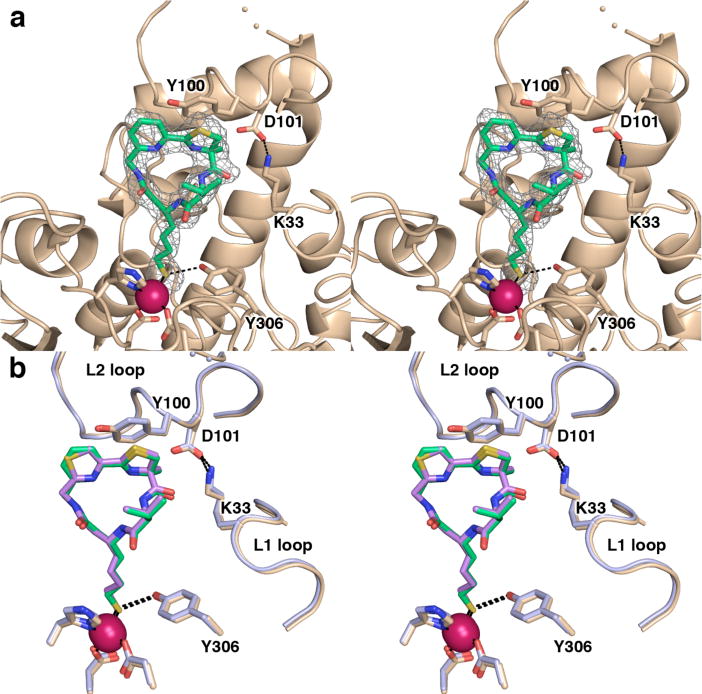
(a) Simulated annealing omit map of 2 bound in the active site of HDAC8 (monomer A, contoured at 2.8σ). Atomic color codes are as follows: C = wheat (protein) or green (inhibitor), N = blue, O = red, S = gold, Zn2+ = magenta sphere. Metal coordination and selected hydrogen bond interactions are shown as solid black or dashed black lines, respectively. Wheat-colored dotted lines indicate the disordered D88-P91 segment in the L2 loop. (b) Superposition of HDAC8 complexes (monomer A) reveals that Largazole analogues 2 and 1 adopt similar binding modes, and the flexible L1 and L2 loops adopt similar conformations. The HDAC8–2 complex is color coded as in (a); for the HDAC8–1 complex, C = light blue (protein) or violet (analogue 1).
Significantly, the thiazole-to-pyridine substitution in 2 does not perturb the overall conformation of the macrocyclic peptide, and the pyridine ring adopts a similar orientation with respect to the thiazoline ring (S–C–C–C dihedral angles are 26°, 22°, and 25° for Largazole, 1, and 2, respectively). The pyridine ring of 2 does not interact with any surrounding residues or water molecules.
Conformational differences observed for the L1 and L2 loops in the HDAC8–1 complex are similarly observed in the HDAC8–2 complex (Figure 4b), including the hydrogen bonds between K33 and D101, D101 and Y154 in monomer A, and K33 of monomer B and the carbonyl group close to the thiazoline ring of 2 in both monomers A and B. Additionally, the L2 loop in monomer B is more disordered than in monomer A, but modeled segments adopt a similar conformation to that observed in the HDAC8–1 complex.
HDAC8–3 Complex
The overall structure of the HDAC8–3 complex determined at 2.39 Å resolution is similar to that of the complex with 2; as these complexes are compared, monomer A in one is overall more similar to monomer B in the other (r.m.s. deviations of 0.19 Å for 347 Cα atoms in monomers A and B and 0.22 Å for 357 Cα atoms in monomers B and A of the complexes with 3 and 2, respectively). The geometry of thiolate-Zn2+ coordination is slightly different, with an average S–Zn2+ distance of 2.4 Å, a C–S–Zn2+ angle of 92°, and a C–C–S–Zn2+ dihedral angle of 108°; ligand–Zn2+–ligand angles range 91°–129° and the metal coordination polyhedron exhibits distorted tetrahedral geometry. An electron density map of the inhibitor is shown in Figure 5a. As observed for 1 and 2, the ester-to-amide substitution rigidifies a planar conformation (amide dihedral angle = 180°), and the alternative substitution pattern on the newly-introduced pyridine ring does not perturb the overall conformation of the peptide macrocycle (the thiazoline-pyridine S–C–C–C dihedral angle is 19°). A superposition of the HDAC8 complexes with 2 and 3 is found in Figure 5b.
