

Abstract
The role of ERβ in prostate cancer is unclear, although its loss of ERβ is associated with aggressive disease. Given that mice deficient in ERβ do not develop prostate cancer, we hypothesized that ERβ loss occurs as a consequence of tumorigenesis caused by other oncogenic mechanisms and that its loss is necessary for tumorigenesis. In support of this hypothesis, we found that ERβ is targeted for repression in prostate cancer caused by PTEN deletion and that loss of ERβ is important for tumor formation. ERβ transcription is repressed by BMI-1, which is induced by PTEN deletion and important for prostate tumorigenesis. This finding provides a mechanism for how ERβ expression is regulated in prostate cancer. Repression of ERβ contributes to tumorigenesis because it enables HIF-1/VEGF signaling that sustains BMI-1 expression. These data reveal a positive feedback loop that is activated in response to PTEN loss and sustains BMI-1.
Keywords: Prostate cancer, PTEN, Estrogen Receptor β, VEGF, BMI-1
INTRODUCTION
The role of estrogen receptors (ERs) in epithelial cell biology and cancer is an emerging area of considerable biological interest and pathological relevance. In the prostate, ERβ is expressed in epithelial cells, while ERα expression is confined to stromal cells (Kuiper et al., 1996; Leav et al., 2001; Thomas and Gustafsson, 2011). The contribution of ERβ to prostate cancer appears to be significant but much remains to be learned (Christoforou et al., 2014; Dey et al., 2013). The inverse correlation between the expression of ERβ and differentiation (Gleason score) (Leav et al., 2001; Mak et al., 2010) is supported by mechanistic cell biology studies demonstrating that one function of ERβ is to impede an epithelial mesenchymal transition (EMT) (Mak et al., 2013; Mak et al., 2010). The mechanism involves the ability of ERβ to sustain prolyl hydroxylase 2 (PHD2) expression and subsequently promote HIF-1α degradation and HIF-1–mediated EMT (Mak et al., 2013).
An important and timely issue is the contribution of ERβ to prostate tumorigenesis. Although the loss of ERβ is associated with higher Gleason grade and more aggressive disease, a causal role for ERβ in impeding the formation of aggressive tumors has not been established. There is evidence that loss of ERβ can increase the incidence of poorly differentiated prostate carcinoma, but the mechanism is not known (Slusarz et al., 2012). This problem has been obscured by the analysis of ERβ knockout (BERKO) mice. These mice do not develop prostate cancer (Antal et al., 2008; Imamov et al., 2004), although some studies have observed prostate hyperplasia in older BERKO mice (Imamov et al., 2004). Furthermore, deletion of ERβ in the FGF8b transgenic model of prostate tumorigenesis did not increase tumor incidence, a finding that has been used to discount a tumor suppressive function for ERβ (Elo et al., 2014).
We approached the problem of the potential role of ERβ in prostate tumorigenesis from a different perspective. Specifically, we hypothesized that ERβ loss occurs as a consequence of tumorigenesis caused by other oncogenic mechanisms and that its loss is necessary for this tumorigenesis. To test this hypothesis, we focused on prostate tumorigenesis induced by PTEN loss for several reasons. Inactivation or loss of PTEN is one of the most common genetic lesions in prostate cancer and its frequency increases with Gleason grade and more aggressive disease (Cairns et al., 1997; Goel et al., 2012; McMenamin et al., 1999). Given that ERβ loss also increases with dedifferentiated, aggressive disease (Mak et al., 2010), these observations suggest a causal relationship between loss of PTEN and loss of ERβ that may be significant for prostate tumorigenesis. The results presented in this study validate this hypothesis and provide a mechanism for how PTEN loss results in the transcriptional repression of ERβ that involves BMI-1, an oncogene that regulates cell proliferation and senescence through the ink4a locus (Jacobs et al., 1999) and has been implicated in prostate tumorigenesis (Lukacs et al., 2010). Importantly, we also establish that the loss of ERβ is necessary for tumorigenesis caused by PTEN loss because it enables autocrine VEGF signaling, which has been implicated in the genesis of several cancers, including prostate [reviewed in (Goel and Mercurio, 2013)].
