

Abstract
Thyroid carcinomas of follicular epithelial derivation are common and generally well-behaved malignancies with excellent cure and survival rates. However, a subset of these carcinomas, whether well-differentiated, poorly differentiated, or anaplastic, is highly aggressive, manifesting with local invasion, recurrence, and distant metastasis. The recognition of dedifferentiation is of paramount importance. In addition, the challenge for Pathologists is to identify the rare aggressive differentiated carcinomas so that treatment may be tailored appropriately. Thus, histological subtyping and documentation of other aggressive features such as widespread invasion and angioinvasion are critical. Mutational analyses in the past decade have delineated the molecular alterations responsible for thyroid carcinogenesis and progression, allowing greater sub-classification and prognostication of thyroid carcinomas. This review article highlights important aggressive morphologic features and molecular mutations associated with thyroid carcinoma.
Keywords: Thyroid carcinoma, Angioinvasion, Mutations, Rearrangements, Epigenetics
Introduction
Most carcinomas derived from the follicular epithelium of the thyroid gland are well differentiated (>90 %) and well-behaved, with a 10-year disease-specific survival of 85–92 % [1, 2]. These indolent cancers are localized to the thyroid gland, do not recur or metastasize beyond local lymph nodes, and are thus easily curable with hemi- or total thyroidectomy, with possible local nodal dissection and/or radioactive iodine ablation. However, a small subset of thyroid carcinomas “go bad” and behave aggressively. Well-differentiated carcinomas may dedifferentiate via a multi-step process of genetic and epigenetic alterations to poorly differentiated and/or undifferentiated/anaplastic thyroid carcinoma (ATC). More intriguingly, well-differentiated carcinomas may behave aggressively without any identifiable dedifferentiated components.
Distant metastasis develops in only 1–15 % of thyroid carcinomas, but when it occurs, it decreases the 10-year survival to 10–40 % [2, 3]. The latest data from Cancer Care Ontario has shown that distant metastasis occurs in <1 % of thyroid cancer patients [3]. The factors predicting a worse outcome in differentiated thyroid carcinomas include clinical parameters, and like other cancers, they vary with tumor stage as defined by the AJCC/UICC [4]. In addition, thyroid tumors in men have tended to be more aggressive and patient age has been identified as an important modifier of risk, hence the development of clinical scoring systems such as AGES (Age, Grade, Extent of disease, Size), AMES (Age, Metastasis, Extent of disease, Size), and MACIS (Metastasis, Age at presentation, Completeness of surgical resection, Invasion, Size) which is one of the more reliable staging methods available [4]. While classically a threshold age of 45 years has been used to upstage patients with thyroid carcinoma, the latest study from The Cancer Genome Atlas (TCGA) Research Network, using whole exome DNA sequencing of papillary thyroid carcinomas (PTC), has shown that mutation density correlates with age [5]. Thus, it is proposed that age should be used as a continuous variable in risk stratification, as opposed to being a static threshold.
The pathways taken to aggressive behavior are complex and manifest a wide variety of morphologic and molecular features. For instance, a well-differentiated thyroid carcinoma without capsular or vascular invasion, whether PTC or follicular thyroid carcinoma (FTC), is an indolent entity, with a 5 year survival of over 95 % [6]. The presence of local invasion and/or lymph node metastasis portends a higher risk of local recurrence, but perhaps because of the availability of highly effective targeted radioactive iodine therapy, long-term outcomes remain favorable [1, 2]. In contrast, the widely invasive FTC with angioinvasion is untreatable and the 5 year survival is dismal at 38 % [6]. The vast amount of molecular data that has emerged in the last decade has allowed greater dissection and identification of those thyroid carcinomas that are likely to behave more aggressively, in terms of local invasion, lymph node metastases, and distant metastases via hematogenous spread. This review was undertaken in light of this emerging molecular data, in order to aid in the early identification of potentially aggressive subtypes of thyroid carcinoma. In addition, the importance of histological subtyping is emphasized here, as certain subtypes of thyroid carcinoma predict more aggressive behavior when compared with molecular mutational characterization alone [7].