Figure 5.
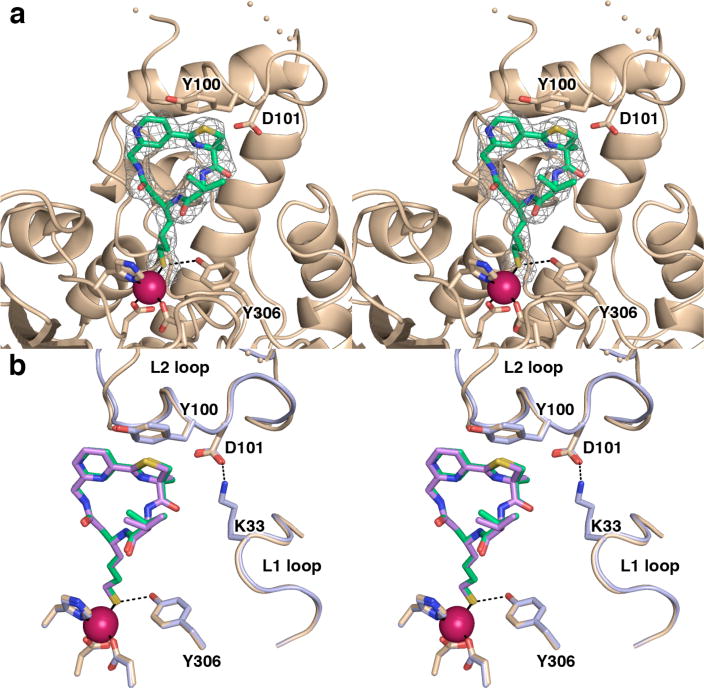
(a) Simulated annealing omit map of 3 bound in the active site of HDAC8 (monomer B, contoured at 2.8σ). Atomic color codes are as follows: C = wheat (protein) or green (inhibitor), N = blue, O = red, and S = gold, Zn2+ = magenta sphere. Metal coordination and selected hydrogen bond interactions are shown as solid black or dashed black lines, respectively. Wheat-colored dotted lines indicate the disordered D87-D92 segment in the L2 loop. (b) Largazole analogues 3 and 2 bind similarly in the active site of HDAC8. While the L2 loops adopt slightly different conformations, Y100 and D101 are positioned similarly in each complex. The HDAC8–3 complex is color coded as in (a); for the HDAC8–2 complex (monomer A), C = light blue (protein) or violet (analogue 2).
As observed in the HDAC8–2 complex, the L2 loop (including Y100 and D101) is positioned similarly to interact with 3 in the HDAC8–3 complex. However, in the complex with 3, the side chain of K33 is disordered in both monomers A and B and therefore does not interact with D101. As observed in the structures of complexes with 1 and 2, the L2 loop exhibits greater disorder in one monomer (monomer A in this structure) compared with the other. Monomer B of the HDAC8–3 complex similarly differs from monomer A in terms of conformational differences in the L1 and L2 loops as well as helix B2, as described above for the complexes with 1 and 2.