RESULTS
ERβ is targeted for repression in prostate tumorigenesis induced by PTEN loss
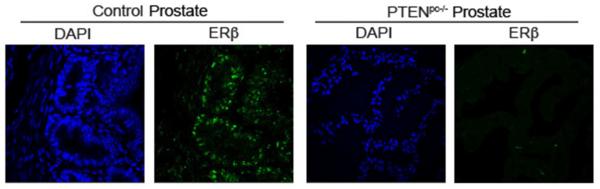
To assess the potential relationship between ERβ and PTEN, we evaluated ERβ expression in a transgenic model of prostate tumorigenesis mice induced by PTEN deletion [Pten loxp/loxp; PB-Cre+ (referred to as Pten pc−/−)]. Prostate-specific deletion of PTEN results in tumors that are invasive and aggressive (Wang et al., 2003). The normal glandular features of the wild-type prostates (control) and tumor formation in age-matched Pten pc−/− prostates are shown in Figure 1A. PTEN expression is apparent in wild-type prostates but absent in Pten pc−/− mice (Figure 1B). Importantly, ERβ expression is lost in Pten pc−/− mice, whereas the age-matched wild-type prostates exhibit ERβ expression (Figure 1C). We also observed that PTEN and ERβ expression correlate in human prostate tumors based on analysis of the cBioportal database (Figures 1D; Table S1). High Gleason grade tumors (primary Gleason grade 5) exhibit uniform loss of PTEN and ERβ (Cairns et al., 1997; Mak et al., 2010). Gleason grade 3 tumors are interesting in this regard because these tumors are characterized by heterogeneity in PTEN expression (McMenamin et al., 1999). Indeed, we quantified PTEN and ERβ mRNA expression in grade 3 tumors and observed a correlation between PTEN and ERβ expression (Figure 1E).
Figure 1. ERβ is targeted for repression in prostate tumorigenesis induced by PTEN loss.

(A) H&E staining of ventral prostates of wild-type (control) and aged-matched Pten pc−/−mice. The expression of PTEN (B) and ERβ (C) in these tissues was examined by immunofluorescence microscopy. Scale bar, 50 µm. (D) A positive correlation between PTEN and ERβ expression in a cohort of 34 prostate tumors determined from the cBioportal database. (E) Four different Grade 3 tumors were analyzed by qPCR for ERβ and PTEN mRNA expression. (F) PTEN-depleted PNT1a cells (shPTEN-1 and shPTEN-2) and control (shGFP) PNT1a cells were analyzed for morphology (phase contrast images) and expression of ERβ and PTEN (immunoblot). PTEN mRNA expression was also quantified by qPCR (bar graph). (G) Immunoblot showing PTEN expression in ERβ-depleted (shERβ-1 and shERβ-2) and control (shGFP) PNT1a cells. Bar graph depicts the lack of an effect of 3β-Adiol and estrogen (E2) treatment on PTEN mRNA expression. Data represent the average of 3 experiments (±SEM). (H) Effects of wortmannin and Akt Inhibitor VIII on cell morphology and ERβ expression in shPTEN PNT1a cells. See also Table S1.
To investigate the relationship between ERβ and PTEN further, we depleted PTEN in PNT1a cells, an immortalized prostate epithelial cell line (Berthon et al., 1995). Depletion of PTEN caused an EMT consistent with previous reports (Mulholland et al., 2012; Song et al., 2009), and it also resulted in a decrease in ERβ mRNA and protein expression compared to control cells indicating that PTEN regulates ERβ expression (Figure 1F). In contrast, ERβ does not appear to impact PTEN expression because depletion of ERβ did not alter PTEN levels (Figure 1G). Furthermore, two physiological ligands of ERβ (3β-Adiol and 17β-estradiol:E2) did not affect PTEN expression (Figure 1G). Interestingly, both a PI3K inhibitor (wortmannin) and Akt inhibitor (Akt Inhibitor VIII) reversed the mesenchymal phenotypes of PTEN depleted PNT1a cells to an epithelial phenotype with a concomitant restoration of ERβ expression (Figure 1H).
In order to support a functional link between PTEN and ERβ expression, we examined the effect of expressing ERβ in PTEN-depleted PNT1a cells. The mesenchymal morphology of PTEN-depleted cells reverted to an epithelial morphology in response to expression of HA-ERβ with a concomitant decrease in mesenchymal markers (N-cadherin and vimentin) (Figure 2A). We also assessed the tumor suppressive activity of ERβ in the context of PTEN by injecting control, PTEN-depleted and PTEN-depleted cells that express ERβ into nude mice. Control cells did not form tumors after 9 weeks, whereas 71% of mice injected with PTEN-depleted cells had tumors at this time (Figure 2B). More importantly, tumor formation was greatly reduced (12% of mice had tumors) when mice were injected with PTEN-depleted cells expressing HA-ERβ (Figure 2B). This observation was substantiated by expressing ERβ in PC3-M cells, a highly tumorigenic, PTEN-prostate cancer cell line (Kozlowski et al., 1984) that expresses low levels of ERβ (Figure 2C). Indeed, ERβ expression dramatically reduced the ability of these cells to form tumors (Figure 2C).
Fig 2. ERβ impedes tumor initiation induced by PTEN loss.