Thyroid Carcinogenesis, Tumor Progression, and the Road to Bad
Morphologic Features Predicting Aggressive Behavior
Papillary Thyroid Carcinoma Variants
Several aggressive variants of PTC have been described, namely tall cell (Fig. 1a), columnar cell (Fig. 1b), hobnail cell (Fig. 1c) and diffuse sclerosing variants. As the name suggests, the tall cell variant is comprised of cells that are at least three times as tall as they are wide. This variant has prominent papillae and usually presents in older patients as large tumors with extrathyroidal extension. The columnar cell variant is rare, resembles secretory endometrium, and is on a continuum with the tall cell variant. New TCGA data have shown that the tall cell variant has the highest mutation density of all PTC variants, correlating with its aggressive behavior [5]. In addition, increased expression of the oncogenic microRNA, miR-21, was found in the tall cell variant [5]. The rare hobnail cell variant consists of cuboidal or oval epithelial cells with apical surface bulging, leading to a hobnail appearance [8]; the molecular changes underlying this recently described variant are unknown. The diffuse sclerosing variant is an exceptionally rare tumor that presents as a diffuse goiter because there is no discrete tumor mass; instead the malignancy infiltrates widely throughout the gland. This variant is most common in children and has a high rate of lung metastasis.
Fig. 1.

Histology of aggressive thyroid carcinomas. a–c Aggressive variants of papillary thyroid carcinoma: tall cell variant (a), columnar cell variant (b), and hobnail cell variant (c). d Angioinvasion (tumor cells with thrombus in vascular channels marked with arrows). e Poorly differentiated thyroid carcinoma arising in a well differentiated follicular variant papillary thyroid carcinoma (bottom left). f Anaplastic thyroid carcinoma
Angioinvasion
Angioinvasion has been a controversial factor in predicting aggressive behavior and distant metastasis, mainly due to loose criteria in judging vascular invasion [3]. Past studies have included any tumor found in or around an endothelial-lined space as evidence of vascular invasion, and these studies have not correlated well with either distant metastases or poor prognosis [3]. Importantly, lymphatic invasion needs to be separated from angioinvasion, since the former leads to lymph node metastasis and a risk of local recurrence as opposed to distant metastasis; while these can often be distinguished based on the presence of red blood cells within the lumen, immunohistochemistry may be required. Rigid criteria that define true angioinvasion, specified as tumor cells in a vascular space with attached thrombus (Fig. 1d) or tumor invasion through a vessel wall with associated thrombus, have been shown to predict distant metastasis and poor outcome in differentiated thyroid cancers in addition to increased rates of angioinvasion with tumor dedifferentiation [3].
The distinction of FTC from follicular adenoma has traditionally been based on capsular or vascular invasion, or on the presence of metastasis. FTCs have a completely follicular architecture and lack the nuclear features of PTC. Follicular variant (FV)-PTC, in contrast, has been defined based on the characteristic nuclear atypia of PTC. These tumors can be divided into two groups, namely minimally invasive (often termed encapsulated) and widely invasive, based on grossly obvious invasion into surrounding parenchyma [9, 10]. Widely invasive tumors have a dismal prognosis [6, 10, 11]. Some authors have separated angioinvasive tumors into a third category but others have persisted in classifying such tumors as minimally invasive because of their encapsulated nature and the need to use a microscope to identify vascular involvement, but these tumors manifest with recurrence and/or distant metastases [3, 9, 10]. These tumors, termed by some as “grossly encapsulated follicular carcinoma with angioinvasion” confer a worse prognosis for the patient [9].
Dedifferentiation
While the solid variant of PTC has prognostic features on par with the classical variant, it is important to recognize this entity and to not confuse it with poorly differentiated thyroid carcinoma (PDTC), which has a much worse prognosis. The solid variant is composed of sheets of large cells with discernible PTC nuclear features, namely nuclear irregularity, crowding, clearing, grooving and pseudo-inclusions; most importantly, it lacks evidence of tumor cell necrosis and has a relatively low mitotic and proliferative index. As per the Turin criteria, PDTCs are defined as having insular, solid or trabecular growth without the florid nuclear features of PTC and exhibiting either tumor cell necrosis (Fig. 1e) or a mitotic count of ≥3/10 high power fields [12]. PDTCs are follicular-cell neoplasms that are morphologically and behaviorally intermediate between the well-differentiated FTC and PTC and the undifferentiated ATC. Interestingly, there are striking regional differences in PDTC incidence rates; Northern Italy has the highest rates (15 %) with much lower rates in the United States (1.8 %) and Japan (<1 %), suggesting a multifactorial causative interaction between genetics, environment, and diet [13]. A recent study has shown that insulin-like growth factor-I mRNA binding protein-3 (IMP3) expression in PDTC appears to be an adverse prognostic factor, in that in univariate analysis, the risk of death was higher for age ≥45, tumors ≥4 cm, and IMP3 positivity by immunohistochemistry [14]. IMP3 expression has been shown to have diagnostic and/or prognostic value in many malignant solid tumors, including thyroid [14].