DISCUSSION
The structures reported herein show that chemical modifications maintaining the 16-membered ring of the macrocyclic skeleton of Largazole are readily accommodated in the active site of HDAC8. The common feature of 1, 2, and 3 is the ester-to-amide substitution that converts the macrocyclic depsipeptide into a peptide. This has relatively little effect on the overall conformation of the macrocyclic skeleton, even though the ester group of Largazole is distorted 9° away from planarity whereas the amide group of each Largazole analogue is nearly perfectly planar with a trans configuration. Although the peptide macrocycle of each analogue pivots by ~25° in the HDAC8 active site relative to Largazole (Figure 2b), this structural change has relatively little consequence for inhibitory potency against HDAC8 since the IC50 values for Largazole and 1 are 228 nM and 255 nM, respectively.26 Additionally, this structural change is readily tolerated with regard to HDAC1, HDAC2, and HDAC3, each of which is inhibited with comparable low-nanomolar potency by Largazole and 1 (Table 2).26
While the amide substitution in the 16-membered macrocyclic ring of Largazole preserves inhibitory potency, further derivatization of the macrocycle skeleton by the substitution of a pyridine ring in 2 and 3 compared with the thiazole ring of 1 moderately or slightly compromises inhibitory activity, respectively, even though this substitution does not appear to perturb the overall conformation of the macrocycle (Figures 4b and 5b). Compound 2 exhibits a ~20–40-fold loss of inhibitory potency against HDAC1, HDAC2, HDAC3, and HDAC8, whereas 3 exhibits a ~6-fold loss of inhibitory potency against these isozymes (Table 2).26 In contrast, a pyridine ring substitution in the parent depsipeptide macrocycle enhances inhibitory potency.19 Perhaps some degree of macrocycle flexibility is required to accommodate the substituted pyridine ring (the macrocycle ester linkage is slightly more flexible than an amide linkage). This could explain why 3 is slightly less potent than 1. However, there is no obvious explanation for the loss of inhibitory potency of 2 relative to 3 (Table 2), since both appear to be readily accommodated in the HDAC8 active site based on the crystal structures shown in Figures 4 and 5.
If macrocycle flexibility is desirable, then the substitution of a more flexible ketone linkage for the depsipeptide ester linkage might enable further derivatization of the macrocycle skeleton with retention or enhancement of inhibitory potency. Additionally, given that substitution of the thiazoline ring by a tetrazole ring yields an analogue with inhibitory activity comparable to that of Largazole,37 it is possible that further derivatization of the thiazoline-thiazole moiety will similarly preserve inhibitory activity as long as the overall macrocycle conformation is retained. Since the thiazoline-thiazole ring system of 1 and the thiazoline-pyridine ring systems of 2 and 3 are solvent-exposed, such derivatization could include the attachment of pendant functional groups to capture additional affinity interactions in the outer active site cleft.
The most important feature of HDAC inhibition by depsipeptide and peptide macrocycles is the strong coordination interaction between the inhibitor thiol group and the active site Zn2+ ion. In the structures of HDAC8 complexes with 1, 2, and 3, the S–Zn2+ coordination distance is 2.3 Å–2.4 Å, which is the ideal coordination bond length36 and is comparable to that observed in the HDAC8–Largazole complex.18 The coordination geometry of the active site Zn2+ ion is tetrahedral or distorted tetrahedral, with average deviations from ideal tetrahedral geometry of 6°, 6°, and 13° for 1, 2, and 3, respectively. For the HDAC8–Largazole complex, the average deviation from ideal tetrahedral Zn2+ coordination geometry is 4°. The deviation from ideal coordination geometry is greatest in the HDAC8–3 complex, but this could be a consequence of the lower resolution of this crystal structure compared with the others.
Some differences are observed in the conformations of the thiol side chain of Largazole and its derivatives, and these differences may influence inhibitory potency. The thiol side chain extends from the macrocycle skeleton into the active site tunnel to enable S–Zn2+ coordination, and the chemical structure of this side chain, which includes a trans double bond between the Cβ and Cγ atoms, is identical in all derivatives (Figure 1). In the HDAC8 complexes with Largazole and 1, the overall side chain conformation is bent such that the Cβ–Cγ–Cδ–Cε torsion angles are anticlinal, with average values of −122° and −124°, respectively. In contrast, the thiol side chains of 2 and 3 tend toward antiperiplanar conformations in their complexes with HDAC8, with average Cβ–Cγ–Cδ–Cε torsion angles of −166° and −168°, respectively. Thus, it appears that more potent inhibitory affinity is achieved for macrocyclic thiols that can bind in the enzyme active site and retain the anticlinal thiol side chain conformation, as initially observed in the HDAC8–Largazole complex.18 If the anticlinal-to-antiperiplanar conformational change of the thiol side chain is caused by the pyridine substitutions in the macrocyclic rings of 2 and 3, it is not clear from the structures of their HDAC8 complexes how this occurs.
