

Abstract
Tissue factor is the primary initiator of coagulation cascade and plays an essential role in hemostasis and thrombosis. In addition, tissue factor and coagulation proteases contribute to the many cellular responses via activation of protease activated receptors. Heart is the organ demonstrating high levels of constitutive tissue factor expression. This review focuses on the role of tissue factor, coagulation proteases and protease activated receptors in heart hemostasis and the pathological heart remodeling associated with myocardial infarction, viral myocarditis and hypertension.
Introduction
Tissue factor
Tissue factor (TF) is a 47-kDa transmembrane cell-surface glycoprotein that belongs to the cytokine receptor superfamily and binds coagulation factor VII/VIIa (FVII/VIIa). The TF:FVIIa binary complex exerts both procoagulant and signaling activities and plays an important role in many biological processes, including hemostasis, thrombosis and inflammation (1). TF is constitutively expressed by perivascular and parenchymal cells. Upon vascular injury, these cellular sources of TF are exposed to the blood and play an essential role in the initiation of hemostasis process. However, during many pathologic conditions increased TF expression on vascular cells (leukocytes and endothelial cells), circulating microparticles or ruptured atherosclerotic plaques triggers thrombosis and disseminated intravascular coagulation (1, 2). In addition, perivascular cell TF exposed to the circulating FVII/FVIIa, as a result of increased vascular permeability, can also participate in these processes (1, 2). The contribution of TF to the cellular responses is mediated via generation of downstream coagulation proteases and subsequent activation of protease activated receptors (PARs).
Protease activated receptors
Protease activated receptors (PARs) are a family of seven transmembrane domain, G protein–coupled receptors activated by proteolytic cleavage (3). After their activation, a new amino terminus peptide is exposed that functions as a tethered ligand (3). The PAR family consists of four members: PAR-1, PAR-2, PAR-3 and PAR-4. Thrombin is the main physiological activator of PAR-1, PAR-3 and PAR-4 (3). In addition, several studies have shown that PAR-1 can be activated by TF:FVIIa:FXa ternary complex, FXa, activated protein C and matrix metalloproteinase-1 (MMP-1) and MMP-13 (4–9). In vitro studies have shown that PAR-2 is activated by several proteases, including trypsin, mast cells tryptase and the coagulation proteases FVIIa and FXa, as well as the TF:FVIIa:FXa ternary complex (3, 10, 11). PARs can also be activated by synthetic agonist peptides corresponding to the tethered ligand sequence (3).
Pathologic heart remodeling
In response to both acute and chronic insults, including coronary artery disease, myocardial infarction, hypertension, viral infections, valve abnormalities and inherited mutations, the heart undergoes an extensive remodeling (12–14). At the early stage, remodeling is an adaptive reaction of the heart to maintain cardiac function but over time leads to maladaptive changes resulting in decreased heart function and increased risk for heart failure. Cardiomyocyte hypertrophy, proliferation of cardiac fibroblasts and cardiac inflammation are the three major processes contributing to pathologic heart remodeling (12, 14). PARs are widely expressed by cells within the cardiovascular system including endothelial cells, smooth muscle cells, cardiac fibroblasts and cardiomyocytes (15). In addition, TF expression can be found on extravascular cells such as fibroblasts, smooth muscle cells (SMCs) and cardiomyocytes(16). Coagulation protease-mediated activation of PARs on these cells contributes to the pathophysiology of heart remodeling.
In this review we will discuss the role of TF and PARs in heart hemostasis as well as their contribution to pathological heart remodeling. Specifically, we will focus on the remodeling of the heart in response to the myocardial infarction, viral infections, activation of adrenergic receptors and pulmonary hypertension. We would like to apologize for not always using the original references due to space and word limitations.
Heart Hemostasis
TF is constitutively expressed by cardiomyocytes and fibroblasts in the heart. We and others have shown that heart hemostasis is severely compromised in mice expressing very low levels of TF (17, 18). These low TF mice uniformly demonstrate hemosiderin deposition and fibrosis in their hearts by 3 months of age. A hemostatic defect was first observed in sub-epicardial and perivascular regions of the heart and over time spread throughout the whole heart (17). Mice expressing low levels of murine FVII exhibited a similar pattern of hemosiderin deposition and fibrosis in their hearts (17, 19). Furthermore, mice with a selective deletion of TF in cardiomyocytes exhibited only mild hemostatic defect under normal conditions but a dramatic impairment of hemostasis after treatment with isoproterenol (20). Importantly, heart hemostasis was restored in low TF mice by overexpression of TF in cardiomyocytes (20). These data strongly suggest that cardiomyocyte TF is essential to maintain heart hemostasis.
It has been proposed that in addition to the important role in heart hemostasis, TF may also have non-hemostatic functions in the heart. For example, the fetal heart expresses much higher TF levels compared to the adult heart (21). This led to the hypothesis that myocardial TF may serve as a morphogenic factor in the developing heart (22). Within cardiomyocytes, TF is found in the intercalated discs and z bands, and co-localized with structural proteins such as desmin and vinculin (23–25). Additionally, the cytoplasmic domain of TF has been demonstrated to interact with the cytoskeleton via filamin A (26, 27) and binding of FVII/FVIIa to TF mediates direct changes of the cytoskeleton (28). In addition, it was shown that TF mediates β1 integrin signaling via both its extracellular and intracellular domains independently of its procoagulant activity (29). These data lead to the hypothesis that TF may contribute to the structural integrity of myocardium. We tested this hypothesis, in part, by examining hearts of mice lacking only the cytoplasmic domain of murine TF. We found no evidence of hemostatic or structural defects in the heart of these mice at 6 months of age under normal conditions (17). However, we cannot exclude that under pathologic conditions the cytoplasmic domain of TF may play a role in maintaining structural integrity.
Myocardial infarction
Myocardial infarction caused by the thrombotic occlusion of coronary vessels is a leading cause of death in the western world (30). Restoration of circulation not only provides oxygen and nutrients to ischemic tissue but also further injures the myocardial tissue by initiating an inflammatory response. The molecular and cellular mechanisms of ischemia/reperfusion (I/R) injury is very complex and has been described elsewhere (31). Myocardial infarction leads to the loss of viable myocardium; as a compensatory response the initial infarct is replaced with collagen rich fibrotic tissue and neighboring cardiomyocytes undergo hypertrophic growth. However, over time the extensive remodeling weakens heart function and leads to heart failure (30).
I/R injury increases TF expression in the heart (32, 33). The inhibition of TF with either active site–inhibited FVIIa, anti-TF antibody or annexin V-Kunitz inhibitor fusion protein (ANV-6L15) reduces infarct size in animal models of heart I/R injury (33–36). We and other showed that inhibition of FXa (37) or thrombin (33, 38) as well as fibrinogen deficiency (39) attenuates myocardial infarction in animal models. In addition, fibrin degradation fragment E1 exacerbates myocardial infarction by facilitating leukocyte infiltration into myocardium (39, 40). Therefore fibrin generation is one mechanism by which the TF-thrombin pathway contributes to myocardial infarction.