ATCs are among the most lethal of all human malignancies and account for less than 3 % of thyroid cancers [13]. ATC is more common in females and in the elderly. Morphologically, ATCs are wildly pleomorphic with abnormal mitoses and giant cells (Fig. 1f) and are widely invasive locally and have high rates of distant metastases. Whether arising de novo or dedifferentiating from well-differentiated thyroid carcinomas, PDTCs and ATCs acquire β-catenin and p53 mutations [13, 15].
Molecular Analysis of Thyroid Carcinogenesis and Tumor Progression
A number of molecular events have been described in both thyroid carcinogenesis and tumor progression. The primary events involve the mitogen activated kinase (MAPK) signaling pathway and the phosphoinositide-3 kinase PI3 K-Akt-mTOR pathway (Fig. 2). Components of the MAPK pathway involve the single-pass transmembrane tyrosine kinase receptor RET (rearranged during transfection), and its downstream signaling molecules RAS and BRAF. Mutations in RET, RAS, and BRAF are usually mutually exclusive, suggesting redundant downstream effects. Neurotrophic kinase receptor 1 (NTRK1) also signals through the MAPK/RAS/BRAF pathway. Important novel data from TCGA have split PTCs into BRAF-like and RAS-like categories, based on exome and RNA sequencing results, RNA and proteomic profiling, and methylation patterns (Fig. 3) [5]. Other factors essential for tumor progression include growth factors and their receptors, cell-cycle regulators, and adhesion molecules (Fig. 2).
Fig. 2.
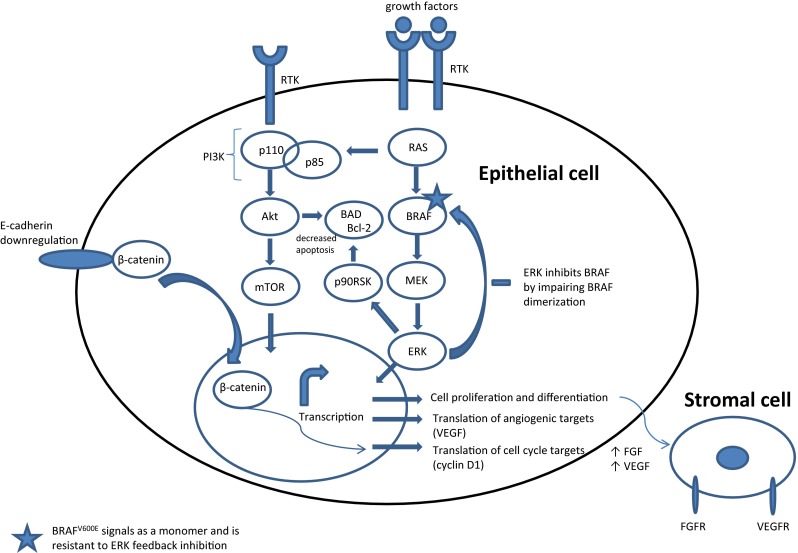
Signaling pathways involved in thyroid carcinogenesis and tumor progression. Growth factors induce receptor tyrosine kinase dimerization, resulting in tyrosine phosphorylation within the cytoplasmic tail, leading to RAS activation. Through the subsequent activation of BRAF and MEK, ERK is activated and translocates to the nucleus, where it activates various transcription factors. Similarly, the mTOR pathway is activated. In aggressive thyroid carcinomas, E-cadherin is down-regulated and β-catenin is stabilized and translocates to the nucleus; this and other epigenetic alterations can upregulate cyclin D1. Adhesion can be altered by multiple mechanisms, including adhesion molecules and growth factors, to impact tumor-stromal interactions. BRAF-like tumors show robust activation of MAPK signaling pathway, while RAS-like tumors activate both MAPK (to a lesser extent than BRAF-like tumors) and Akt-mTOR pathway, using p90RSK as a crucial cross-link to bcl-2 signaling [5]. Abbreviations: Akt protein kinase B, BAD bcl-2 associated death promoter, bcl-2 B cell lymphoma 2, ERK extracellular-signal-related kinase, FGF fibroblast growth factor, FGFR FGF receptor, MEK mitogen activated protein kinase kinase, mTOR mechanistic target of rapamycin, RTK receptor tyrosine kinase, VEGF vascular endothelial growth factor, VEGFR VEGF receptor
Fig. 3.