(A) HA-ERβ was expressed in PTEN-depleted cells and the impact on cell morphology (phase contrast images) and expression of ERβ (immunoblot) was evaluated. Scale bar, 50 µm. (B) The indicated cells (105) were injected subcutaneously into nu/nu mice (n = 7) and tumor formation was assessed by palpation. The curve comparison was done using log rank test p <0.05. (C) HA-ERβ was expressed in PC3-M cells, which are PTEN- and express very low level of ERβ. The indicated cells (105) were injected subcutaneously into nu/nu mice (n = 7) and tumor formation was assessed by palpation. The curve comparison was done using log rank test p <0.05. (D) Effects of wortmannin and Akt Inhibitor VIII on BMI-1 expression in shPTEN PNT1a cells. (E) Expression of BMI-1 in PTEN depleted (shPTEN) and control (shGFP) PNT1a cells (F) BMI-1 was expressed in PNT1a cells and the effect on ERβ expression was evaluated by immunoblotting. (G) BMI-1 expression was diminished in PC3-M cells using shRNAs (shBMI-1 and shBMI-2) and the expression of ERβ was assessed (immunoblots). (H) Effect of estrogen (E2: 10 nM)) on BMI-1 expression in shPTEN cells. (I) Schematic of the ERβ promoter showing the primer sets that were used to assess binding activity of BMI-1 in the eight regions depicted by the black boxes. ChIP was performed using a BMI-1 Ab and the line graph depicts the quantitation of the ChIP results by qPCR normalized to IgG. Data represent the average of two separate experiments. (J) Luciferase reporter constructs containing regions 1 and 2 of the ERβ promoter (left graph) or region 1 (right graph) were expressed in PC3-M cells. Luciferase activity was normalized to Renilla and the experiment was repeated three times with similar results. See also Figure S1.
BMI-1 is a transcriptional repressor of ERβ
To define the mechanism by which loss of PTEN diminishes ERβ expression, we focused on BMI-1, the key regulatory component of the polycomb repressive complex-1 that modulates chromatin structure and represses the transcription of a number of genes (Cao et al., 2005; Jacobs et al., 1999; Miyazaki et al., 2008), for several reasons. BMI-1 has been implicated in prostate hyperplasia and tumorigenesis (Lukacs et al., 2010; van Leenders et al., 2007). We also found that expression of BMI-1 in ERβ expressing PTEN-depleted cells promoted tumor formation (Figure 2C). Furthermore, PTEN loss induces BMI-1 expression (Goel et al., 2012), as evident in PNT1a cells (Figure 2D). The ability of both wortmannin and Akt Inhibitor VIII to attenuate BMI-1 expression in shPTEN cells supports our previous finding on their ability to restore ERβ expression in these cells (Figure 2E). For these reasons, we evaluated the possibility that BMI-1 represses ERβ. Expression of BMI-1 in PNT1a cells suppressed ERβ compared to control cells (Figure 2F). Conversely, we depleted BMI-1 in PC3-M cells and observed an induction of ERβ expression compared to the control (Figure 2G). We also observed that 17β-estradiol (E2) had no effect on the expression of BMI-1 on PTEN depleted cells suggesting ERα does not regulate BMI-1 (Figure 2H). The multiple bands observed in the BMI-1 immunoblots may represent phosphorylated forms of the protein (Nacerddine et al., 2012).
The inverse functional relationship between BMI-1 and ERβ prompted us to test the possibility that BMI-1 is a transcriptional repressor of ERβ. Therefore, we performed ChIP analysis on the ERβ promoter to assess BMI-1 binding. We examined 8 regions within the 3 kb spanning from +331 to − 2996 base pairs and detected one major BMI-1 binding locus in region 1 proximal to the transcription start site (Figures 2I and Figure S1A). The impact of BMI-1 on ERβ promoter activity was evaluated by expressing luciferase reporter constructs containing two regions of the promoter (region 1 and regions 1+2) in control and BMI-1-depleted PC3-M cells. Diminishing BMI-1 expression resulted in a significant increase in luciferase activity in both constructs compared to the control (Figure 2J). Moreover, this activity is concentrated in region 1 supporting our ChIP data. Given that ERβ expression is regulated by two promoters, 0N and 0K (Hirata et al., 2001), we sought to determine which promoter is utilized by BMI-1 to exert its repressive function. The BMI-1 binding locus of region 1 in our ChIP assays lined up perfectly within the 0N promoter but not within the 0K promoter (Figure S1B). These data indicate that ERβ transcription is repressed by BMI-1 on the 0N promoter.
Role of ERβ repression in prostate tumorigenesis
The critical issue that arises from the foregoing observations is why ERβ repression is important for prostate tumorigenesis induced by PTEN loss (see Figure 1A). Previously, we reported that loss of ERβ stabilizes HIF-1α and enables autocrine VEGF signaling in prostate cancer cells (Mak et al., 2010). This finding is relevant because autocrine VEGF signaling has emerged as an important component of tumorigenesis (Goel and Mercurio, 2013). Of particular relevance, we reported that autocrine VEGF signaling results in the enhancement of BMI-1 expression by a mechanism that involves Neuropilin-2 (NRP2) and Gli-1 (Goel et al., 2012). Together, these observations support the hypothesis that ERβ repression is important for prostate tumorigenesis induced by PTEN loss because it enables autocrine VEGF signaling via HIF-1α stabilization that sustains BMI-1 expression. Initially, we assessed HIF-1α and VEGF expression in wild-type and Pten pc−/− prostates. Deletion of PTEN induced expression of HIF-1α, VEGF, as well as BMI-1 (Figures 3A, 3B, 3C), in tumor cells. This observation is supported by analysis of PTEN-depleted PNT1a cells, which express high levels of HIF-1α, VEGF-A and BMI-1 compared to control cells (shGFP) (Figure 3D). To demonstrate that the increase in VEGF and BMI-1 expression observed in response to PTEN deletion is dependent on ERβ repression, we expressed ERβ in PTEN-depleted PNT1a cells and observed a reduction in VEGF and BMI-1 expression compared to controls (Figure 3E).
