

Abstract
This review discusses hemoglobin D-Punjab, also known as hemoglobin D-Los Angeles, one of the most common hemoglobin variants worldwide. It is derived from a point mutation in the beta-globin gene (HBB: c.364G>C; rs33946267) prevalent in the Punjab region, Northwestern Indian. Hemoglobin D-Punjab can be inherited in heterozygosis with hemoglobin A causing no clinical or hematological alterations, or in homozygosis, the rarest form of inheritance, a condition that is commonly not related to clinical symptomatology. Moreover, this variant can exist in association with other hemoglobinopathies, such as thalassemias; the most noticeable clinical alterations occur when hemoglobin D-Punjab is associated to hemoglobin S. The clinical manifestations of this association can be similar to homozygosis for hemoglobin S. Although hemoglobin D-Punjab is a common variant globally with clinical importance especially in cases of double heterozygosis, hemoglobin S/D-Punjab is still understudied. In Brazil, for example, hemoglobin D-Punjab is the third most common hemoglobin variant. Thus, this paper summarizes information about the origin, geographic distribution, characterization and occurrence of hemoglobin D-Punjab haplotypes to try to improve our knowledge of this variant. Moreover, a list of the main techniques used in its identification is provided emphasizing the importance of complementary molecular analysis for accurate diagnosis.
Keywords: Hemoglobins, Haplotypes, Diagnosis, Hemoglobinopathies
Introduction
Mutations in genes encoding hemoglobin chains are present in about 7% of worldwide population.1 These genetic alterations can affect the production rate of globin chains and cause thalassemia, or they can modify molecule structure and generate hemoglobin variants.1,2
Hemoglobin variants usually are the consequence of single amino acid substitutions caused by point mutations in genes encoding globin chains, resulting in a tetramer with different physicochemical characteristics.3 According to the Globin Gene Server database (http://globin.cse.psu.edu/), 1198 hemoglobin variants were described until September 2014. Most of the hemoglobin variants described do not cause symptomatic clinical manifestations; however, in some cases, they can be associated to relevant pathophysiology, e.g., hemoglobin S (Hb S). Hb S is the most frequent hemoglobin variant in the world; its clinical outcome is severe in homozygous or in association with other relatively common hemoglobinopathies, such as beta-thalassemia, Hb C or Hb D.3,4 Different to the other hemoglobinopathies, this last one is still poorly studied, especially in Brazil, where there are few recent studies about its prevalence, diagnosis and clinical aspects. Moreover, Hb D presents considerable geographic distribution and can be associated with Hb S, forming a heterozygous composite with peculiar clinical severity.
Thus, this study aimed to gather information available in the literature about type D hemoglobins, more specifically about Hb D-Punjab, in order to draw attention to its importance and to encourage the scientific community to take interest in the subject, particularly in Brazil where studies on this variant are scarce.
History
Until the early 1950s, only four types of hemoglobin had been described including adult hemoglobin (Hb A) and two variants (Hb S and Hb C). The fourth identified hemoglobin was Hb D which was first described by Itano in 1951. In his study, Itano was analyzing a multiethnic family from the Los-Angeles region with British, American and Indian features, and found a molecule with characteristics different to the other known hemoglobins. This hemoglobin had electrophoretic mobility similar to Hb S in alkaline pH, however, in acid pH, its migration resembled Hb A. Furthermore, Hb D exhibited normal solubility when in its reduced state and did not undergo sickling.5,6
Since the discovery of this new variant and the knowledge of its molecular structure, other hemoglobins with similar patterns discovered in populations from different ethnicities have been nominated according to the regions in which they were described. In a study performed in 1962, Baglioni analyzed the molecular structure of five hemoglobins, Hb D-Chicago, Hb D-North Carolina, Hb D-Punjab, Hb D-Portugal and Hb D-Oak Ridge and found that all exhibited the same chemical composition as the first one discovered, Hb D-Los Angeles.6,7 The most frequent denominations for this mutant hemoglobin found in the literature are Hb D-Los Angeles or Hb D-Punjab.