The contribution of thrombin-mediated activation of PAR-1 to myocardial infarction is less clear. We found that PAR-1 deficiency has no effect on the infarct size after cardiac I/R injury (38). In contrast, Strande and colleagues reported that the PAR-1 antagonist SCH 79797 reduces infarct size during myocardial I/R injury in rats (41). The possible off-target effect of SCH 79797 or PAR-4 mediated compensation for PAR-1 deficiency may explain these different findings (42, 43). Interestingly, we found that PAR-1 deficient mice had reduced dilatation of the left ventricle and better preservation of heart function two weeks after I/R injury. In vitro studies demonstrated that activation of PAR-1 with either thrombin or agonist peptide leads to a series of molecular and morphological changes that lead to hypertrophic growth of cardiomyocytes (44, 45). Consistent with these observations, we showed that cardiomyocyte-specific overexpression of PAR-1 induced eccentric heart hypertrophy and decreased heart function (38). Importantly, cardiomyocyte-specific deletion of TF reduced eccentric hypertrophy in PAR-1 transgenic mice, indicating that activation of PAR-1 on cardiomyocytes was mediated by local TF-dependent thrombin generation (38). Interestingly, Loubele and colleagues demonstrated that protective effects of exogenous administration of activated protein C on cardiac I/R injury required PAR-1 (46) suggesting PAR-1 mediated responses may be ligand specific.
The protective effect of TF:FVIIa complex inhibition on I/R-induced myocardial infarction could be also mediated via attenuation of PAR-2 signaling. We showed that PAR-2 deficiency reduces infarct size, oxidative stress, and expression of inflammatory cytokines caused by I/R injury (47). Furthermore, 4 weeks after I/R injury hypertrophy and dilatation of left ventricle were significantly attenuated whereas heart function was better preserved in PAR-2 deficient mice (47). However, this protection could simply be the result of smaller initial infarct size observed in PAR-2 deficient mice. Therefore in the follow up studies we investigated the “infarct independent” contribution of PAR-2 to heart remodeling. Consistent with previous observations, we demonstrated that activation of PAR-2 induced hypertrophic growth and of cultured rat neonatal cardiomyocytes (48). In addition, PAR-2 activation on mouse cardiomyocytes increased expression of the pro-fibrotic chemokine MCP-1. Furthermore, cardiomyocyte-specific overexpression of PAR-2 in mice induced heart hypertrophy, cardiac fibrosis, inflammation and heart failure (48). Finally, in a mouse model of myocardial infarction induced by permanent ligation of the left anterior descending coronary artery, PAR-2 deficiency reduced heart remodeling and improved heart function independently of its contribution to the size of the initial infarct (48). Further studies are required to determine if these effects are mediated solely by TF:FVIIa–dependent activation of PAR-2 or other PAR-2 ligands, for example mast cell tryptase (49, 50).
In contrast to our studies, several recent papers reported that activation of PAR-2 with a PAR-2 agonist peptide has a beneficial effect in ex vivo as well in vivo models of heart I/R injury (51–53). The protective mechanism involved endothelial cell-dependent vasodilation of coronary vessels (53). The similar differences between genetic deficiency and pharmacologic inhibition of the receptor have been also reported for PAR-4 (54–56). The possible explanations for these contradictory results may include so called “biased agonist” signaling (57, 58), cell type specific responses or accessibility of the inhibitors to the target cells. Importantly, a recent study indicated that commonly used PAR-2 agonist peptide SLIGRL-NH2 facilitates PAR-2 independent effects (59). We have discussed that in detail in our recent papers (48, 56).
Viral Myocarditis
Myocarditis is defined as inflammation within the myocardium in response to different insults, such as viral infections, autoimmune reactions, toxins and adverse drug reactions. Coxsackievirus B3 (CVB3) was one of the first viruses described and investigated to cause viral myocarditis (60). Viral myocarditis can be divided in three phases. The early phase with virus replication in the heart and innate immune responses, the acute phase with declining virus load but extensive myocardial inflammation and the late stage with virus clearance, declining myocardial inflammation and ongoing cardiac remodeling leading to dilated cardiomyopathy and heart failure. Viral infections induce TF expression and activate coagulation (61, 62). Interestingly, heart failure induced by viral myocarditis is associated with increased risk of thrombosis, demonstrated by increased incidence of ventricular fibrin deposition and presence of thrombi (63). Data by us and others suggest that the early/acute inflammatory response after infection, but not late/chronic inflammation, leads to increased TF expression and subsequent thrombosis in the heart (23, 63–65).
Thrombin mediated PAR-1 activation was linked to increased susceptibility of endothelial cells to herpes viruses infection in vitro (66). However, we have recently shown that PAR-1 deficiency reduces levels of interferon (IFN)-β and CXCL10 during the early phase of infection in mice, resulting in higher viral loads and increased inflammation at day 8 after CVB3 infection (67). As a consequence, pathological heart remodeling was increased in the heart of PAR-1 deficient mice infected with CVB3, as demonstrated by increased fibrosis, hypertrophy and significant reduction in heart function compared to wild type mice. Inhibition of either TF or thrombin in wild type mice also significantly increased CVB3 levels in the heart and cardiac injury, linking TF-dependent thrombin generation to PAR-1 activation (67). Our study revealed an unexpected protective role for the TF/thrombin/PAR-1 pathway during CVB3 infection-induced myocarditis in mice.
With regard to PAR-2 in myocarditis, Weithauser and collaborators observed that PAR-2 reduced toll-like receptor 3 (TLR3) mediated immune responses in vitro and in vivo during CVB3 infection (68). PAR-2 deficient mice exhibited significantly reduced CVB3 myocarditis associated with increased activation of the IFN-β pathway. The authors further showed that activation of PAR-2 in isolated cardiac fibroblasts reduced TLR3 mediated signaling, resulting in reduced IFN-β expression and STAT1 activation (68). The authors explained this observation due to direct interaction of PAR-2 with TLR3.
Collectively, these observations suggest that targeting TF/thrombin/PAR-1 pathway may impair the innate immune response to CVB3 infection whereas inhibition of PAR-2 may be a new strategy to reduce viral myocarditis induced heart remodeling. However, the PAR-2 activating protease in CVB3 myocarditis has yet to be determined. A possible candidate would be mast cell tryptase (61).
Renin-angiotensin system, hypertension and cardiac remodeling
The renin-angiotensin system (RAS) plays a key role in the development and pathophysiology of hypertension. The major consequences of hypertension are end-organ damage and cardiovascular complications including atrial fibrillation, left ventricular hypertrophy and congestive heart failure (69–71). Angiotensin II (Ang II) is the main effector of the RAS, and most of its effects are mediated by angiotensin type 1 (AT1) receptors (69, 70). Long-term exposure to Ang II leads to development of cardiovascular remodeling, fibrosis and heart hypertrophy. The mechanism of Ang II-induced heart remodeling may involve the direct action of Ang II on target tissues or may be mediated by an Ang II-induced increase in blood pressure (69, 70). One consequence of AT1 receptor activation by Ang II is upregulation of TF expression. Several in vitro studies demonstrated that Ang II induces TF expression in smooth muscle cells, endothelial cells and monocytes (72–74). TF expression was also upregulated in hypertensive rats throughout the endothelium and media of blood vessels and blocking the AT1 receptor with valsartan inhibited the upregulation of TF expression (75). Importantly, elevated levels of plasma TF are observed in patient with hypertension and AT1 receptor blockage significantly reduced TF activity (76). These data indicate that upregulation of TF expression during hypertension is mediated via the AT1 receptor and leads to systemic activation of coagulation cascade. Consistent with these observations our preliminary data demonstrate increased levels of thrombin-antithrombin (TAT) complexes in the circulation of Ang II treated mice (Antoniak and Pawlinski, unpublished data). In addition, in mice subjected to chronic Ang II infusion, PAR-1 deficiency reduced the expression of pro-fibrotic genes and attenuated collagen deposition in the hearts and aortas. These effects were independent from changes in blood pressure. The reduced pro-fibrotic phenotype in PAR-1 deficient mice resulted in a better preservation of heart function compared to wild type controls (Antoniak and Pawlinski, unpublished data). In addition, PAR-1 expression in the murine lungs is associated with angiotensin-converting enzyme activity (77).