Figure 3. Role of ERβ repression in prostate tumorigenesis.

Ventral prostates of wild-type (control) and aged-matched Pten pc−/−mice were stained for (A) HIF-1α, (B) VEGF-A and (C) BMI-1 and analyzed by immunofluorescence microscopy. Scale bar, 50 µm. (D) Immunoblot showing the expression of HIF1α, VEGF-A and BMI-1 in PTEN-depleted (shPTEN) and control (shGFP) PNT1a cells. (E) Expression of VEGF-A and BMI-1 in ERβ expressing PTEN depleted cells (shPTEN-2+HA-ERβ) compared to PTEN depleted cells (shPTEN).
The results reported so far indicate that PTEN loss results in the BMI-1-mediated repression of ERβ and that repression of ERβ enables VEGF signaling that sustains BMI-1 expression. In essence, the data reveal a positive feedback loop that functions to maintain BMI-1 expression and is activated in response to PTEN loss. This hypothesis infers that loss of ERβ should induce BMI-1 expression by a mechanism that involves HIF-1/VEGF signaling. Indeed, depletion of ERβ in PNT1a cells induced HIF-1α, VEGF-A and BMI-1 expression compared to the controls (Figure 4A). We also observed an induction of NRP2 in ERβ-depleted cells with a concomitant increase in BMI-1 (Figure S2). Similar results were obtained by depleting proyl hydroxylase 2 (PHD2), which is sustained by ERβ (Mak et al., 2013) (Figure 4B). The PHD2-depleted cells also had high expression levels of N-cadherin and vimentin compared to the control cells supporting our previous observation that loss of PHD2 induced an EMT in PNT1a cells (Mak et al., 2013). In order to establish a causal role for HIF-1α in regulating BMI-1 expression, we knocked down HIF-1α in ERβ-depleted cells and observed a substantial decrease in VEGF-A and BMI-1 expression compared to ERβ-depleted cells alone (Figure 4C). These expression differences were also manifested in cell morphology. Control (shGFP) and shERβ/shHIF-1α cells exhibited an epithelial morphology compared to the mesenchymal morphology of shERβ/shGFP cells (Figure 4C). Based on these data, the possibility existed that BMI-1 is a HIF-1α target gene. However, promoter activity analyses did not support this possibility (data not shown). For this reason, we focused on the role of VEGF-A signaling in regulating BMI-1 in the context of ERβ. Specifically, knocking down VEGF-A in ERβ-depleted cells attenuated BMI-1 expression and induced an epithelial morphology (Figure 4D).
Fig 4. ERβ represses BMI-1 by a HIF-1α/VEGF-mediated mechanism.

(A) Expression of HIF-1α VEGF-A and BMI-1 in ERβ depleted PNT1a cells (shERβ-1 and shERβ-2) and control cells (shGFP) was assessed by immunoblotting. (B) Expression of N-cadherin, vimentin, HIF-1α, VEGF-A, and BMI-1 in PHD2-depleted cells (shPHD2-1 and shPHD2-2) compared to control cells (shGFP) as assessed by immunoblotting. (C) HIF-1α expression was diminished in ERβ-depleted PNT1a cells using shRNA and the impact on cell morphology and expression of HIF-1α, VEGF-A and BMI-1 was determined. Scale bar, 50 µm. (D) VEGF-A expression was diminished in ERβ-depleted PNT1a cells using shRNA and the impact on cell morphology (phase contrast images) and expression of VEGF and BMI-1 was determined by immunoblotting. (E) H&E staining of ventral prostates from 10 month old wild-type (control) and BERKO mice. Arrows indicate areas of hyperplasia. These tissues were stained for ERβ, HIF-1α, VEGF-A, BMI-1 and PTEN and analyzed by immunofluorescence microscopy. Scale bar, 50 µm. (F) An inverse correlation between BMI-1 and ERβ in a cohort of 87 prostate tumors determined from analysis of the cBioportal database (Figure S1B) (Taylor et al., 2010). (G) Schematic summarizing the major conclusions of the study. See also Figure S2 and Table S2.
Subsequently, we analyzed the prostates of BERKO mice to assess the impact of ERβ loss on HIF-1α, VEGF and BMI-1 expression. Ventral prostates of wild-type mice (control) exhibit normal glandular structure with ERβ expression in epithelial cells and undetectable HIF-1α, BMI-1 expression and very low VEGF-A (Figure 4E). In contrast, BERKO mice of the same age and genetic background exhibited hyperplasia and decreased epithelial differentiation with high HIF-1α, VEGF-A and BMI-1 expression in the absence of ERβ (Figure 4E). Interestingly, both control and BERKO prostates exhibited a similar intensity of PTEN staining (Figure 4E). Five BERKO mice and their control counterparts were examined with similar findings. These observations were substantiated by the observation that a negative correlation between ERβ and BMI-1 expression exists in a cohort of 87 human prostate tumors based on analysis of the cBioportal database (Figures 4F; Table S2).