Mutation β121 Glu→Gln (GAA→CAA)
Hb D-Punjab is a variant derived from a point mutation in the beta-globin gene (HBB) in the first base of the 121 codon (GAA→CAA) with the substitution of glutamine for glutamic acid (Glu>Gln) in the beta globin chain.8 According to the Globin Gene Server database, besides Hb D-Punjab, there are seven other types of Hb D, caused by different point mutations in HBB. They are Hb D-Agri (HBB:c.29C>A;364G>C), Hb D-Bushman (HBB:c.49G>C), Hb D-Ouled Rabah (HBB:c.60C>A or 60C>G), Hb D-Iran (HBB:c.67G>C), Hb D-Granada (HBB:c.68A>T), Hb D-Ibadan (HBB:c.263C>A) and Hb D-Neath (HBB:c.365A>C).
Origin and geographic distribution
Hb D-Punjab is one of the most common hemoglobin variants worldwide, after Hb S and Hb C. It is prevalent in Punjab region, Northwest Indian, with an estimated frequency of 2.0%. In western India, more specifically in the Gujarat region, its frequency drops by one half.9
Hb D-Punjab is also common in countries such as Italy,10 Belgium,11 Austria12 and Turkey.13 Hb D-Punjab is the second most common hemoglobin alteration in Turkey; it occurs in 0.2% of the population of Denizli province in southeastern Turkey, where it is the most common variant. A total of 57.9% of abnormal hemoglobins observed in this province are Hb D-Punjab, followed by Hb S, with 21.9%.13 A similar frequency occurs in Xinjiang province, northwestern China, where 55.6% of variant hemoglobins are Hb D-Punjab.14,15
In Brazil there are no recent studies reporting the frequency of Hb D-Punjab in the population, but in a large survey conducted in 1993 by Bonini-Domingos, which evaluated the prevalence of hemoglobinopathies in about 100,000 Brazilian individuals from every state in the country, Hb D-Punjab was observed to be the third most common hemoglobin variant in the population.16 Posteriorly, in a screening for hemoglobinopathies performed in a hospital-based population in southeastern Brazil by Sonati et al., the authors found hemoglobin alterations in 34.4% of the individuals, of which 0.4% corresponded to Hb D-Punjab. Despite the low frequency, Hb D-Punjab was also the third most frequent variant identified in this population.17
Initially, the geographic distribution of Hb D-Punjab suggests that this mutation originated in the central region of Asia, as it is prevalent in Punjab, a region of India, and Northwestern China. Then it spread to neighboring countries by migration.11,18,19 However, haplotype analysis of beta-globin clusters provides another insight into the origin of this hemoglobin. The analysis of polymorphic sites, 5′-ɛ HincII, γG HindIII, γA HindIII, ψβ HincII, 3′-ψβ HincII, 5′-β HinfI, β AvaII, 3′-β HinfI, 3′-β BamHI (Figure 1), showed certain haplotypes associated to Hb D-Punjab, suggesting a multicentric origin for this mutation.19–24
Figure 1.
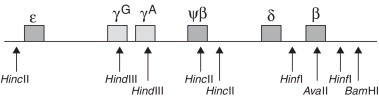
Restriction polymorphic sites in cluster beta-globin for haplotypes screening in association with Hb D-Punjab
Source: Modified from Yavarian et al.19
The study of Atalay et al. considered seven beta-globin gene polymorphic sites (5′-ɛ HincII, γG HindIII, γA HindIII, ψβ HincII, 3′-ψβ HincII, β AvaII and 3′-β HinfI) and identified three haplotypes. One of them is the Mediterranean Haplotype I (+ − − − − + +) which is the most common and found in populations of Mexico,22 Italy10 and Iran.23 A second recently discovered pattern was associated to Hb D-Punjab cases in Thailand (− + + − + + +)8 and the third haplotype was found in Turkish populations (− + − − + + +).20
In 2009, Yavarian et al. analyzed seven polymorphic sites (5′-ɛ HincII, γG HindIII, γA HindIII, ψβ HincII, 3′-ψβ HincII, 5′-β HinfI and 3′-β HinfI) and found four haplotype patterns associated to Hb D-Punjab in the Indian population. The first and most common was the Mediterranean Haplotype I (+ − − − − + +), the second seems to be a product of the Mediterranean Haplotype I variation probably due to an independent mutation (+ − − − − − +), the third was the same haplotype found in Turkey (− + − − + + +), and the fourth a common haplotype in Indian and English populations (− + − + + + +).19
Considering the same restriction sites assessed by Atalay et al. in 2007 and in 2009, Bahadir et al. reported the occurrence of another haplotype in the Turkish population (− − + − + + +).20,24 Due to lack of studies in Chinese and Indian populations, Patel et al. evaluated individuals with Hb D-Punjab from Agharia in India and found another haplotype (− − − − − + +) in addition to haplotype I (+ − − − − + +).21 Table 1 summarizes all these results.