McGuire and coworkers found that PAR-2 deficiency had no effect on diastolic pressures and only moderately reduced systolic pressures in hypertension induced by a high salt diet (78). In a more recent study the group showed that chronic PAR-2 stimulation in mice causes endothelial and vascular smooth muscle cell dysfunction (79). Interestingly, this effect was partly due to PAR-2 desensitization. PAR-2 activation affects small arteries independently of NO whereas the PAR-2 dependent vasodilation in larger arteries and the aorta is entirely NO-dependent (79). PAR-2 mediated vasodilation of arteries was protective against the negative effects of Ang II-induced dysfunction. The authors hypothesized that Ang II-induced cyclooxygenases increased the sensitivity of the arteries to the PAR-2 agonist (80). However, the effects of PAR-2 activation were very subtle, suggesting that PAR-2 is not a major regulator of blood pressure and vascular tone in vivo.
Activation of β-adrenergic receptors with isoproterenol also causes hypertension, activation of coagulation and induces pathologic heart remodeling (81). Sato et al. observed that PAR-2 activation has no effect on isoproterenol stimulation of human airway epithelial cells (82). Further, mast cell tryptase contributed to the cardiac fibrosis but did not influence blood pressure or ventricular dilatation in hypertensive rats (83). Isoproterenol treatment of rat cardiomyocytes and cardiac fibroblasts increased PAR-1 activation that was dependent on MMP-13 activity. MMP-13 inhibition significantly attenuated isoproterenol-induced PAR-1 activation and reduced PAR-1 dependent heart dysfunction (9). In addition, cardiac cells express MMP-1 and its activity is increased after chronic isoproterenol infusion in the rat heart in vivo (84). This suggests that not only MMP-13 but also MMP-1 may lead to PAR-1 dependent cardiac pathology in chronic β-adrenergic receptor stimulation.
Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is a complex cardiovascular disease with multiple etiologies. Elevated levels of circulating TF-positive microparticles (85) and increased TF expression have been observed in the lungs of patients with PAH (86, 87) as well as in rat and mouse models of the disease (88–90). Despite the prevalence of in situ thrombi in the lungs, biomarkers of coagulation are only modestly elevated in the plasma of PAH patients (91).
Pathologic right ventricle (RV) remodeling occurs downstream of increased pulmonary vascular resistance caused by thrombosis, vasoconstriction, and remodeling of the pulmonary vessel wall (92). In PAH, pulmonary vascular remodeling is caused by endothelial cell proliferation and dysfunction, SMC proliferation and migration, and fibroblast proliferation and differentiation into SMC (93). The presence of thrombi in the lungs might also contribute to the vasoconstriction and increased pulmonary vascular resistance (86). The increased pulmonary artery pressure and workload on the heart causes patients to develop progressive RV hypertrophy and eventual RV failure. A direct role for TF in PAH has yet to be established, however several animal models have implicated the TF-dependent pathways in pulmonary vascular remodeling, vasoconstriction, and RV hypertrophy. The emerging hypothesis is that FXa and thrombin may exert these effects by activating PAR-2 and PAR-1 on pulmonary vascular cells.
Overexpression of tissue factor pathway inhibitor on pulmonary SMCs attenuated pulmonary vascular remodeling, RV hypertrophy and heart function in the mouse model of PAH induced by chronic hypoxia (94). FXa inhibition with rivaroxaban significantly reduced RV hypertrophy and function in the rat monocrotaline model of PAH (95). Interestingly, FXa inhibition had no effect on pulmonary artery muscularization (95). FXa can activate PAR-2, the expression of which is increased in the pulmonary arteries of patients with PAH and in rodent models (96). A recent study by Kwapiszewska and colleagues demonstrated that PAR-2 deficient mice were protected from chronic hypoxia-mediated remodeling of the pulmonary artery and RV hypertrophy and dysfunction (96). In vitro studies demonstrated that proliferation of pulmonary artery SMCs was mediated by mast cell tryptase-dependent PAR-2 activation, suggesting that mast cell tryptase may be another ligand activating PAR-2 during PAH (96).
Vasoconstriction that results from dysregulation of the nitric oxide-cGMP pathway is implicated in the pathogenesis of PAH (97). In vitro experiments using human pulmonary artery endothelial cells revealed that thrombin treatment dose-dependently decreased expression of endothelial nitric oxide synthase, guanyl cyclase and phosphodiesterase 5. The effects of thrombin on the nitric oxide-cGMP pathway were PAR-1 dependent (98). Moreover, the direct thrombin inhibitor dabigatran prevented RV remodeling and hypertrophy in monocrotaline-treated rats (98). These data suggest that thrombin-dependent activation of PAR-1 might contribute to PAH by downregulating nitric oxide-cGMP pathway and causing vasoconstriction. Thrombin might also contribute to excessive vasoconstriction in PAH by directly activating vascular smooth muscle cell contraction via PAR-1. In endothelial cell-denuded porcine pulmonary arteries, thrombin increased intracellular calcium and caused phosphorylation of myosin light chain 20 (MLC20), which lead to a sustained contraction of the vessel that was attenuated by PAR-1 antagonism (99, 100). The use of PAR-1 agonist peptide had similar effects as thrombin (99, 100). PAR-1 and PAR-2 can also be activated by the TF:FVIIa:FXa ternary complex on endothelial cells (4) but a direct link between ternary complex-dependent PAR activation and PAH has yet to be established.
Summary
TF plays an important role in the heart during both physiologic and pathologic conditions. Under normal conditions, TF is required to maintain heart hemostasis and protect this vital organ from bleeding. Furthermore, TF-thrombin-PAR-1 pathway also plays an important role in protecting the heart against pathologic remodeling induced by viral myocarditis. The effects are mediated by enhancing anti-viral innate immune response at the initial phase of infection, reducing the viral load in the heart and minimizing an inflammation-mediated damage of myocardial tissue. In contrast, PAR-2 signaling seems to enhance viral myocarditis and heart damage (Figure 1).
Figure 1. Role of TF and coagulation proteases in the heart hemostasis and heart remodeling after the myocardial infarction and myocarditis.