Strikingly, the binding of Largazole analogues in the active site of HDAC8 reveals new features regarding the conformational flexibility of loops flanking the active site and the role of these loops in accommodating the binding of large, structurally-similar ligands. Such conformational flexibility presumably accompanies the binding of protein substrates in vivo and as such may play an important role in governing substrate specificity. Surprisingly, the binding of all three Largazole analogues triggers significant conformational changes of the L2 loop that propagate through to the L1 loop and helix B2; moreover, these conformational changes differ from those observed in the HDAC8–Largazole complex.18 It is particularly noteworthy to see such differences in protein conformational changes that accommodate the binding of isosteric ligands, and it is even more noteworthy to see such differences in the binding of the same ligand to independent monomers in the same crystal.
The L2 loop of HDAC8 is already known to undergo conformational changes to accommodate the binding of different ligands, and residues contained in this loop are usually characterized by higher thermal B factors and/or lack of clear electron density. However, in the present array of structures, the rearrangement of this loop is more pronounced than usually observed. For example, the side chain of D101, which usually hydrogen bonds with bound substrates or inhibitors, is shifted by ~7 Å away from the macrocycle, such that adjacent residue Y100 now associates more closely with the macrocycle (Figures 2a, 4a, and 5a). Interestingly, recent molecular dynamics simulations suggest that the L1 and L2 loops can interact through K33 and D87/D88/D89.38 Here, we show that a direct interaction between the L1 and L2 loops can also be made between K33 and D101, as observed in monomers A of the complexes with 1 and 2. Presumably, such L1-L2 loop interactions play an important role in governing specificity and affinity in the binding of substrates and inhibitors.
These results are consistent with the high conformational flexibility of the L1 and L2 loops of HDAC8 observed in crystallographic18,27,39,40 and molecular dynamics (MD) studies.38,41 In comparison, the loops surrounding the active site of HDAC1, which is a more promising drug target and binds Largazole and its analogues ~200 times more tightly, are much less flexible in MD simulations.42 In the absence of a crystal structure of a complex between HDAC1 and a Largazole derivative, we analyzed the structural relationship between the HDAC8-1 complex and the structure of HDAC1 complexed with the ELM2 and SANT domains of metastasis-associated protein 1 (MTA1).43 Figure 6a shows the surface representation of the overlay of HDAC1 in blue mesh and chain A of the HDAC8–1 complex in light brown in the area around the binding site; the cartoon representation in Figure 6b is similarly color-coded. It can be seen that the L1 and L2 loops adopt different conformations that enhance the shape complementarity to 1, even though the overall structural similarity is very high. The L1 loop in HDAC1, which is four residues longer and more rigid than the corresponding L1 loop in HDAC8, protrudes on one side of the active site mouth as indicated by the blue mesh in Figure 6a. Additional interactions with the isopropyl group and the amide functionality of Largazole analogues, which are maintained during extensive MD simulations,26 rationalize the increased binding affinity of the Largazole analogues to HDAC1. It is possible, too, that decreased flexibility of the L1 and L2 loops of HDAC1 may decrease the conformational entropic cost of inhibitor binding. These results reinforce the importance of HDAC surface interactions for the binding and isozyme selectivity of Largazole and related depsipeptide inhibitors.
Figure 6.
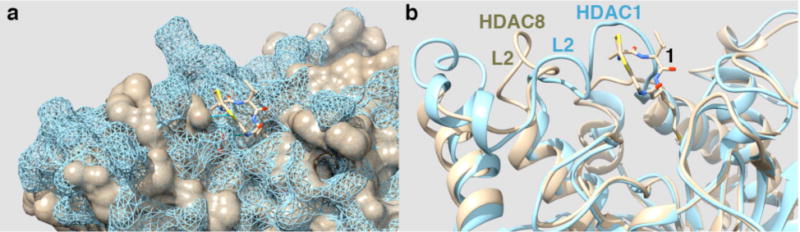
(a) Surface representation of the HDAC8–1 complex (brown) and HDAC1 (blue mesh). (b) Cartoon representation of the superposition in (a), with the position of the flexible loop L2 in HDAC1 and HDAC8 indicated.