Separation of well-differentiated papillary thyroid carcinomas by molecular signatures, based on the BRAF-RAS score (BRS), −1 to +1, generated by The Cancer Genome Atlas Research Network data [5]. The BRS is based on a 71 gene signature. TDS: thyroid differentiation score based on 16 thyroid metabolism and function genes, including TFF3, KIT, PVRL4, FHL1, miR-21 (oncogenic miR in several tumor types); TDS correlated with grade, risk of recurrence, and MACIS [5]
BRAFV600E Mutation
The BRAFV600E mutation is the most commonly detected genetic alteration in thyroid cancer; its incidence varies from 29 to 70 % of PTCs, depending on whether follicular variant lesions were included in the studies (reviewed in ref. [16]). This mutation leads to constitutive activation of BRAF and upregulation of the downstream MAPK pathway [16].Tumors driven by BRAFV600E exhibit high MAPK-signaling; they do not respond to the negative feedback from ERK, as ERK inhibits dimerized BRAF, while BRAFV600E signals as a monomer [17]. The BRAFV600E mutation is also associated with impaired function of the sodium-iodide symporter, leading to impaired uptake of iodine by thyroid follicular cells and resistance to radio-iodide therapy [2]. A chromosomal rearrangement resulting in the fusion protein AKAP9-BRAF that also activates BRAF signaling has been demonstrated in radiation-induced PTCs [16].
BRAFV600E mutations in PTC correlate with lymph node metastases, extrathyroidal extension, and distant metastases [7, 18]. This was interpreted to imply that PTCs with BRAF mutations behave more aggressively. However, this only holds true for studies in which all PTC variants were analyzed together, irrespective of histotype. In contrast, studies in which the classical variant (the variant that harbors BRAFV600E mutations) was analyzed separately, BRAF mutations were not predictive of aggressive features [7]. Importantly, when all PTCs were examined as a group, morphologic classification was found to be a better predictor of aggressive behavior than BRAF mutation status, with classical variant PTCs being at higher risk for lymph node metastases, extra-thyroidal extension, and vascular invasion than follicular variant lesions [7]. Novel molecular data from the TCGA indicate that PTCs with BRAFV600E mutations are a diverse group of tumors, consisting of at least four molecular subtypes and varying degrees of thyroid differentiation and behavior (Fig. 3) [5].
BRAF mutations are also found in 13 % of PDTCs and up to 35 % of ATCs [18, 19]. This is consistent with morphologic studies that have shown these tumors to arise by progressive dedifferentiation from more differentiated tumors, including classical variant PTCs [16].
RET Mutations and Rearrangements
Well-documented mutations of RET result in constitutive activation of the protein, leading to medullary thyroid carcinoma, including sporadic forms and those associated with familial medullary thyroid carcinoma and multiple endocrine neoplasia 2 (MEN2) syndromes [16]. RET is normally expressed in neural crest cell derivatives and the genitourinary tract, where it is involved in renal organogenesis and spermatogenesis [16]. It is not a normal component of follicular epithelial cells, however RET/PTC, which is a rearranged form of RET, was the first genetic alteration identified in the neoplastic follicular cells of PTC.
More than 15 gene fusions in this family have been described and expression of the various fusion proteins relies on promoters of genes expressed by differentiated thyroid follicular cells [16]. RET/PTC rearrangements are early events in thyroid carcinogenesis and are highly specific for PTC [16], and thus may be useful in the diagnosis of borderline cases. However, because RET/PTC rearrangements rely on promoters found in differentiated thyroid cells, expression of these fusion proteins is often lost in PDTCs and ATCs. The majority (50–90 %) of PTCs following the Chernobyl disaster were found to have RET/PTC rearrangements [16, 20]. Specifically, RET/PTC3 was associated with the solid variant and aggressive behavior [20, 21].