Table 1.
Haplotypes described in the literature associated with Hb D-Punjab and the locations where they were first found.
| Polymorphic sites of cluster beta |
Occurrence | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 5′-ɛ HincII |
γG HindIII |
γA HindIII |
ψβ HincII |
3′-ψβ HincII |
5′-β HinfI |
β AvaII |
3′-β HinfI |
3′-β BamHI |
|
| + | − | − | − | − | NR | + | NR | + | Mexico,18,20 Italy,10,18 Iran18,21 |
| + | − | − | − | − | NR | + | + | NR | Turkey,18,22 India19 |
| − | + | + | − | + | NR | + | NR | + | Thailand8,18 |
| − | + | + | − | + | NR | + | + | NR | Turkey18 |
| − | + | − | − | + | NR | + | + | NR | Turkey18 |
| − | − | + | − | + | NR | + | + | NR | Turkey22 |
| + | − | − | − | − | + | NR | + | NR | Iran, the Netherlands, Belgium17 |
| + | − | − | − | − | − | NR | + | NR | Iran17 |
| − | + | − | − | + | + | NR | + | NR | Iran17 |
| − | + | − | + | + | + | NR | + | NR | Iran, India, England17 |
NR: not reported.
As the haplotypes described in association with Hb D-Punjab are prevalent in different regions, it is suggested that the origin of the mutation in codon β121 may be multicentric. According to Yavarian et al., one important aspect is that the mutation site is located in a short palindromic region, where many mutations are described, suggesting that codon β121 exhibits a higher mutation rate than is expected. Thus, there is a chance that mutational events give rise to the same hemoglobin in different regions.19 However, it is not yet possible to confirm the origin and distribution of this hemoglobin as there are few studies about haplotypes in populations from China20 and from other continents such as North, South and Central America and Africa. In Brazil, no study has been performed to screen haplotypes associated to Hb D-Punjab.
Inheritance of Hb D-Punjab and associated clinical features
The Hb D-Punjab can be inherited in heterozygosis with normal Hb A, characterizing the heterozygous trait. This condition presents no clinical or hematological alterations. The homozygous component Hb DD, the rarest form of inheritance, is not commonly related to symptomatic cases, but occasionally individuals with this profile can develop mild to moderate hemolytic anemia.25,26
The association of this variant with other abnormal hemoglobins, such as Hb S or thalassemia can also occur. Usually, interactions between Hb D-Punjab and beta thalassemia course with mild microcytic and hypochromic anemia, but do not present relevant clinical or hematological changes.27
However, when Hb D-Punjab is associated to Hb S, the double heterozygous Hb S/D, the result is moderate to severe clinical manifestations; this association can be clinically similar to homozygous Hb SS.25 Pain, due to vaso-occlusive events, is one of the most common complications. Furthermore, stroke and acute chest syndrome can occur in carriers of this genotype.28–30
Recently, Patel et al. published a study on Hb S/D-Punjab patients with moderate to severe clinical symptomatology similar to the Hb SS genotype who had vaso-occlusion episodes or required blood transfusions. The authors observed greater vulnerability for red cell lysis in Hb S/D-Punjab patients than with the Hb SS genotype.31 Clinical severity in Hb S/D-Punjab individuals could be related to the fact that Hb D favors polymerization of Hb S.32
Polymerization of Hb S molecules is triggered by hydrophobic interactions between the Val-β6 amino acid of an Hb S molecule and the Phe-β85 and Leu-β88 amino acids of adjacent hemoglobin molecules. Subsequently, these hydrophobically associated Hb S molecules interact with other contact sites of Hb S polymers, such as β73 and β121, resulting in the formation of a complex polymer structure. Thus, mutations in these contact sites can affect the biochemical properties of the Hb S polymers formed. It is believed that the amino acid changes in the β121 Glu→Gln position encourage the interaction of Hb S molecules and consequently its polymerization.33,34 Adachi et al. showed that the Hb S polymerization rate is higher when Hb D-Punjab is present. This may explain the clinical severity of sickle cell disease in double heterozygous Hb S/D-Punjab, as Hb D-Punjab increases the probability of sickling due to a shorter transit time of these cells in the blood capillaries.32
Hydroxyurea (HU) administration is a potential therapeutic strategy for sickle cell anemia (Hb SS), showing efficacy in increasing Hb F levels and consequently reducing the number of painful crises, acute chest syndrome, transfusions, and hospitalizations. However, HU therapy for Hb S/D-Punjab has not been established. Patel et al. treated Hb S/D-Punjab patients using low doses (10 mg/kg/day) of HU noticed an effective reduction in the incidence of vaso-occlusion and the frequency of blood transfusions. These results suggest that HU is a promising therapy for clinical symptomatology in Hb S/D-Punjab individuals.31
Laboratory diagnosis
Numerous laboratory methods are used to diagnose hemoglobin variants and thalassemias.