CONCLUDING REMARKS
As observed in previous structural studies,27,29,39,40 active site loops – especially the L2 loop – play an important role in the binding of small hydroxamate inhibitors to HDAC8. These loops play an even more important role in accommodating the 16-membered ring of the macrocyclic depsipeptide inhibitor Largazole and its amide derivatives 1, 2, and 3. While residues in the L2 loop such as D101 are capable of hydrogen bonding with the Largazole macrocycle, the binding of the three amide derivatives shows that a hydrogen bond interaction with D101 need not be conserved to maintain inhibitory potency. Indeed, the current study shows that the L2 loop exhibits remarkable flexibility to better accommodate structural variations in the macrocycle skeleton, all while maintaining ideal S-Zn2+ coordination geometry by the thiol side chain. Strikingly, alternative loop conformations accommodate the binding of isosteric ligands, or even the same ligand, to independent monomers in the same crystal. Such flexibility in both the L1 and L2 loops accommodates diverse protein substrates in the HDAC8 active site. Crystal structures of HDAC8 complexes with Largazole and its macrocyclic analogues provide an unprecedented view of how such molecular accommodation is achieved.
Acknowledgments
We thank the National Synchrotron Light Source at Brookhaven National Laboratory for access to beamline X29 for X-ray crystallographic data collection. Additionally, we thank Dr A. Lebedev for helpful discussions.
FUNDING: We thank the NIH for grants GM49758 (to D.W.C.) and CA152314 (to R.M.W. and O.W.) in support of this research.
Footnotes
Accession Codes
The atomic coordinates and the crystallographic structure factors of HDAC8 complexes with 1, 2, and 3 have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 4RN0, 4RN1, and 4RN2, respectively.
The authors declare no competing conflicts of interest.
References
- 1.De Ruijter AJM, van Gennip AH, Caron HN, Kemp S, van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 4.Genshaft A, Moser JAS, D’Antonio EL, Bowman CM, Christianson DW. Energetically unfavorable amide conformations for N6-acetyllysine side chains in refined protein structures. Proteins: Struct, Funct, Bioinf. 2013;81:1051–1057. doi: 10.1002/prot.24262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Opin Struct Biol. 2011;21:735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfson NA, Pitcairn CA, Fierke CA. HDAC8 substrates: Histones and beyond. Biopolymers. 2013;99:112–126. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emanuele S, Lauricella M, Tesoriere G. Histone deacetylase inhibitors: apoptotic effects and clinical implications. Int J Oncol. 2008;33:637–646. [PubMed] [Google Scholar]
- 9.Marks PA. Histone deacetylase inhibitors: a chemical genetics approach to understanding cellular functions. Biochim Biophys Acta. 2010;1799:717–725. doi: 10.1016/j.bbagrm.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol. 2012;6:637–656. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenet. 2012;4:5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 13.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 14.Guan P, Fang H. Clinical development of histone deacetylase inhibitor romidepsin. Drug Discov Ther. 2010;4:388–391. [PubMed] [Google Scholar]
- 15.Furumai R, Matsuyama A, Kobashi N, Lee K-H, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- 16.Taori K, Paul VJ, Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J Am Chem Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 17.Ying Y, Taori K, Kim H, Hong J, Luesch H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J Am Chem Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 18.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J Am Chem Soc. 2011;133:12474–12477. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bowers AA, West N, Newkirk TL, Troutman-Youngman AE, Schreiber SL, Wiest O, Bradner JE, Williams RM. Synthesis and histone deacetylase inhibitory activity of largazole analogs: alteration of the zinc-binding domain and macrocyclic scaffold. Org Lett. 2009;11:1301–1304. doi: 10.1021/ol900078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowers A, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM. Total synthesis and biological mode of action of largazole: a potent class I histone deacetylase inhibitor. J Am Chem Soc. 2008;130:11219–11222. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nasveschuk CG, Ungermannova D, Liu X, Phillips AJ. A concise total synthesis of largazole, solution structure, and some preliminary structure-activity relationships. Org Lett. 2008;10:3595–3598. doi: 10.1021/ol8013478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh AK, Kulkarni S. Enantioselective total synthesis of (+)-largazole, a potent inhibitor of histone deacetylase. Org Lett. 2008;10:3907–3909. doi: 10.1021/ol8014623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guerra-Bubb JM, Bowers AA, Smith WB, Paranal R, Estiu G, Wiest O, Bradner JE, Williams RM. Synthesis and HDAC inhibitory activity of isosteric thiazoline-oxazole largazole analogs. Bioorg Med Chem Lett. 2013;23:6025–6028. doi: 10.1016/j.bmcl.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowers AA, Greshock TJ, West N, Estiu G, Schreiber SL, Wiest O, Williams RM, Bradner JE. Synthesis and conformation-activity relationships of the peptide isosteres of FK228 and largazole. J Am Chem Soc. 2009;131:2900–2905. doi: 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rettig I, Koeneke E, Trippel F, Mueller WC, Burhenne J, Kopp-Schneider A, Fabian J, Schober A, Fernekorn U, von Deimling A, Deubzer HE, Milde T, Witt O, Oehme I. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015;6:e1657. doi: 10.1038/cddis.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clausen DJ, Smith WB, Haines BE, Wiest O, Bradner JE, Williams RM. Modular synthesis and biological activity of pyridyl-based analogs of the potent class I histone deacetylase inhibitor Largazole. Bioorg Med Chem. 2015 doi: 10.1016/j.bmc.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Decroos C, Bowman CM, Moser JAS, Christianson KE, Deardorff MA, Christianson DW. Compromised structure and function of HDAC8 mutants identified in Cornelia de Lange Syndrome spectrum disorders. ACS Chem Biol. 2014;9:2157–2164. doi: 10.1021/cb5003762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfí A, De Francesco R, Steinkühler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Barends TRM, Dijkstra BW. Acetobacter turbidans α-amino acid ester hydrolase: merohedral twinning in P21 obscured by pseudo-translational NCS. Acta Crystallogr D. 2003;59:2237–2241. doi: 10.1107/s0907444903020729. [DOI] [PubMed] [Google Scholar]
- 32.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee H, Rezai-Zadeh N, Seto E. Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol Cell Biol. 2004;24:765–773. doi: 10.1128/MCB.24.2.765-773.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chakrabarti P. Geometry of interaction of metal ions with sulfur-containing ligands in protein structures. Biochemistry. 1989;28:6081–6085. doi: 10.1021/bi00440a052. [DOI] [PubMed] [Google Scholar]
- 37.Li X, Tu Z, Li H, Liu C, Li Z, Sun Q, Yao Y, Liu J, Jiang S. Biological evaluation of new largazole analogues: alteration of macrocyclic scaffold with click chemistry. ACS Med Chem Lett. 2013;4:132–136. doi: 10.1021/ml300371t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunze MBA, Wright DW, Werbeck ND, Kirkpatrick J, Coveney PV, Hansen DF. Loop interactions and dynamics tune the enzymatic activity of the human histone deacetylase 8. J Am Chem Soc. 2013;135:17862–17868. doi: 10.1021/ja408184x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang B-C, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 40.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Estiu G, West N, Mazitschek R, Greenberg E, Bradner JE, Wiest O. On the inhibition of histone deacetylase 8. Bioorg Med Chem. 2010;18:4103–4110. doi: 10.1016/j.bmc.2010.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weerasinghe SVW, Estiu G, Wiest O, Pflum MKH. Residues in the 11 Å channel of histone deacetylase 1 promote catalytic activity: implications for designing isoform-selective histone deacetylase inhibitors. J Med Chem. 2008;51:5542–5551. doi: 10.1021/jm800081j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, Fairall L, Schwabe JWR. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell. 2013;51:57–67. doi: 10.1016/j.molcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]