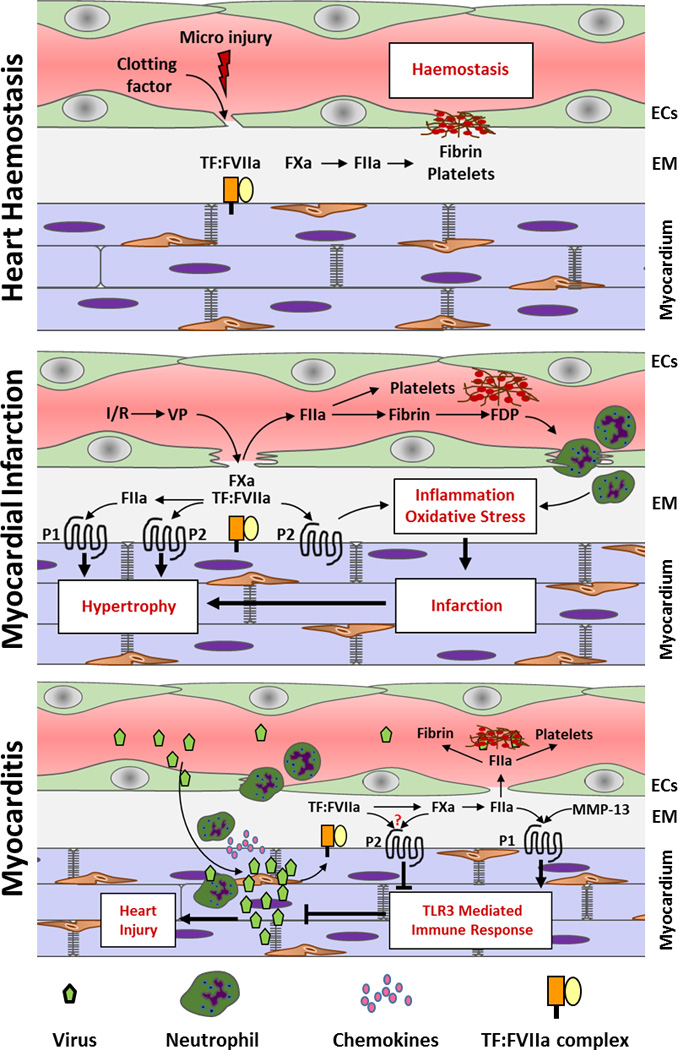
Abbreviations: FIIa – thrombin, VP – vascular permeability, P1 – PAR-1, P2 – PAR2, EM – extracellular matrix
During myocardial infarction-induced heart remodeling, TF dependent activation of coagulation and activation of PARs contribute to both initial size of the infarct as well as cellular process associated with pathologic heart remodeling including hypertrophic growth of cardiomyocytes and fibrosis (Figure 1).
TF-dependent activation of coagulation may also contribute to the heart remodeling associated with hypertension. Activation of PAR-1 and PAR-2 promote fibrosis, loss of heart function, and vasodilation caused by Ang II and isoproterenol. Similarly, PAR-1 and PAR-2 contribute to PAH via endothelial and smooth muscle cell-mediated vasoconstriction of pulmonary arteries. More studies are required to investigate if coagulation proteases and PARs can directly contribute to the remodeling of RV during PAH via affecting cardiomyocytes and cardiac fibroblast responses (Figure 2).
Figure 2. Contribution of TF and coagulation protease to the heart remodeling induced by systemic and pulmonary hypertension.
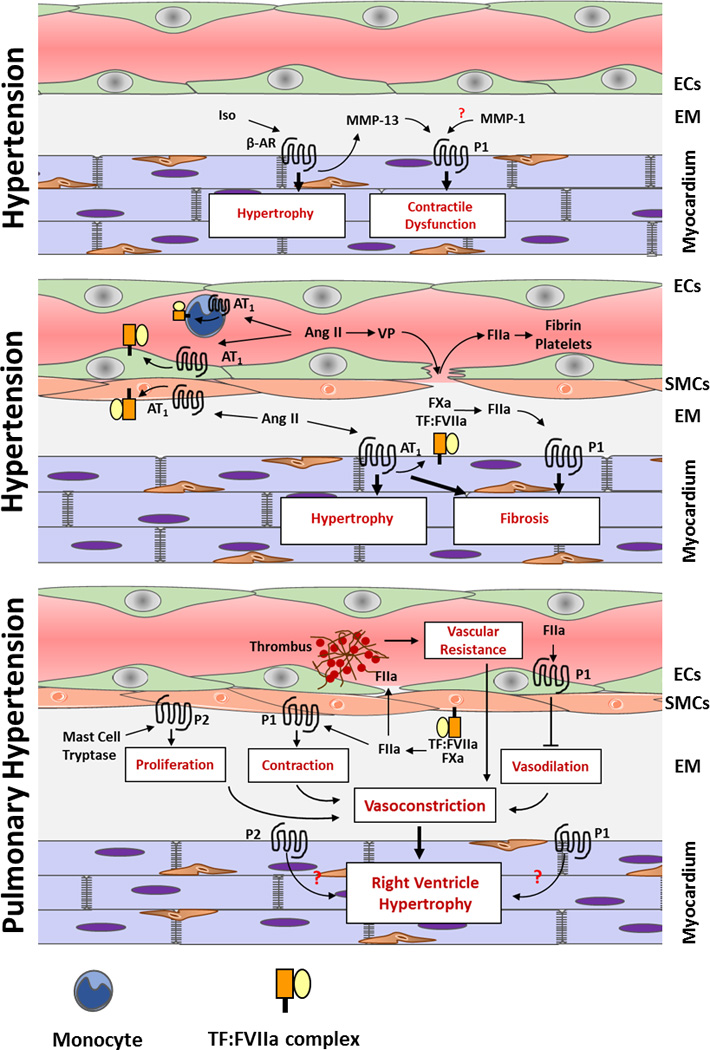
Abbreviations: Iso – Isoproterenol, FIIa – thrombin, VP – vascular permeability, P1 – PAR-1, P2 – PAR2, EM – extracellular matrix
Acknowledgments
This work was supported by grants from the Myocarditis Foundation (S.A.) and the National Institutes of Health E.M.S (HL121990-01) and R.P. (HL096679).