NTRK1 Rearrangements
NTRK1 is a receptor tyrosine kinase, which binds neural growth factor, and regulates nervous system development. PTCs have been shown to harbor rearrangements of this proto-oncogene, resulting in ectopic expression and constitutive activation (Fig. 2) [16]. To date, no definite correlation has been established between NTRK1 rearrangements and PTC behavior (Fig. 3) [5].
RAS Mutations
RAS proteins are GTP-binding proteins that regulate cell growth via several pathways, including MAPK and PI3 K. In thyroid cancer, RAS mutations lead to constitutive protein activation. RAS mutations have been identified in 45 % of FTCs and 35 % of FV-PTCs [16, 22]. While initially thought to promote tumorigenesis due to their presence in “follicular adenomas”, difficulties in distinguishing follicular adenomas from FV-PTC or FTC may explain these findings [23]. As with BRAF, tumors with RAS mutations have a distinct signature obtained from TCGA data. A BRAFK601E mutation, also found in FV-PTC, has been grouped into the RAS-like category in the new TCGA study [5]. The common molecular alterations and expression profiles of FTC and FV-PTC have raised the question of whether these are truly different entities [24].
Some have considered RAS mutations to be important for tumor progression, rather than carcinogenesis, and this is supported by their increased incidence in poorly differentiated and anaplastic carcinomas compared with well-differentiated tumors [16, 25]. The prevalence of RAS mutations ranges from 18 to 55 % in PDTC and from 4 to 60 % in ATC [12–14]. RAS mutations stimulate chromosomal instability and, thus, may predispose to tumor dedifferentiation [13, 14]. However, since the majority of FTCs and FV-PTCs harbor RAS mutations and are not aggressive cancers, it is more likely that these are early events that are not of prognostic significance. Dedifferentiation is likely not driven by BRAF or RAS mutations individually, but rather by the combined effort of multiple genetic alterations [5, 15].
PPARγ Rearrangements
A balanced translocation that fuses PAX8, a thyroid differentiation transcription factor, to peroxisome-proliferator activated receptor gamma (PPARγ), results in the PAX8-PPARγ fusion oncogene. The PAX8 promoter is responsible for driving expression of this fusion protein [26, 27]. The nuclear receptor PPARγ regulates metabolism, cell growth and differentiation, but is thought to be inactivated by rearrangement.
Well-differentiated thyroid tumors, mainly FTCs, FV-PTCs, and tumors classified as “follicular adenomas” when nuclear features were not examined in pure follicular-patterned tumors, express this fusion protein. These rearrangements have been implicated in angioinvasive thyroid carcinomas [26]. Interestingly, immunohistochemical localization studies show that PPARγ expression is decreased in carcinomas with an invasive component [7]. Expression is lost with dedifferentiation of thyroid tumors, likely due to promoter loss in dedifferentiated cells.
TERT Promoter Dysregulation
Telomerase reverse transcriptase (TERT) is the catalytic subunit of telomerase, the enzyme responsible for maintaining telomere length at the end of chromosomes. Many cancers overexpress TERT, and TERT induction in mouse models leads to tumorigenesis. Liu et al. [28] recently reported C228T and C250T TERT promoter mutations in thyroid carcinoma; they found that TERT mutations frequently occurred together with BRAFV600E mutations, and were most prevalent in aggressive, dedifferentiated thyroid carcinomas.The recent TCGA study identified C228T, C228A, and C250T TERT promoter mutations in 9.4 % of PTCs, with C228T being the most common mutation (7.0 %) [5]. These TERT mutations were present in all histological subtypes of PTC examined, but occurred more frequently in less-differentiated PTCs and were strongly associated with older age, MACIS scores, and high risk of recurrence [5].
Growth Factors and Their Receptors
The epidermal growth factor receptor (EGFR) family transmits signals involved in control of cell growth and differentiation. There are four members, also known as ERBB or human epidermal growth factor receptor (HER) [1–4]. The overexpression of HER1 has been shown to confer a poor prognosis to thyroid carcinomas [16]. HER2 is overexpressed in ATCs [29]. Four distinct fibroblast growth factor receptor (FGFR) genes have been reported (FGFR1 through four), with at least 23 different ligands that signal through a complex family of receptor-tyrosine kinases, and ultimately impinge on ERK to affect the MAPK and Akt pathways [16, 30]. Basic fibroblast growth factor two is increased in thyroid cancer, and increases cell proliferation in rat thyroid follicular cells [31]. Normal and neoplastic follicular epithelial cells express different FGFRs, namely FGFR2 in normal thyroid and FGFR1, FGFR3, and FGFR4 in thyroid carcinoma, with FGFR4 being expressed in more aggressive cancers [32]. FGFR2 is down-regulated in thyroid tumors as a result of promoter methylation [30]. To demonstrate its role as a tumor suppressor gene, restoration of FGFR-IIIb has been shown to inhibit BRAF phosphorylation, resulting in diminished MAPK activation, even in the presence of constitutively activated BRAF signaling due to point mutation [30].