DISCUSSION
This study provides insight into the role of ERβ in prostate tumorigenesis and the mechanisms that regulate its expression. First and foremost, we demonstrate that prostate tumorigenesis caused by PTEN deletion involves BMI-1-mediated repression of ERβ and that repression of ERβ enables HIF-1/VEGF signaling that sustains BMI-1 expression. These findings should help to clarify the issue of why prostate cancer has not been observed in BERKO mice. Specifically, we argue that loss of ERβ is not sufficient to promote tumorigenesis in the absence of an oncogenic stimulus despite the fact that BMI-1 expression is increased. This hypothesis is consistent with the report that BMI-1 inhibition slows the growth of PTEN-deletion-induced prostate cancer but it does not prevent tumorigenesis (Lukacs et al., 2010). In fact, we found that BERKO prostates retain PTEN expression. A reasonable hypothesis going forward is that PTEN loss involves additional events such as enhancement of PI3K/Akt signaling that are essential for tumorigenesis (Worby and Dixon, 2014).
The fact that ERβ expression is lost during tumorigenesis caused by PTEN deletion is significant and relevant to other studies that have investigated the consequences of ERβ loss in the prostate. Specifically, it was reported recently that deletion of ERβ in the FGF8b transgenic model of prostate tumorigenesis did not increase tumor incidence (Elo et al., 2014). Although the authors discounted a tumor suppressive role for ERβ based on these data, this conclusion should be tempered by the likely possibility that FGF8b-mediated tumorigenesis involves repression of ERβ, similar to our finding with prostate tumorigenesis caused by PTEN deletion. For this reason, deleting ERβ in either the FGF8b or PTEN models would not be expected to increase tumor incidence.
This study also addresses the mechanism by which ERβ is regulated in prostate cancer. Several studies have observed an inverse correlation between ERβ expression and differentiation (Gleason grade) but the mechanisms that contribute to the loss of ERβ in high-grade cancers are not well understood. Some reports indicated that hypermethylation of the ERβ promoter is associated with loss of expression (Lau et al., 2000; Zhu et al., 2004). Although our data do not discount the contribution of promoter methylation, compelling evidence now exists that BMI-1 expression correlates with Gleason grade and that BMI-1 is induced as a direct consequence of PTEN loss or inactivation (Goel et al., 2012). Moreover, we detected an inverse correlation between ERβ and BMI-1 in a cohort of human prostate tumors. These observations, coupled with our demonstration that BMI-1 can bind to the ERβ 0N promoter and repress transcription, strongly implicate BMI-1 in the repression of ERβ in prostate cancer. Paradoxically, ERβ is expressed in prostate cancer metastases (Fixemer et al., 2003; Lai et al., 2004). It is tempting to speculate that this ERβ expression is regulated by the 0K promoter, which is not repressed by BMI-1. From a different perspective, these findings add a new dimension to our understanding of how BMI-1 contributes to prostate tumorigenesis. Although BMI-1 has also been reported to suppress PTEN expression in nasopharyngeal epithelial cells (Song et al., 2009), we did not observe this phenomenon in the prostate epithelial and carcinoma cells that we analyzed.
Our finding that ERβ functions to suppress BMI-1 is significant because it forms the basis of our hypothesis that a positive feedback loop exists that maintains BMI-1 expression. Although ERβ has been implicated as a ‘gatekeeper’ that impedes prostate tumorigenesis (Dey et al., 2013; Hussain et al., 2012; Slusarz et al., 2012), the mechanisms involved are not known. Clearly, its ability to repress BMI-1 is one such mechanism. Moreover, these findings add to our understanding of how BMI-1 is regulated in prostate cancer. Previous work by our group demonstrated that autocrine VEGF signaling in tumor cells sustains BMI-1 expression (Goel et al., 2012), but it was not apparent that this pathway is subject to inhibition by ERβ. As mentioned, autocrine VEGF signaling in tumor cells is emerging as an important mechanism that sustains the function of cancer stem cells and promotes tumor initiation as evidenced by data obtained from several different cancers including prostate (Goel and Mercurio, 2013). Moreover, the ability of VEGF signaling to sustain BMI-1 expression accounts for how this pathway contributes to de-differentiation and tumorigenesis. The ability of ERβ to promote HIF-1α degradation and, consequently, repress VEGF expression and signaling provides a mechanism for suppressing the tumorigenic potential of BMI-1. Indeed, the induction of HIF-1α, VEGF and BMI-1 expression in BERKO mice, in concert with the inverse correlation observed between ERβ and these molecules in human prostate tumors, provide support for this hypothesis. These findings also reinforce the hypothesis that loss of ERβ in prostate cancer mimics hypoxia by enabling HIF-1α/VEGF signaling. Interestingly, a tumor suppressive function for ERβ in breast cancer was reported recently (Yuan et al., 2014).