References
- 1.Williams JC, Mackman N. Tissue factor in health and disease. Front Biosci (Elite Ed) 2012;4:358–372. doi: 10.2741/383. Epub 2011/12/29. [DOI] [PubMed] [Google Scholar]
- 2.Pawlinski R, Mackman N. Cellular sources of tissue factor in endotoxemia and sepsis. Thromb Res. 2010;125(Suppl 1):S70–S73. doi: 10.1016/j.thromres.2010.01.042. Epub 2010/02/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–264. doi: 10.1038/35025229. Epub 2000/09/23. [DOI] [PubMed] [Google Scholar]
- 4.Riewald M, Kravchenko VV, Petrovan RJ, O'Brien PJ, Brass LF, Ulevitch RJ, et al. Gene induction by coagulation factor Xa is mediated by activation of protease-activated receptor 1. Blood. 2001;97(10):3109–3116. doi: 10.1182/blood.v97.10.3109. Epub 2001/05/09. [DOI] [PubMed] [Google Scholar]
- 5.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296(5574):1880–1882. doi: 10.1126/science.1071699. Epub 2002/06/08. [DOI] [PubMed] [Google Scholar]
- 6.Camerer E, Kataoka H, Kahn M, Lease K, Coughlin SR. Genetic evidence that protease-activated receptors mediate factor Xa signaling in endothelial cells. J Biol Chem. 2002;277(18):16081–16087. doi: 10.1074/jbc.M108555200. Epub 2002/02/19. [DOI] [PubMed] [Google Scholar]
- 7.Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120(3):303–313. doi: 10.1016/j.cell.2004.12.018. Epub 2005/02/15. [DOI] [PubMed] [Google Scholar]
- 8.Austin KM, Covic L, Kuliopulos A. Matrix metalloproteases and PAR1 activation. Blood. 2013;121(3):431–439. doi: 10.1182/blood-2012-09-355958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaffre F, Friedman AE, Hu Z, Mackman N, Blaxall BC. Beta-adrenergic receptor stimulation transactivates protease-activated receptor 1 via matrix metalloproteinase 13 in cardiac cells. Circulation. 2012;125(24):2993–3003. doi: 10.1161/CIRCULATIONAHA.111.066787. Epub 2012/05/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A. 2000;97(10):5255–5260. doi: 10.1073/pnas.97.10.5255. Epub 2000/05/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rao LV, Pendurthi UR. Tissue factor-factor VIIa signaling. Arterioscler Thromb Vasc Biol. 2005;25(1):47–56. doi: 10.1161/01.ATV.0000151624.45775.13. Epub 2004/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annual review of pharmacology and toxicology. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. Epub 2005/04/12. [DOI] [PubMed] [Google Scholar]
- 13.Chien KR, Olson EN. Converging pathways and principles in heart development and disease: CV@CSH. Cell. 2002;110(2):153–162. doi: 10.1016/s0092-8674(02)00834-6. Epub 2002/08/02. [DOI] [PubMed] [Google Scholar]
- 14.Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348(20):2007–2018. doi: 10.1056/NEJMra021498. Epub 2003/05/16. [DOI] [PubMed] [Google Scholar]
- 15.Steinberg SF. The cardiovascular actions of protease-activated receptors. Molecular pharmacology. 2005;67(1):2–11. doi: 10.1124/mol.104.003103. Epub 2004/09/17. [DOI] [PubMed] [Google Scholar]
- 16.Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24(6):1015–1022. doi: 10.1161/01.ATV.0000130465.23430.74. Epub 2004/05/01. [DOI] [PubMed] [Google Scholar]
- 17.Pawlinski R, Fernandes A, Kehrle B, Pedersen B, Parry G, Erlich J, et al. Tissue factor deficiency causes cardiac fibrosis and left ventricular dysfunction. Proc Natl Acad Sci U S A. 2002;99(24):15333–15338. doi: 10.1073/pnas.242501899. Epub 2002/11/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parry GC, Erlich JH, Carmeliet P, Luther T, Mackman N. Low levels of tissue factor are compatible with development and hemostasis in mice. J Clin Invest. 1998;101(3):560–569. doi: 10.1172/JCI814. Epub 1998/03/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu H, Noria F, Sandoval-Cooper MJ, Menchen H, Donahue DL, Ploplis VA, et al. Severe deficiency of coagulation Factor VII results in spontaneous cardiac fibrosis in mice. The Journal of pathology. 2009;217(3):362–371. doi: 10.1002/path.2454. Epub 2008/11/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pawlinski R, Tencati M, Holscher T, Pedersen B, Voet T, Tilley RE, et al. Role of cardiac myocyte tissue factor in heart hemostasis. J Thromb Haemost. 2007;5(8):1693–1700. doi: 10.1111/j.1538-7836.2007.02649.x. Epub 2007/08/01. [DOI] [PubMed] [Google Scholar]
- 21.Luther T, Flossel C, Mackman N, Bierhaus A, Kasper M, Albrecht S, et al. Tissue factor expression during human and mouse development. The American journal of pathology. 1996;149(1):101–113. [PMC free article] [PubMed] [Google Scholar]
- 22.Luther T, Mackman N. Tissue factor in the heart. Multiple roles in hemostasis, thrombosis, and inflammation. Trends in cardiovascular medicine. 2001;11(8):307–312. doi: 10.1016/s1050-1738(01)00129-3. [DOI] [PubMed] [Google Scholar]
- 23.Szotowski B, Goldin-Lang P, Antoniak S, Bogdanov VY, Pathirana D, Pauschinger M, et al. Alterations in myocardial tissue factor expression and cellular localization in dilated cardiomyopathy. Journal of the American College of Cardiology. 2005;45(7):1081–1089. doi: 10.1016/j.jacc.2004.12.061. Epub 2005/04/06. [DOI] [PubMed] [Google Scholar]
- 24.Luther T, Dittert DD, Kotzsch M, Erlich J, Albrecht S, Mackman N, et al. Functional implications of tissue factor localization to cell-cell contacts in myocardium. The Journal of pathology. 2000;192(1):121–130. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH667>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 25.Rauch U. Tissue factor and cardiomyocytes. Thrombosis research. 2012;129(Suppl 2):S41–S43. doi: 10.1016/j.thromres.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 26.Pena E, Arderiu G, Badimon L. Subcellular localization of tissue factor and human coronary artery smooth muscle cell migration. Journal of thrombosis and haemostasis : JTH. 2012;10(11):2373–2382. doi: 10.1111/j.1538-7836.2012.04910.x. [DOI] [PubMed] [Google Scholar]
- 27.Ott I, Fischer EG, Miyagi Y, Mueller BM, Ruf W. A role for tissue factor in cell adhesion and migration mediated by interaction with actin-binding protein 280. The Journal of cell biology. 1998;140(5):1241–1253. doi: 10.1083/jcb.140.5.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Versteeg HH, Hoedemaeker I, Diks SH, Stam JC, Spaargaren M, van Bergen En Henegouwen PM, et al. Factor VIIa/tissue factor-induced signaling via activation of Src-like kinases, phosphatidylinositol 3-kinase, and Rac. The Journal of biological chemistry. 2000;275(37):28750–28756. doi: 10.1074/jbc.M907635199. [DOI] [PubMed] [Google Scholar]