Vascular endothelial growth factor (VEGF) has been shown to be overexpressed in PTC, where it is correlated with increased tumor size, extrathyroidal extension, and BRAF mutations [33]. EGFR, FGFR, and VEGF signal in part through the PI3 K-Akt-mTOR pathway, which is important in thyroid carcinogenesis and tumor progression (Fig. 2). PI3 K may be activated by tyrosine kinase receptors such as RET/PTC and may include RAS as a second messenger. Akt mutations have been reported in radioactive iodine-refractory, metastatic thyroid carcinomas [16].
Cell Cycle Regulators
Cell-cycle progression is controlled by cyclin–cyclin dependent kinase complexes. Cyclin D1 expression is observed in PTC, not in normal thyroid tissue, and is specifically correlated with lymph node metastases [34]. The reverse is true for the tumor suppressor, p27; this protein that acts as the “brakes” of cell cycle progression is lost through epigenetic mechanisms in differentiated PTCs that develop lymph node metastases [34, 35].
Inactivating mutations in p53, the guardian of the genome, are implicated in tumor progression and are found mainly in ATCs [16].
Adhesion and Stromal Interactions
Dysadhesion is a major feature of aggressive thyroid carcinomas. Adhesion molecules are lost during the invasive process, as exemplified by disruption of membrane bound β-catenin, as well as E-cadherin, fibronectin and the Carcinoembryonic Antigen-related Cell Adhesion Molecule 1 (CEACAM1) [16]. β-catenin is involved both in cell adhesion by forming cell membrane complexes with E-cadherin and in signal transduction through the Wnt pathway which activates cell cycle progression genes. Mutations in the CTNNB1 gene, found in PDTCs and ATCs, alter β-catenin phosphorylation sites and prevent its degradation, leading to Wnt signaling activation. Such CTNNB1 mutations are rare or absent in well differentiated thyroid cancers. Additionally, ATCs have reduced E-cadherin expression, which also leads to accumulation of nuclear β-catenin and Wnt signaling activation [16]. Cells harboring RET point mutations or RET/PTC rearrangements are also able to mobilize β-catenin from the membrane and increase its cytosolic pool [16]. The functions of CAECAM1 (reviewed in ref. [16]) are controversial, in terms of being an oncogene or a tumor suppressor gene. Its putative ligand, osteopontin, has been shown to be overexpressed in PTCs and positively correlated with lymph node metastases [16]. Many other molecules and mutations have been recently identified and shown to play important roles in thyroid tumorigenesis and progression [5], but a comprehensive analysis is beyond the scope of this brief review.
Conclusions
Well-differentiated and dedifferentiated thyroid carcinomas are a heterogeneous group, with varying molecular signatures and behaviors. The road to aggressive behavior may be predicted based on both histological and molecular subtyping. Aggressive tumors may be well differentiated and in these tumors, histological subtyping and accurate morphology, specifically angioinvasion, are pertinent features to highlight as they bear prognostic implications. Finally, a multitude of molecular players are involved in tumorigenesis, progression, and aggressive behavior, including point mutations, rearrangements, and epigenetic alterations that target growth factors and their receptors, tyrosine kinases, angiogenesis mediators, cell-cycle regulators, and adhesion molecules.