Hemoglobins can be separated by electrophoresis in cellulose acetate at pH 8.635 and in agarose gel at pH 6.2.36 In cellulose acetate, the migration of Hb D is slower than Hb A with a position similar to Hb S, but in acid pH its migration position is the same as Hb A. These electrophoretic patterns, however, are not specific for variants of the Hb D group, as there are other hemoglobin variants with similar behavior, such as Hb G and Hb Korle-bu.
The structural characterization of hemoglobin variants by electrophoretic separation of hemoglobin polypeptide chains is used in cases in which the results of the initial tests are inconclusive.37,38 The mutant βD chain migrates similar to the βS chain using this method, but slightly below this fraction in alkaline pH, while it migrates to the same position as the normal βA chain in acid pH (Figure 2).
Figure 2.
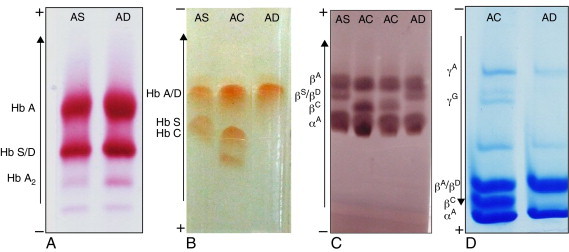
Electrophoretic mobility of Hb D-Punjab compared to other known hemoglobins, such as Hb A, S and C. Images correspond to hemoglobin electrophoresis in cellulose acetate (A), hemoglobin electrophoresis in agarose gel (B), polypeptide chains electrophoresis in cellulose acetate (C) and polypeptide chains electrophoresis in polyacrylamide gel (D). The genotypes of each sample are at the top of the figure. Current direction and position of the different hemoglobins are beside each method.
It is also possible to separate hemoglobins according to their isoelectric point (pI) by isoelectric focusing (IEF).39 In the case of Hb D-Punjab, its pI is near to pH 7.1, a position similar to other hemoglobin variants such as Hb Korle-Bu (Figure 3). Although it has great resolution, this technique requires specific kits and is more expensive than conventional electrophoresis.
Figure 3.
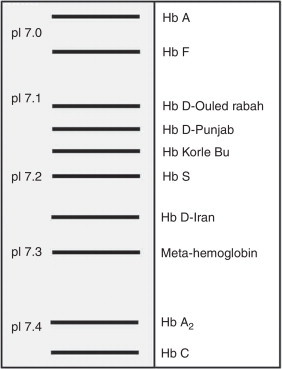
Schematic representation of relative mobilities of different hemoglobins using the isoelectric focusing method. Hb D-Punjab is shown in the figure with isoelectric point (pI) between 7.1 and 7.2.
High performance liquid chromatography (HPLC) methods have been developed for hemoglobin fractionation with high sensitivity and specificity. This technique is an integrated method based on ion-exchange to separate and identify the relative percentage of specific hemoglobins in whole blood samples (Figure 4). Another method employed to detect hemoglobin fractions is capillary electrophoresis (CE) based on the principle of electrokinetic separation. Despite the excellent resolution and reproducibility, this technique is only now arriving in some countries, including Brazil.
Figure 4.