- 29.Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-Habermann B, et al. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008;111(1):190–199. doi: 10.1182/blood-2007-07-101048. Epub 2007/09/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White HD, Chew DP. Acute myocardial infarction. Lancet. 2008;372(9638):570–584. doi: 10.1016/S0140-6736(08)61237-4. Epub 2008/08/19. [DOI] [PubMed] [Google Scholar]
- 31.Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest. 1985;76(5):1713–1719. doi: 10.1172/JCI112160. Epub 1985/11/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golino P, Ragni M, Cirillo P, Avvedimento VE, Feliciello A, Esposito N, et al. Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nat Med. 1996;2(1):35–40. doi: 10.1038/nm0196-35. Epub 1996/01/01. [DOI] [PubMed] [Google Scholar]
- 33.Erlich JH, Boyle EM, Labriola J, Kovacich JC, Santucci RA, Fearns C, et al. Inhibition of the tissue factor-thrombin pathway limits infarct size after myocardial ischemia-reperfusion injury by reducing inflammation. Am J Pathol. 2000;157(6):1849–1862. doi: 10.1016/S0002-9440(10)64824-9. Epub 2000/12/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Golino P, Ragni M, Cirillo P, Scognamiglio A, Ravera A, Buono C, et al. Recombinant human, active site-blocked factor VIIa reduces infarct size and no-reflow phenomenon in rabbits. American journal of physiology Heart and circulatory physiology. 2000;278(5):H1507–H1516. doi: 10.1152/ajpheart.2000.278.5.H1507. Epub 2000/04/25. [DOI] [PubMed] [Google Scholar]
- 35.Loubele ST, Spek CA, Leenders P, van Oerle R, Aberson HL, van der Voort D, et al. Active site inhibited factor VIIa attenuates myocardial ischemia/reperfusion injury in mice. J Thromb Haemost. 2009;7(2):290–298. doi: 10.1111/j.1538-7836.2008.03232.x. Epub 2008/11/28. [DOI] [PubMed] [Google Scholar]
- 36.Yeh CH, Chen TP, Wang YC, Fang SW, Wun TC. Potent cardioprotection from ischemia-reperfusion injury by a two-domain fusion protein comprising annexin V and Kunitz protease inhibitor. J Thromb Haemost. 2013;11(8):1454–1463. doi: 10.1111/jth.12314. Epub 2013/06/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macchi L, Moussa WB, Guillou S, Tamareille S, Lamon D, Prunier D, et al. The synthetic pentasaccharide fondaparinux attenuates myocardial ischemia-reperfusion injury in rats via STAT-3. Shock. 2014;41(2):166–171. doi: 10.1097/SHK.0000000000000072. Epub 2013/12/05. [DOI] [PubMed] [Google Scholar]
- 38.Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM, et al. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circulation. 2007;116(20):2298–2306. doi: 10.1161/CIRCULATIONAHA.107.692764. Epub 2007/10/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petzelbauer P, Zacharowski PA, Miyazaki Y, Friedl P, Wickenhauser G, Castellino FJ, et al. The fibrin-derived peptide Bbeta15-42 protects the myocardium against ischemia-reperfusion injury. Nature Medicine. 2005;11(3):298–304. doi: 10.1038/nm1198. Epub 2005/02/22. [DOI] [PubMed] [Google Scholar]
- 40.Zacharowski K, Zacharowski P, Reingruber S, Petzelbauer P. Fibrin(ogen) and its fragments in the pathophysiology and treatment of myocardial infarction. J Mol Med. 2006;84(6):469–477. doi: 10.1007/s00109-006-0051-7. Epub 2006/05/31. [DOI] [PubMed] [Google Scholar]
- 41.Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE. SCH 79797, a selective PAR1 antagonist, limits myocardial ischemia/reperfusion injury in rat hearts. Basic research in cardiology. 2007;102(4):350–358. doi: 10.1007/s00395-007-0653-4. Epub 2007/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Serio C, Pellerito S, Duarte M, Massi D, Naldini A, Cirino G, et al. Protease-activated receptor 1-selective antagonist SCH79797 inhibits cell proliferation and induces apoptosis by a protease-activated receptor 1-independent mechanism. Basic & clinical pharmacology & toxicology. 2007;101(1):63–69. doi: 10.1111/j.1742-7843.2007.00078.x. Epub 2007/06/20. [DOI] [PubMed] [Google Scholar]
- 43.Strande JL. Letter by Strande regarding article"Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy". Circulation. 2008;117(24):e495. doi: 10.1161/CIRCULATIONAHA.107.758219. author reply e6. Epub 2008/06/19. [DOI] [PubMed] [Google Scholar]
- 44.Glembotski CC, Irons CE, Krown KA, Murray SF, Sprenkle AB, Sei CA. Myocardial alpha-thrombin receptor activation induces hypertrophy and increases atrial natriuretic factor gene expression. J Biol Chem. 1993;268(27):20646–20652. Epub 1993/09/25. [PubMed] [Google Scholar]
- 45.Sabri A, Muske G, Zhang H, Pak E, Darrow A, Andrade-Gordon P, et al. Signaling properties and functions of two distinct cardiomyocyte protease-activated receptors. Circulation research. 2000;86(10):1054–1061. doi: 10.1161/01.res.86.10.1054. Epub 2000/05/29. [DOI] [PubMed] [Google Scholar]
- 46.Loubele ST, Spek CA, Leenders P, van Oerle R, Aberson HL, Hamulyak K, et al. Activated protein C protects against myocardial ischemia/ reperfusion injury via inhibition of apoptosis and inflammation. Arteriosclerosis, thrombosis, and vascular biology. 2009;29(7):1087–1092. doi: 10.1161/ATVBAHA.109.188656. Epub 2009/04/18. [DOI] [PubMed] [Google Scholar]
- 47.Antoniak S, Rojas M, Spring D, Bullard TA, Verrier ED, Blaxall BC, et al. Protease-activated receptor 2 deficiency reduces cardiac ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2010;30(11):2136–2142. doi: 10.1161/ATVBAHA.110.213280. Epub 2010/08/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antoniak S, Sparkenbaugh EM, Tencati M, Rojas M, Mackman N, Pawlinski R. Protease activated receptor-2 contributes to heart failure. PLoS One. 2013;8(11):e81733. doi: 10.1371/journal.pone.0081733. Epub 2013/12/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhattacharya K, Farwell K, Huang M, Kempuraj D, Donelan J, Papaliodis D, et al. Mast cell deficient W/Wv mice have lower serum IL-6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia-reperfusion. International journal of immunopathology and pharmacology. 2007;20(1):69–74. doi: 10.1177/039463200702000108. Epub 2007/03/10. [DOI] [PubMed] [Google Scholar]
- 50.Somasundaram P, Ren G, Nagar H, Kraemer D, Mendoza L, Michael LH, et al. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. The Journal of pathology. 2005;205(1):102–111. doi: 10.1002/path.1690. Epub 2004/12/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Napoli C, Cicala C, Wallace JL, de Nigris F, Santagada V, Caliendo G, et al. Protease-activated receptor-2 modulates myocardial ischemia-reperfusion injury in the rat heart. Proc Natl Acad Sci U S A. 2000;97(7):3678–3683. doi: 10.1073/pnas.97.7.3678. Epub 2000/03/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang R, Zatta A, Kin H, Wang N, Reeves JG, Mykytenko J, et al. PAR-2 activation at the time of reperfusion salvages myocardium via an ERK1/2 pathway in in vivo rat hearts. Am J Physiol Heart Circ Physiol. 2007;293(5):H2845–H2852. doi: 10.1152/ajpheart.00209.2007. Epub 2007/08/28. [DOI] [PubMed] [Google Scholar]