References
- 1.Albores-Saavedra J, Henson DE, Glazer E, et al. Changing patterns in the incidence and survival of thyroid cancer with follicular phenotype–papillary, follicular, and anaplastic: a morphological and epidemiological study. Endocr Pathol. 2007;18:1–7. doi: 10.1007/s12022-007-0002-z. [DOI] [PubMed] [Google Scholar]
- 2.O’Neill CJ, Oucharek J, Learoyd D, et al. Standard and emerging therapies for metastatic differentiated thyroid cancer. Oncologist. 2010;15:146–156. doi: 10.1634/theoncologist.2009-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mete O, Asa SL. Pathological definition and clinical significance of vascular invasion in thyroid carcinomas of follicular epithelial derivation. Mod Pathol. 2011;24:1545–1552. doi: 10.1038/modpathol.2011.119. [DOI] [PubMed] [Google Scholar]
- 4.Brierley JD, Asa SL. Thyroid cancer. Chapter 11. In: Gospodarowicz MK, O’Sullivan B, Sobin L (Eds): Prognostic factors in cancer. Third edition, Wiley-Liss Inc. 2006;119–122.
- 5.The Cancer Genome Atlas ResearchNetwork Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676–690. doi: 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Avanzo A, Treseler P, Ituarte PHG, et al. Follicular thyroid carcinoma: histology and prognosis. Cancer. 2004;100:1123–1129. doi: 10.1002/cncr.20081. [DOI] [PubMed] [Google Scholar]
- 7.Cheng S, Serra S, Mercado M, et al. A high-throughput proteomic approach provides distinct signatures for thyroid cancer behavior. Clin Cancer Res. 2011;17:2385–2394. doi: 10.1158/1078-0432.CCR-10-2837. [DOI] [PubMed] [Google Scholar]
- 8.Asioli S, Erickson LA, Sebo TJ, et al. Papillary thyroid carcinoma with prominent hobnail features: a new aggressive variant of moderately differentiated papillary carcinoma. A clinicopathologic, immunohistochemical, and molecular study of eight cases. Am J Surg Pathol. 2010;34:44–52. doi: 10.1097/PAS.0b013e3181c46677. [DOI] [PubMed] [Google Scholar]
- 9.Ghossein R. Encapsulated malignant follicular cell-derived thyroid tumors. Endocr Pathol. 2010;21:212–218. doi: 10.1007/s12022-010-9141-8. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Z, Gandhi M, Nikiforova MN, et al. Molecular profile and clinical-pathologic features of the follicular variant of papillary thyroid carcinoma. An unusually high prevalence of RAS mutations. Am J Clin Pathol. 2003;120:71–77. doi: 10.1309/ND8D9LAJTRCTG6QD. [DOI] [PubMed] [Google Scholar]
- 11.Rivera M, Ricarte-Filho J, Knauf J, et al. Molecular genotyping of papillary thyroid carcinoma follicular variant according to its histologic subtypes (encapsulated vs non-encapsulated infiltrative) reveals distinct BRAF and RAS mutations. Mod Pathol. 2010;23:1191–1200. doi: 10.1038/modpathol.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volante M, Collini P, Nikiforov YE, et al. Poorly differentiated thyroid carcinoma: the Turin proposal for the use of uniform diagnostic criteria and an algorithmic diagnostic approach. Am J Surg Pathol. 2007;31:1256–1264. doi: 10.1097/PAS.0b013e3180309e6a. [DOI] [PubMed] [Google Scholar]
- 13.Hannallah J, Rose J, Guerrero MA. Comprehensive literature review: recent advances in diagnosing and managing patients with poorly differentiated thyroid carcinoma. Int J Endocrinol. 2013;Article ID 317487. [DOI] [PMC free article] [PubMed]
- 14.Asioli S, Erickson LA, Righi A, et al. Poorly differentiated carcinoma of the thyroid: validation of the Turin proposal and analysis of IMP3 expression. Mod Pathol. 2010;23:1269–1278. doi: 10.1038/modpathol.2010.117. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Rostan G, Sobrinho-Simões M. Poorly differentiated thyroid carcinoma: an evolving entity. Diagn Histopathol. 2011;17:114–123. doi: 10.1016/j.mpdhp.2010.12.001. [DOI] [Google Scholar]
- 16.Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev. 2006;6:292–306. doi: 10.1038/nrc1836. [DOI] [PubMed] [Google Scholar]
- 17.Pratilas CA, Taylor BS, Ye Q, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci USA. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Namba H, et al. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J Clin Endocrinol Metab. 2003;88:4393–4397. doi: 10.1210/jc.2003-030305. [DOI] [PubMed] [Google Scholar]