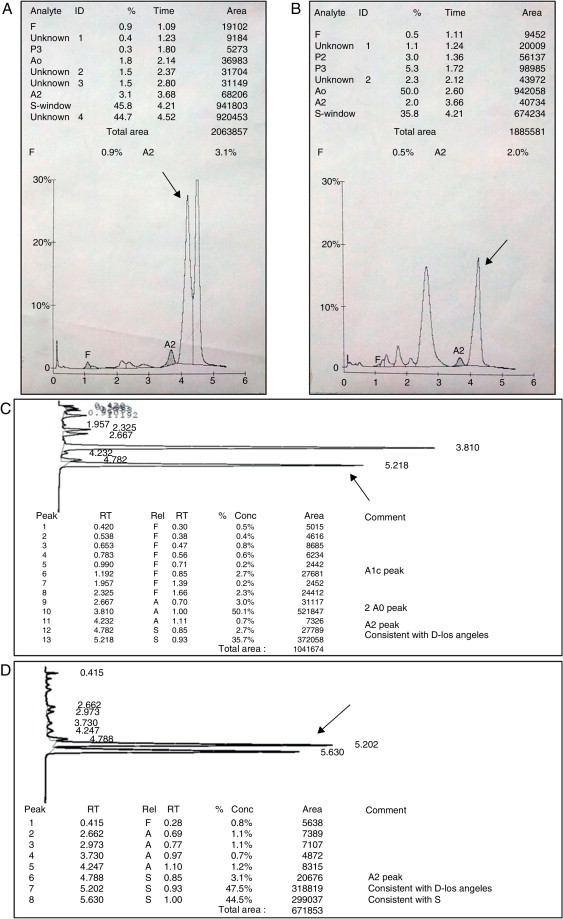
Chromatographic profile of a heterozygous trait for Hb D-Punjab (B and C) and a double heterozygous Hb S/D-Los Angeles A and D). (A and B) The VARIANT I system chromatogram with heterozygous beta-thalassemia analysis kit. The arrow indicates the pike corresponding to Hb D-Los Angeles with average retention time of 4.1–4.3 min. It is possible to note decreases in Hb A2 values, a characteristic of samples with this hemoglobin variant. (C and D) The ultra2 Resolution system chromatogram with Genesys analysis kit. The arrow indicates the Hb D-Punjab spike, with retention time between 0.92 and 0.96 min related to Hb S.
Reverse-phase HPLC (RP-HPLC) of human globin chains has been reported as an important tool for detecting hemoglobinopathies. Nevertheless, although it is widely used for the detection, quantification and purification of many proteins from biological materials, its clinical application for hemoglobins is still limited due to the long analytical time, complex sample preparation and resolution problems.40,41 Recently, studies have had success in optimizing the method,40,41 but in many laboratories the technique is still employed exclusively for research.
Although all these tests aim to diagnose Hb D-Punjab, they are not considered conclusive because of the large number of hemoglobin variants that are similar to each other. It is always recommended to perform molecular analysis for genotype confirmation with PCR-RFLP as an alternative42 (Figure 5).
Figure 5.
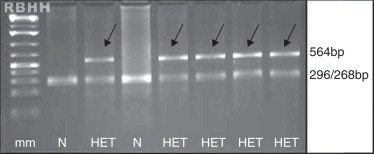
Agarose gel visualization of fragments generated by the PCR-RFLP technique for screening of β121 Glu→Gln (GAA→CAA) mutation. Normal alleles generate two fragments and mutant alleles maintain the 564 bp fragment, indicated by the arrows. N: absent mutation; HET: heterozygous for Hb D-Los Angeles; mm: molecular marker of 100 bp. Restriction enzyme: EcoRI (G↓AATTC).
When the Hb D variant is not identified by the abovementioned methodologies, sequencing of beta-globin gene or other techniques that discriminate amino acid changes are recommended.
Using these methodologies over the last 10 years, 70 cases of type D hemoglobins from southeast Brazil were diagnosed in the Hemoglobin and Hematological Genetic Diseases Laboratory of the Universidade Estadual Paulista (UNESP) a referral center for hemoglobin diagnosis. Of these, 66 (94.3%) had a profile compatible to Hb D-Punjab, eight (12.1%) were compound heterozygous Hb S/D-Punjab and 58 (87.9%) heterozygous Hb A/D-Punjab, all of which were confirmed by molecular tests. The other four remaining cases presented electrophoretic and chromatographic profiles compatible with the “Hb A/D-non-Los Angeles” trait, possibly Hb A/D-Iran (data not published).