In summary, the data we report advance our understanding of how ERβ functions in prostate cancer as both a gatekeeper of epithelial differentiation and tumorigenesis and a target of oncogenic stimuli as depicted in Fig. 4G.
MATERIALS AND METHODS
Cells and Reagents
PNT1a cells were obtained from M. Littmann (Baylor College of Medicine, Houston). The human prostate cancer cell line, LNCaP was obtained from American Type Culture Collection (ATCC). PC3-M cells were obtained from R. C. Bergan (Northwestern University, Chicago). 3β-androstane-diol (3β-adiol) and 17β-estradiol (E2) experiments were performed by incubating cells with 3β-adiol (5 µM; Sigma) or E2 (10 nM; Sigma) for 2–3 days. Wortmannin and Akt inhibitor VIII were obtained from Calbiochem. Cells were incubated with these inhibitors (5 µM) for 18 -20 hours prior to subsequent analyses. The generation of ERβ and PHD2-ablated PNT1a cells using shRNAs has been described previously (Mak et al., 2013). Lentiviruses (pLKO.1) containing BMI-1 shRNA oligonucleotides (TRCN0000020154, TRCN0000020156, TRCN0000012565), VEGF-A shRNA oligonucleotides (TRCN0000003343), HIF-1α shRNA (TRCN0000054449), PTEN shRNA (TRCN0000028989, TRCN0000028990) or pLKO-shGFP control were purchased from Open Biosystems and used to infect cells following standard protocols. Stable cell transfectants were generated by puromycin or hygromycin selection (0.5 µg/mL for PNT1a and 2 µg/mL for PC3-M cells). The resultant ERβ, PHD2, BMI-1, HIF-1α or VEGF-A-ablated cells were used for subsequent experiments. A lentiviral plasmid (FUGW) expressing BMI-1 and the control vector (FUGW) carrying the empty vector (EV) were obtained from Addgene.
Biochemical analyses
For immunoblotting, the following Abs were used ERβ and PTEN (GeneTex), BMI-1 (Cell Signaling), vimentin (Dako), HIF-1α (Novus), PHD2 (Abcam), α-tubulin and β-actin (Sigma). Immune complexes were detected using enhanced chemiluminescence (ECL; Pierce). For quantitative real-time RT-PCR (qPCR), total RNA was extracted from cells using the TRI reagent (Sigma) and was reverse transcribed using reverse transcription reagents (Applied Biosystems) and analyzed by SYBR Green Master (Rox) (Roche) using a real-time PCR system (ABI; Prism 7900HT Sequence Detection system). The expression of target genes was normalized to 18s RNA and analyzed by the comparative cycle threshold method (ΔΔCt).
ChIP was performed using the ChIP-IT Express kit (53008; Active Motif) with minor modifications (46). Briefly, the attached cells were cross-linked using 1% formaldehyde for 15 min at RT with rotation. Subsequent steps for ChIP analysis were performed according to the manufacturer’s protocol. For chromatin precipitation, 3 µg of BMI-1 antibody (Cell Signaling) or human isotype IgG (eBioscience; 16-4301-81) was used. End-point real-time PCR was performed using the primer pairs listed in Figure S1E. For luciferase assays, PC3-M cells were transfected with the desired plasmids and the Renilla luciferase construct to normalize for transfection efficiency. Luciferase assays were performed using Dual Glo luciferase assay system (Promega). Relative luciferase activity was calculated as the ratio of firefly luciferase to Renilla luciferase activity.
Xenograft experiments
Cells were mixed with Matrigel (30%) and injected subcutaneously into nu/nu mice (Jackson Laboratories; 6-week-old) using a single dose: PNT1a (106) and PC3-M (105). Animals were monitored three times per week for tumor formation by palpation. All animal experiments were in accordance with institutional guidelines and were approved by the Institutional Animal Care and Use Committee at the University of Massachusetts Medical School.
Transgenic mice
ERβ knockout (BERKO) mice were generated by the Korach laboratory (Krege et al., 1998) and were purchased from The Jackson Laboratory. The knockout allele was maintained on a C57BL/6 background. The mice were used in these studies were 10 months old. Sections from these prostates and age-matched controls were processed for immunostaining as described below. A similar approach was used for specimens obtained from prostate tissue obtained from Ptenloxp/loxp; PB-Cre+ mice (prostate cancer) and age-matched Pten+/+; PB-Cre+ mice (normal prostate) (Hubner et al., 2012).