- 53.McLean PG, Aston D, Sarkar D, Ahluwalia A. Protease-activated receptor-2 activation causes EDHF-like coronary vasodilation: selective preservation in ischemia/reperfusion injury: involvement of lipoxygenase products, VR1 receptors, and C-fibers. Circ Res. 2002;90(4):465–472. doi: 10.1161/hh0402.105372. Epub 2002/03/09. [DOI] [PubMed] [Google Scholar]
- 54.Seqqat R, Rafiq K, Hanscom M, Kunapuli SP, Steinberg SF, Houser SR, et al. Protease activated receptor-4 regulates post-infarction ventricular remodeling and cardiac function. Circulation. 2007;116(16):45-. [Google Scholar]
- 55.Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE. Inhibiting protease-activated receptor 4 limits myocardial ischemia/reperfusion injury in rat hearts by unmasking adenosine signaling. J Pharmacol Exp Ther. 2008;324(3):1045–1054. doi: 10.1124/jpet.107.133595. Epub 2007/12/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Antoniak S, Pawlinski R, Mackman N. Protease-activated receptors and myocardial infarction. IUBMB life. 2011;63(6):383–389. doi: 10.1002/iub.441. Epub 2011/03/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nature reviews Drug discovery. 2010;9(5):373–386. doi: 10.1038/nrd3024. Epub 2010/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, et al. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Molecular pharmacology. 2009;76(4):791–801. doi: 10.1124/mol.109.055509. Epub 2009/07/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Betts RJ, Mann TS, Henry PJ. Inhibitory influence of the hexapeptidic sequence SLIGRL on influenza A virus infection in mice. The Journal of pharmacology and experimental therapeutics. 2012;343(3):725–735. doi: 10.1124/jpet.112.196485. Epub 2012/09/19. [DOI] [PubMed] [Google Scholar]
- 60.Shauer A, Gotsman I, Keren A, Zwas DR, Hellman Y, Durst R, et al. Acute viral myocarditis: current concepts in diagnosis and treatment. The Israel Medical Association journal : IMAJ. 2013;15(3):180–185. Epub 2013/05/15. [PubMed] [Google Scholar]
- 61.Antoniak S, Mackman N. Coagulation, protease-activated receptors, and viral myocarditis. Journal of cardiovascular translational research. 2014;7(2):203–211. doi: 10.1007/s12265-013-9515-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Antoniak S, Mackman N. Multiple roles of the coagulation protease cascade during virus infection. Blood. 2014;123(17):2605–2613. doi: 10.1182/blood-2013-09-526277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tomioka N, Kishimoto C, Matsumori A, Kawai C. Mural thrombi in mice with acute viral myocarditis. Japanese circulation journal. 1985;49(12):1277–1279. doi: 10.1253/jcj.49.1277. Epub 1985/12/01. [DOI] [PubMed] [Google Scholar]
- 64.Antoniak S, Boltzen U, Riad A, Kallwellis-Opara A, Rohde M, Dorner A, et al. Viral myocarditis and coagulopathy: increased tissue factor expression and plasma thrombogenicity. Journal of molecular and cellular cardiology. 2008;45(1):118–126. doi: 10.1016/j.yjmcc.2008.03.013. Epub 2008/05/23. [DOI] [PubMed] [Google Scholar]
- 65.Antoniak S, Boltzen U, Eisenreich A, Stellbaum C, Poller W, Schultheiss HP, et al. Regulation of cardiomyocyte full-length tissue factor expression and microparticle release under inflammatory conditions in vitro. J Thromb Haemost. 2009;7(5):871–878. doi: 10.1111/j.1538-7836.2009.03323.x. Epub 2009/02/21. [DOI] [PubMed] [Google Scholar]
- 66.Sutherland MR, Friedman HM, Pryzdial EL. Thrombin enhances herpes simplex virus infection of cells involving protease-activated receptor 1. Journal of thrombosis and haemostasis : JTH. 2007;5(5):1055–1061. doi: 10.1111/j.1538-7836.2007.02441.x. Epub 2007/04/28. [DOI] [PubMed] [Google Scholar]
- 67.Antoniak S, Owens AP, 3rd, Baunacke M, Williams JC, Lee RD, Weithauser A, et al. PAR-1 contributes to the innate immune response during viral infection. The Journal of clinical investigation. 2013;123(3):1310–1322. doi: 10.1172/JCI66125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weithauser A, Bobbert P, Antoniak S, Bohm A, Rauch BH, Klingel K, et al. Protease-activated receptor 2 regulates the Innate Immune Response to Viral Infection in a CVB3-induced Myocarditis. Journal of the American College of Cardiology. 2013;62(19):1737–1745. doi: 10.1016/j.jacc.2013.05.076. Epub 2013/07/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292(1):C82–C97. doi: 10.1152/ajpcell.00287.2006. Epub 2006/07/28. [DOI] [PubMed] [Google Scholar]
- 70.Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29(7):367–374. doi: 10.1016/j.tips.2008.05.003. Epub 2008/06/27. [DOI] [PubMed] [Google Scholar]
- 71.Varughese GI, Lip GY. Is hypertension a prothrombotic state? Current hypertension reports. 2005;7(3):168–173. doi: 10.1007/s11906-005-0005-4. [DOI] [PubMed] [Google Scholar]
- 72.Taubman MB, Marmur JD, Rosenfield CL, Guha A, Nichtberger S, Nemerson Y. Agonist-mediated tissue factor expression in cultured vascular smooth muscle cells. Role of Ca2+ mobilization and protein kinase C activation. J Clin Invest. 1993;91(2):547–552. doi: 10.1172/JCI116234. Epub 1993/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sugano T, Tsuji H, Masuda H, Nishimura H, Yoshizumi M, Kawano H, et al. Adrenomedullin inhibits angiotensin II-induced expression of tissue factor and plasminogen activator inhibitor-1 in cultured rat aortic endothelial cells. Arterioscler Thromb Vasc Biol. 2001;21(6):1078–1083. doi: 10.1161/01.atv.21.6.1078. Epub 2001/06/09. [DOI] [PubMed] [Google Scholar]
- 74.Napoleone E, Di Santo A, Camera M, Tremoli E, Lorenzet R. Angiotensin-converting enzyme inhibitors downregulate tissue factor synthesis in monocytes. Circ Res. 2000;86(2):139–143. doi: 10.1161/01.res.86.2.139. Epub 2000/02/10. [DOI] [PubMed] [Google Scholar]
- 75.Muller DN, Mervaala EM, Dechend R, Fiebeler A, Park JK, Schmidt F, et al. Angiotensin II (AT(1)) receptor blockade reduces vascular tissue factor in angiotensin II-induced cardiac vasculopathy. Am J Pathol. 2000;157(1):111–122. doi: 10.1016/S0002-9440(10)64523-3. Epub 2000/07/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koh KK, Chung WJ, Ahn JY, Han SH, Kang WC, Seo YH, et al. Angiotensin II type 1 receptor blockers reduce tissue factor activity and plasminogen activator inhibitor type-1 antigen in hypertensive patients: a randomized, double-blind, placebo-controlled study. Atherosclerosis. 2004;177(1):155–160. doi: 10.1016/j.atherosclerosis.2004.07.008. Epub 2004/10/19. [DOI] [PubMed] [Google Scholar]