- 19.Nikiforova MN, et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab. 2003;88:5399–5404. doi: 10.1210/jc.2003-030838. [DOI] [PubMed] [Google Scholar]
- 20.Nikiforov YE, Rowland JM, Bove KE, et al. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997;57:1690–1694. [PubMed] [Google Scholar]
- 21.Williams D. Cancer after nuclear fallout: lessons from the Chernobyl accident. Nat Rev Cancer. 2002;2:543–549. doi: 10.1038/nrc845. [DOI] [PubMed] [Google Scholar]
- 22.Park JY, Kim WY, Hwang TS, et al. BRAF and RAS mutations in follicular variants of papillary thyroid carcinoma. Endocr Pathol. 2013;24:69–76. doi: 10.1007/s12022-013-9244-0. [DOI] [PubMed] [Google Scholar]
- 23.Elsheikh TM, Asa SL, Chan JK, et al. Interobserver and intraobserver variation among experts in the diagnosis of thyroid follicular lesions with borderline nuclear features of papillary carcinoma. Am J Clin Pathol. 2008;130:736–744. doi: 10.1309/AJCPKP2QUVN4RCCP. [DOI] [PubMed] [Google Scholar]
- 24.Asa SL, Giordano TJ, LiVolsi VA. Implications of the TCGA genomic characterization of papillary thyroid carcinoma for thyroid pathology: does follicular variant papillary thyroid carcinoma exist? Thyroid. 2014;Online, ahead of print. PMID:25409450. [DOI] [PubMed]
- 25.Volante M, Rapa I, Gandhi M, et al. RAS mutations are the predominant molecular alteration in poorly differentiated thyroid carcinomas and bear prognostic impact. J Clin Endocrinol Metab. 2009;94:4735–4741. doi: 10.1210/jc.2009-1233. [DOI] [PubMed] [Google Scholar]
- 26.Eberhardt NL, Grebe SK, McIver B, et al. The role of the PAX8/PPARgamma fusion oncogene in the pathogenesis of follicular thyroid cancer. Mol Cell Endocrinol. 2010;321:50–56. doi: 10.1016/j.mce.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kroll TG. Molecular rearrangements and morphology in thyroid cancer. Am J Pathol. 2002;160:1941–1944. doi: 10.1016/S0002-9440(10)61142-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Bishop J, Shan Y, et al. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer. 2013;20:603–610. doi: 10.1530/ERC-13-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elliott DD, Sherman SI, Bisaidy NL, et al. Growth factor receptors expression in anaplastic thyroid carcinoma: potential markers for therapeutic stratification. Hum Pathol. 2008;39:15–20. doi: 10.1016/j.humpath.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 30.Kondo T, Zheng L, Liu W, et al. Epigenetically controlled fibroblast growth factor receptor 2 signaling imposes on the RAS/BRAF/mitogen-activated protein kinase pathway to modulate thyroid cancer progression. Cancer Res. 2007;67:5461–5470. doi: 10.1158/0008-5472.CAN-06-4477. [DOI] [PubMed] [Google Scholar]
- 31.Logan A, Black EG, Gonzalez AM, et al. Basic fibroblast growth factor: an autocrine mitogen of rat thyroid follicular cells? Endocrinology. 1992;130:2363–2372. doi: 10.1210/endo.130.4.1312454. [DOI] [PubMed] [Google Scholar]
- 32.St. Bernard R, Zheng L, Liu W, et al. Fibroblast growth factor receptors as molecular targets in thyroid carcinoma. Endocrinology. 2005;146:1145–1153. doi: 10.1210/en.2004-1134. [DOI] [PubMed] [Google Scholar]
- 33.Jo YS, Li S, Song JH, et al. Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer. J Clin Endocrinol Metab. 2006;91:3667–3670. doi: 10.1210/jc.2005-2836. [DOI] [PubMed] [Google Scholar]
- 34.Khoo ML, Beasley NJ, Ezzat S, et al. Overexpression of cyclin D1 and underexpression of p27 predict lymph node metastases in papillary thyroid carcinoma. J Clin Endocrinol Metab. 2002;87:1814–1818. doi: 10.1210/jcem.87.4.8353. [DOI] [PubMed] [Google Scholar]
- 35.Khoo ML, Ezzat S, Freeman JL, et al. Cyclin D1 protein expression predicts metastatic behavior in thyroid papillary microcarcinomas but is not associated with gene amplification. J Clin Endocrinol Metab. 2002;87:1810–1813. doi: 10.1210/jcem.87.4.8352. [DOI] [PubMed] [Google Scholar]