Immunostaining
Murine prostate specimens from transgenic mice (see above) and human prostate cancer specimens, which were obtained from the Tissue Bank at the University of Massachusetts Medical School, were fixed in paraformaldehyde (4%), embedded in paraffin, sectioned (5 µM) and used for hematoxylin and eosin (H&E) and immunofluorescence staining. Immunoflurorescence staining was conducted according to the manufacturer’s instructions (Invitrogen, Life Sciences). After antigen unmasking, the specimens were incubated in 10% serum in PBS for 30 minutes, washed for 3 min in PBST, and incubated with rabbit polyclonal ERβ antibody (Gene Tex, Cat GTX 112927) or rabbit BMI-1 mAb (Cell Signaling, Cat 5856S) overnight at 4°C. The slides were washed 5 min with PBST and incubated 45 minutes in a dark chamber with the fluorochrome-conjugated secondary antibody (goat anti-rabbit conjugated Alexa Fluor 488, Life Sciences A-11008). Slides were washed and counterstained in the dark with DAPI (Invitrogen) for 10 minutes, washed with three changes of PBST and mounted under coverslips with aqueous mounting medium (Thermo Electron corp. Pittsburgh, PA). Results were analyzed with an LSM 710 Meta confocal microscope (Carl Zeiss MicroImaging Gmbh, Munich, Germany).
Statistical analysis
Data are presented as the mean from three separate experiments ± SD. The Student t test was used to determine the significance of independent experiments. The criterion P < 0.05 was used to determine statistical significance.
Supplementary Material
Highlights.
Prostate tumorigenesis caused by PTEN deletion involves loss of estrogen receptor β
ERβ transcription is repressed by BMI-1, which is induced by PTEN deletion
ERβ repression is needed for tumorigenesis because it enables HIF/VEGF signaling
HIF/VEGF signaling sustains BMI-1 expression resulting in a positive feedback loop
SIGNIFICANCE.
Understanding how nuclear hormone receptors contribute to the etiology of prostate cancer is significant for deciphering the biology of this disease and improving therapy. In this study, we demonstrate that repression of estrogen receptor beta (ERβ) is an integral component of prostate cancer caused by loss of the tumor suppressor PTEN, which is one of the most common genetic lesions in this cancer. Repression of ERβ is important for prostate tumorigenesis because it enables a signaling pathway that sustains expression of BMI-1, a transcriptional repressor that is a critical oncogenic factor in prostate and other cancers.
Acknowledgements
NIH Grant CA159865 supported this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Antal MC, Krust A, Chambon P, Mark M. Sterility and absence of histopathological defects in nonreproductive organs of a mouse ERbeta-null mutant. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2433–2438. doi: 10.1073/pnas.0712029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthon P, Cussenot O, Hopwood L, Leduc A, Maitland N. Functional expression of sv40 in normal human prostatic epithelial and fibroblastic cells - differentiation pattern of nontumorigenic cell-lines. Int J Oncol. 1995;6:333–343. doi: 10.3892/ijo.6.2.333. [DOI] [PubMed] [Google Scholar]
- Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer research. 1997;57:4997–5000. [PubMed] [Google Scholar]
- Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Molecular cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Christoforou P, Christopoulos PF, Koutsilieris M. The role of Estrogen Receptor Beta in Prostate Cancer. Mol Med. 2014 doi: 10.2119/molmed.2014.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey P, Barros RP, Warner M, Strom A, Gustafsson JA. Insight into the mechanisms of action of estrogen receptor beta in the breast, prostate, colon, and CNS. Journal of molecular endocrinology. 2013;51:T61–74. doi: 10.1530/JME-13-0150. [DOI] [PubMed] [Google Scholar]
- Elo T, Yu L, Valve E, Makela S, Harkonen P. Deficiency of ERbeta and prostate tumorigenesis in FGF8b transgenic mice. Endocrine-related cancer. 2014;21:677–690. doi: 10.1530/ERC-13-0480. [DOI] [PubMed] [Google Scholar]
- Fixemer T, Remberger K, Bonkhoff H. Differential expression of the estrogen receptor beta (ERbeta) in human prostate tissue, premalignant changes, and in primary, metastatic, and recurrent prostatic adenocarcinoma. The Prostate. 2003;54:79–87. doi: 10.1002/pros.10171. [DOI] [PubMed] [Google Scholar]
- Goel HL, Chang C, Pursell B, Leav I, Lyle S, Xi HS, Hsieh CC, Adisetiyo H, Roy-Burman P, Coleman IM, et al. VEGF/Neuropilin-2 Regulation of Bmi-1 and Consequent Repression of IGF-IR Define a Novel Mechanism of Aggressive Prostate Cancer. Cancer Discov. 2012;2:906–921. doi: 10.1158/2159-8290.CD-12-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel HL, Mercurio AM. VEGF targets the tumour cell. Nature reviews Cancer. 2013;13:871–882. doi: 10.1038/nrc3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata S, Shoda T, Kato J, Hoshi K. The multiple untranslated first exons system of the human estrogen receptor beta (ER beta) gene. The Journal of steroid biochemistry and molecular biology. 2001;78:33–40. doi: 10.1016/s0960-0760(01)00071-1. [DOI] [PubMed] [Google Scholar]