Conclusion
Although not always associated with relevant clinical history, Hb D-Punjab is a relatively common hemoglobin worldwide; it is the third most common hemoglobin variant in Brazil. Greater attention is given when the association of Hb D-Punjab with another abnormal hemoglobin or thalassemia leads to clinical symptoms. However, this issue is still understudied especially in Brazil and the pathophysiological mechanisms caused by Hb D-Punjab are poorly known. There is no recent information about the prevalence of Hb D-Punjab, association with other hemoglobin variants, or characterization of typical haplotypes in the Brazilian population. Therefore, it is important to conduct studies about this hemoglobin variant.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
The authors would like to thank Fundação de Amparo à Pesquisa do Estado de São Paulo (Foundation for Research Support of the State of Sao Paulo) – FAPESP, by supporting research in our laboratory (process number 2012/19653-1). In addition, to thank Lucas Pereira Ramos for English text revision.
References
- 1.Weatherall D.J., Clegg J.B. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704–712. [PMC free article] [PubMed] [Google Scholar]
- 2.Kohne E. Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Dtsch Arztebl Int. 2011;108(31/32):532–540. doi: 10.3238/arztebl.2011.0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thom C.S., Dickson C.F., Gell D.A., Weiss M.J. Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harb Perspect Med. 2013;3(3):a011858. doi: 10.1101/cshperspect.a011858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frenette P.S., Atweh G.F. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest. 2007;117(4):850–858. doi: 10.1172/JCI30920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itano H.A. A third abnormal hemoglobin associated with hereditary hemolytic anemia. Proc Natl Acad Sci U S A. 1951;37(12):775–784. doi: 10.1073/pnas.37.12.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vella F., Lehmann H. Haemoglobin D Punjab (D Los Angeles) J Med Genet. 1974;11(4):341–348. doi: 10.1136/jmg.11.4.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baglioni C. Abnormal human haemoglobins. VII. Chemical studies on haemoglobin D. Biochim Biophys Acta. 1962;59(2):437–449. doi: 10.1016/0006-3002(62)90194-4. [DOI] [PubMed] [Google Scholar]
- 8.Fucharoen S., Changtrakun Y., Surapot S., Fucharoen G., Sanchaisuriya K. Molecular characterization of Hb D-Punjab β121(GH4)Glu→Gln in Thailand. Hemoglobin. 2002;26(3):261–269. doi: 10.1081/hem-120015030. [DOI] [PubMed] [Google Scholar]
- 9.Köseler A., Öztürk O., Atalay A., Atalay E.Ö. Allele frequency of VNTR locus D1S80 observed in Hb D-Los Angeles carriers. Mol Biol Rep. 2012;39(12):10747–10750. doi: 10.1007/s11033-012-1966-4. [DOI] [PubMed] [Google Scholar]
- 10.Fioretti G., De Angioletti M., Pagano L., Lacerra G., Viola A., de Bonis C. DNA polymorphisms associated with Hb D-Los Angeles b121(GH4)Glu→Gln in Southern Italy. Hemoglobin. 1993;17(1):9–17. doi: 10.3109/03630269308998881. [DOI] [PubMed] [Google Scholar]
- 11.Husquinet H., Parent M.T., Schoos-Barbette S., Dodinval-Versie J., Lambotte C., Galacteros F. Hemoglobin D-Los Angeles β121(GH4)Glu→Gln in the province of Liege Belgium. Hemoglobin. 1986;10(6):587–592. doi: 10.3109/03630268609036563. [DOI] [PubMed] [Google Scholar]
- 12.Lischka A., Pollak A., Bauer K., Aschauer H., Braunitzer G. Hemoglobin D “Los Angeles” in an Austrian family: biochemical identification, clinical aspects, and kindred study. Hemoglobin. 1984;8(4):353–361. doi: 10.3109/03630268408991718. [DOI] [PubMed] [Google Scholar]