- 77.Matej R, Housa D, Pouckova P, Zadinova M, Olejar T. Radiation-induced production of PAR-1 and TGF-beta 1 mRNA in lung of C57Bl6 and C3H murine strains and influence of pharmacoprophylaxis by ACE inhibitors. Pathology, research and practice. 2007;203(2):107–114. doi: 10.1016/j.prp.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 78.McGuire JJ, Van Vliet BN, Halfyard SJ. Blood pressures, heart rate and locomotor activity during salt loading and angiotensin II infusion in protease-activated receptor 2 (PAR2) knockout mice. BMC physiology. 2008;8:20. doi: 10.1186/1472-6793-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hughes KH, Wijekoon EP, Valcour JE, Chia EW, McGuire JJ. Effects of chronic in-vivo treatments with protease-activated receptor 2 agonist on endothelium function and blood pressures in mice. Canadian journal of physiology and pharmacology. 2013;91(4):295–305. doi: 10.1139/cjpp-2012-0266. [DOI] [PubMed] [Google Scholar]
- 80.Chia E, Kagota S, Wijekoon EP, McGuire JJ. Protection of protease-activated receptor 2 mediated vasodilatation against angiotensin II-induced vascular dysfunction in mice. BMC pharmacology. 2011;11:10. doi: 10.1186/1471-2210-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pinelli A, Trivulzio S, Tomasoni L, Bertolini B, Brenna S, Bonacina E, et al. Myocardial infarction non-invasively induced in rabbits by administering isoproterenol and vasopressin: protective effects exerted by verapamil. Fundamental & clinical pharmacology. 2004;18(6):657–667. doi: 10.1111/j.1472-8206.2004.00296.x. [DOI] [PubMed] [Google Scholar]
- 82.Sato S, Ito Y, Kondo M, Ohashi T, Ito S, Nakayama S, et al. Ion transport regulated by protease-activated receptor 2 in human airway Calu-3 epithelia. British journal of pharmacology. 2005;146(3):397–407. doi: 10.1038/sj.bjp.0706330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension. 2009;53(6):1041–1047. doi: 10.1161/HYPERTENSIONAHA.108.123158. [DOI] [PubMed] [Google Scholar]
- 84.Miura S, Ohno I, Suzuki J, Suzuki K, Okada S, Okuyama A, et al. Inhibition of matrix metalloproteinases prevents cardiac hypertrophy induced by beta-adrenergic stimulation in rats. Journal of cardiovascular pharmacology. 2003;42(2):174–181. doi: 10.1097/00005344-200308000-00004. [DOI] [PubMed] [Google Scholar]
- 85.Bakouboula B, Morel O, Faure A, Zobairi F, Jesel L, Trinh A, et al. Procoagulant membrane microparticles correlate with the severity of pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2008;177(5):536–543. doi: 10.1164/rccm.200706-840OC. Epub 2007/11/17. [DOI] [PubMed] [Google Scholar]
- 86.Berger G, Azzam ZS, Hoffman R, Yigla M. Coagulation and anticoagulation in pulmonary arterial hypertension. The Israel Medical Association journal : IMAJ. 2009;11(6):376–379. Epub 2009/08/25. [PubMed] [Google Scholar]
- 87.Altman R, Scazziota A, Rouvier J, Gurfinkel E, Favaloro R, Perrone S, et al. Coagulation and fibrinolytic parameters in patients with pulmonary hypertension. Clinical cardiology. 1996;19(7):549–554. doi: 10.1002/clc.4960190706. Epub 1996/07/01. [DOI] [PubMed] [Google Scholar]
- 88.White RJ, Meoli DF, Swarthout RF, Kallop DY, Galaria II, Harvey JL, et al. Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. American journal of physiology Lung cellular and molecular physiology. 2007;293(3):L583–L590. doi: 10.1152/ajplung.00321.2006. Epub 2007/06/26. [DOI] [PubMed] [Google Scholar]
- 89.White RJ, Galaria II, Harvey J, Blaxall BC, Cool CD, Taubman MB. Tissue factor is induced in a rodent model of severe pulmonary hypertension characterized by neointimal lesions typical of human disease. Chest. 2005;128(6 Suppl):612S–613S. doi: 10.1378/chest.128.6_suppl.612S-a. Epub 2005/12/24. [DOI] [PubMed] [Google Scholar]
- 90.Bauer EM, Zheng H, Comhair S, Erzurum S, Billiar TR, Bauer PM. Complement C3 deficiency attenuates chronic hypoxia-induced pulmonary hypertension in mice. PloS one. 2011;6(12):e28578. doi: 10.1371/journal.pone.0028578. Epub 2011/12/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Delbeck M, Nickel KF, Perzborn E, Ellinghaus P, Strassburger J, Kast R, et al. A role for coagulation factor Xa in experimental pulmonary arterial hypertension. Cardiovascular research. 2011;92(1):159–168. doi: 10.1093/cvr/cvr168. Epub 2011/06/17. [DOI] [PubMed] [Google Scholar]
- 92.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):13S–24S. doi: 10.1016/j.jacc.2004.02.029. Epub 2004/06/15. [DOI] [PubMed] [Google Scholar]
- 93.Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004;68(2):75–103. doi: 10.1016/j.mvr.2004.06.001. Epub 2004/08/18. [DOI] [PubMed] [Google Scholar]
- 94.White TA, Witt TA, Pan S, Mueske CS, Kleppe LS, Holroyd EW, et al. Tissue factor pathway inhibitor overexpression inhibits hypoxia-induced pulmonary hypertension. American journal of respiratory cell and molecular biology. 2010;43(1):35–45. doi: 10.1165/rcmb.2009-0144OC. Epub 2009/08/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Delbeck M, Nickel KF, Perzborn E, Ellinghaus P, Strassburger J, Kast R, et al. A role for coagulation factor Xa in experimental pulmonary arterial hypertension. Cardiovasc Res. 2011;92(1):159–168. doi: 10.1093/cvr/cvr168. Epub 2011/06/17. [DOI] [PubMed] [Google Scholar]
- 96.Kwapiszewska G, Markart P, Dahal BK, Kojonazarov B, Marsh LM, Schermuly RT, et al. PAR-2 inhibition reverses experimental pulmonary hypertension. Circulation research. 2012;110(9):1179–1191. doi: 10.1161/CIRCRESAHA.111.257568. Epub 2012/03/31. [DOI] [PubMed] [Google Scholar]
- 97.Farber HW, Loscalzo J. Pulmonary arterial hypertension. The New England journal of medicine. 2004;351(16):1655–1665. doi: 10.1056/NEJMra035488. Epub 2004/10/16. [DOI] [PubMed] [Google Scholar]
- 98.Nickel KF, Laux V, Heumann R, von Degenfeld G. Thrombin has biphasic effects on the nitric oxide-cGMP pathway in endothelial cells and contributes to experimental pulmonary hypertension. PloS one. 2013;8(6):e63504. doi: 10.1371/journal.pone.0063504. Epub 2013/06/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maki J, Hirano M, Hoka S, Kanaide H, Hirano K. Involvement of reactive oxygen species in thrombin-induced pulmonary vasoconstriction. Am J Respir Crit Care Med. 2010;182(11):1435–1444. doi: 10.1164/rccm.201002-0255OC. Epub 2010/07/20. [DOI] [PubMed] [Google Scholar]
- 100.Maki J, Hirano M, Hoka S, Kanaide H, Hirano K. Thrombin activation of proteinase-activated receptor 1 potentiates the myofilament Ca2+ sensitivity and induces vasoconstriction in porcine pulmonary arteries. Br J Pharmacol. 2010;159(4):919–927. doi: 10.1111/j.1476-5381.2009.00591.x. Epub 2010/02/05. [DOI] [PMC free article] [PubMed] [Google Scholar]