- Hubner A, Mulholland DJ, Standen CL, Karasarides M, Cavanagh-Kyros J, Barrett T, Chi H, Greiner DL, Tournier C, Sawyers CL, et al. JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:12046–12051. doi: 10.1073/pnas.1209660109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S, Lawrence MG, Taylor RA, Lo CY, BioResource APC, Frydenberg M, Ellem SJ, Furic L, Risbridger GP. Estrogen receptor beta activation impairs prostatic regeneration by inducing apoptosis in murine and human stem/progenitor enriched cell populations. PloS one. 2012;7:e40732. doi: 10.1371/journal.pone.0040732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamov O, Morani A, Shim GJ, Omoto Y, Thulin-Andersson C, Warner M, Gustafsson JA. Estrogen receptor beta regulates epithelial cellular differentiation in the mouse ventral prostate. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9375–9380. doi: 10.1073/pnas.0403041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- Kozlowski JM, Fidler IJ, Campbell D, Xu ZL, Kaighn ME, Hart IR. Metastatic behavior of human tumor cell lines grown in the nude mouse. Cancer research. 1984;44:3522–3529. [PubMed] [Google Scholar]
- Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15677–15682. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JS, Brown LG, True LD, Hawley SJ, Etzioni RB, Higano CS, Ho SM, Vessella RL, Corey E. Metastases of prostate cancer express estrogen receptor-beta. Urology. 2004;64:814–820. doi: 10.1016/j.urology.2004.05.036. [DOI] [PubMed] [Google Scholar]
- Lau KM, LaSpina M, Long J, Ho SM. Expression of estrogen receptor (ER)-alpha and ER-beta in normal and malignant prostatic epithelial cells: regulation by methylation and involvement in growth regulation. Cancer research. 2000;60:3175–3182. [PubMed] [Google Scholar]
- Leav I, Lau KM, Adams JY, McNeal JE, Taplin ME, Wang J, Singh H, Ho SM. Comparative studies of the estrogen receptors beta and alpha and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. The American journal of pathology. 2001;159:79–92. doi: 10.1016/s0002-9440(10)61676-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs RU, Memarzadeh S, Wu H, Witte ON. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell stem cell. 2010;7:682–693. doi: 10.1016/j.stem.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak P, Chang C, Pursell B, Mercurio AM. Estrogen receptor beta sustains epithelial differentiation by regulating prolyl hydroxylase 2 transcription. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4708–4713. doi: 10.1073/pnas.1221654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, Gouvin LM, Sharma VM, Mercurio AM. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer cell. 2010;17:319–332. doi: 10.1016/j.ccr.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer research. 1999;59:4291–4296. [PubMed] [Google Scholar]
- Miyazaki M, Miyazaki K, Itoi M, Katoh Y, Guo Y, Kanno R, Katoh-Fukui Y, Honda H, Amagai T, van Lohuizen M, et al. Thymocyte proliferation induced by pre-T cell receptor signaling is maintained through polycomb gene product Bmi-1-mediated Cdkn2a repression. Immunity. 2008;28:231–245. doi: 10.1016/j.immuni.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, Gleave M, Wu H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer research. 2012;72:1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacerddine K, Beaudry JB, Ginjala V, Westerman B, Mattiroli F, Song JY, van der Poel H, Ponz OB, Pritchard C, Cornelissen-Steijger P, et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. The Journal of clinical investigation. 2012;122:1920–1932. doi: 10.1172/JCI57477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarz A, Jackson GA, Day JK, Shenouda NS, Bogener JL, Browning JD, Fritsche KL, MacDonald RS, Besch-Williford CL, Lubahn DB. Aggressive prostate cancer is prevented in ERalphaKO mice and stimulated in ERbetaKO TRAMP mice. Endocrinology. 2012;153:4160–4170. doi: 10.1210/en.2012-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song LB, Li J, Liao WT, Feng Y, Yu CP, Hu LJ, Kong QL, Xu LH, Zhang X, Liu WL, et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. The Journal of clinical investigation. 2009;119:3626–3636. doi: 10.1172/JCI39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nature reviews Cancer. 2011;11:597–608. doi: 10.1038/nrc3093. [DOI] [PubMed] [Google Scholar]
- van Leenders GJ, Dukers D, Hessels D, van den Kieboom SW, Hulsbergen CA, Witjes JA, Otte AP, Meijer CJ, Raaphorst FM. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. European urology. 2007;52:455–463. doi: 10.1016/j.eururo.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- Worby CA, Dixon JE. Pten. Annual review of biochemistry. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- Yuan B, Cheng L, Chiang HC, Xu X, Han Y, Su H, Wang L, Zhang B, Lin J, Li X, et al. A phosphotyrosine switch determines the antitumor activity of ERbeta. The Journal of clinical investigation. 2014;124:3378–3390. doi: 10.1172/JCI74085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Leav I, Leung YK, Wu M, Liu Q, Gao Y, McNeal JE, Ho SM. Dynamic regulation of estrogen receptor-beta expression by DNA methylation during prostate cancer development and metastasis. The American journal of pathology. 2004;164:2003–2012. doi: 10.1016/s0002-9440(10)63760-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.