- 13.Atalay E.Ö., Koyuncu H., Turgut B., Atalay A., Yildiz S., Bahadir A. High incidence of Hb D-Los Angeles β121(GH4) Glu→Gln in Denizli province Aegean region of Turkey. Hemoglobin. 2005;29(4):307–310. doi: 10.1080/03630260500311685. [DOI] [PubMed] [Google Scholar]
- 14.Li H.J., Zhao X.N., Qin F., Li H.W., Li L., He X.J. Abnormal hemoglobins in the silk road region of China. Hum Genet. 1990;86(2):231–235. doi: 10.1007/BF00197711. [DOI] [PubMed] [Google Scholar]
- 15.Pandey S., Mishra R.M., Pandey S., Shah V., Saxena R. Molecular characterization of hemoglobin D Punjab traits and clinical–hematological profile of the patients. Sao Paulo Med J. 2012;130(4):248–251. doi: 10.1590/S1516-31802012000400008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonini-Domingos C.R. Univ. Estadual Paulista – Unesp; São José do Rio Preto, SP: 1993. Prevenção das hemoglobinopatias no Brasil – Diversidade Genética e Metodologia Laboratorial. 144f. Tese (Doutorado em Ciências) – Instituto de Biociências, Letras e Ciência Exatas. [Google Scholar]
- 17.Sonati M.F., Kimura E.M., Grotto H.Z., Gervasio S.A., Costa F.F. Hereditary hemoglobinopathies in a population from southeast Brazil. Hemoglobin. 1996;20(2):175–179. doi: 10.3109/03630269609027926. [DOI] [PubMed] [Google Scholar]
- 18.Brittenham G.M. Globin gene variants and polymorphism in India. In: Winter W.P., editor. Hemoglobin variants in human populations. CRC Press Inc.; Florida: 1988. pp. 79–110. [Google Scholar]
- 19.Yavarian M., Karimi M., Paran F., Neven C., Harteveld C.L., Giordano P.C. Multi Centric origin of Hb D-Punjab β121(GH4)Glu→Gln, GAA>CAA. Hemoglobin. 2009;33(6):399–405. doi: 10.3109/03630260903344598. [DOI] [PubMed] [Google Scholar]
- 20.Atalay E.Ö., Atalay A., Ustel E., Yildiz S., Oztürk O., Köseler A. Genetic origin of Hb D-Los Angeles β121(GH4)Glu→Gln, GAA→CAA according to the β-globin gene cluster haplotypes. Hemoglobin. 2007;31(3):387–391. doi: 10.1080/03630260701459416. [DOI] [PubMed] [Google Scholar]
- 21.Patel D.K., Mashon R.S., Patel S., Dash P.M., Das B.S. β-Globin gene haplotypes linked with the Hb D-Punjab β121(GH4)Glu→Gln, GAA→CAA mutation in eastern India. Hemoglobin. 2010;34(6):530–537. doi: 10.3109/01676830.2010.525900. [DOI] [PubMed] [Google Scholar]
- 22.Perea F.J., Casas-Castañeda M., Villalobos-Arámbula A.R., Barajas H., Alvarez F., Camacho A. Hb D-Los Angeles associated with Hb S or β-thalassemia in four Mexican Mestizo families. Hemoglobin. 1999;23(3):231–237. doi: 10.3109/03630269909005703. [DOI] [PubMed] [Google Scholar]
- 23.Rahimi Z., Akramipour R., Nagel R.L., Ahmadi A.S., Merat A., Bahrehmand F. The β-globin gene haplotypes associated with Hb D-Los Angeles β121(GH4)Glu→Gln in Western Iran. Hemoglobin. 2006;30(1):39–44. doi: 10.1080/03630260500454105. [DOI] [PubMed] [Google Scholar]
- 24.Bahadir A. Hb D-Los Angeles beta121(GH4)Glu→Gn and Hb Beograd beta121(GH4)Glu 1 Val: implications for their laboratory diagnosis and genetic origins. Turk J Hematol. 2009;26(1):17–20. [PubMed] [Google Scholar]
- 25.Adekile A., Muah-AlI A., Akar N.A. Does elevated hemoglobin F modulate the phenotype in Hb SD-Los Angeles? Acta Haematol. 2010;123(3):135–139. doi: 10.1159/000276998. [DOI] [PubMed] [Google Scholar]
- 26.Taghavi Basmanj M., Karimipoor M., Amirian A., Jafarinejad M., Katouzian L., Valaei A. Co-inheritance of hemoglobin D and β-thalassemia traits in three families: clinical relevance. Arch Iran Med. 2011;14(1):61–63. [PubMed] [Google Scholar]
- 27.Naoum P.C., Moraes M.S., Radispiel J., Cavalheri P.P., Valeri F.F. Hb D/Talassemia beta associada à anemia crônica. Rev Bras Hematol Hemoter. 2002;24(1):51–52. [Google Scholar]
- 28.Ohene-Frempong K., Weiner S.J., Sleeper L.A., Miller S.T., Embury S., Moohr J.W. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–294. [PubMed] [Google Scholar]
- 29.Castro O., Brambilla D.J., Thorington B., Reindorf C.A., Scott R.B., Gillette P. The acute chest syndrome in sickle cell disease: incidence and risk factors. The cooperative study of sickle cell disease. Blood. 1994;84(2):643–649. [PubMed] [Google Scholar]
- 30.Hamidieh A.A., Jalili M., Khojasteh O., Ghavamzadeh A. First report of successful stem cell transplantation in a patient with sickle cell hemoglobin D disease. J Pediatr Hematol Oncol. 2010;32(5):397–399. doi: 10.1097/MPH.0b013e3181df614b. [DOI] [PubMed] [Google Scholar]
- 31.Patel S., Purohit P., Ranjeet S.M., Dehury S., Meher S., Sahoo S. The effect of hydroxyurea on compound heterozygotes for sickle cell-hemoglobin D-Punjab—a single centre experience in eastern India. Pediatr Blood Cancer. 2014;61(8):1341–1346. doi: 10.1002/pbc.25004. [DOI] [PubMed] [Google Scholar]
- 32.Adachi K., Kim J., Ballas S., Surrey S., Asakura T. Facilitation of Hb S polymerization by the substitution of Glu for Gln at β121. J Biol Chem. 1988;263(12):5607–5610. [PubMed] [Google Scholar]
- 33.Charache S., Conley C.L. Rate of sickle cells during deoxygenation of blood from persons with various sickling disorders. Blood. 1964;24:25–48. [PubMed] [Google Scholar]
- 34.Benesch R.E., Kwong S., Benesch R., Edalji R. Location and bond type of intermolecular contacts in the polymerization of haemoglobin S. Nature. 1977;269(5631):772–775. doi: 10.1038/269772a0. [DOI] [PubMed] [Google Scholar]
- 35.Marengo-Rowe A.J. Rapid electrophoresis and quantitation of haemoglobins on cellulose acetate. J Clin Pathol. 1965;18(6):790–792. doi: 10.1136/jcp.18.6.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vella F. Acid-agar gel electrophoresis of human hemoglobins. Am J Clin Pathol. 1968;49(3):440–442. doi: 10.1093/ajcp/49.3_ts.440. [DOI] [PubMed] [Google Scholar]
- 37.Schneider R.G. Differentiation of electrophoretically hemoglobins – such as S, D, G and P or A2, C, E, and O – by electrophoresis of the globin chains. Clin Chem. 1974;20(9):1111–1115. [PubMed] [Google Scholar]
- 38.Alter B.P., Goff S.C., Efremov G.D., Gravely M.E., Huisman T.H. Globin chain electrophoresis: a new approach to the determination of the G/A ratio in fetal haemoglobin and to studies of globin synthesis. Br J Haematol. 1980;44(4):527–532. doi: 10.1111/j.1365-2141.1980.tb08706.x. [DOI] [PubMed] [Google Scholar]
- 39.Naoum P.C. Livraria Santos Editora Ltda; São Paulo: 1990. Eletroforese – Técnicas e Diagnósticos. [Google Scholar]
- 40.Nemati H., Bahrami G., Rahimi Z. Rapid separation of human globin chains in normal and thalassemia patients by RP-HPLC. Mol Biol Rep. 2011;38(5):3213–3218. doi: 10.1007/s11033-010-9994-4. [DOI] [PubMed] [Google Scholar]
- 41.Wan J.H., Tian P.L., Luo W.H., Wu B.Y., Xiong F., Zhou W.J. Rapid determination of human globin chains using reversed-phase high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;15(901):53–58. doi: 10.1016/j.jchromb.2012.05.041. [DOI] [PubMed] [Google Scholar]
- 42.Saiki R.K., Gelfand D.H., Stoffel S., Scharf S.J., Higuchi R., Horn G.T. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239(4839):487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]