

Abstract
We present LuciPHOr2, a site localization tool for generic post-translational modifications (PTMs) using tandem mass spectrometry data. As an extension of the original LuciPHOr (version 1) for phosphorylation site localization, the new software provides a site-level localization score for generic PTMs and associated false discovery rate called the false localization rate. We describe several novel features such as operating system independence and reduced computation time through multiple threading. We also discuss optimal parameters for different types of data and illustrate the new tool on a human skeletal muscle dataset for lysine-acetylation.
Availability and implementation: The software is freely available on the SourceForge website http://luciphor2.sourceforge.net.
Contact: hyung_won_choi@nuhs.edu.sg, nesvi@med.umich.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Post-translational modifications (PTMs) induce conformational changes in proteins that influence their enzymatic activity, subcellular localization and/or their binding affinity (Hoffman et al., 2008; Silva et al., 2013). High-throughput analysis of PTMs is typically achieved through mass spectrometry (MS) (Silva et al., 2013), where tandem (MS/MS) mass spectra are searched against a sequence database allowing for PTMs. A major challenge in the interpretation of MS/MS data for PTMs is that database search engines can correctly match a peptide sequence with incorrectly localized PTM(s). These errors are not controlled by the type I error of peptide identification because each peptide has a different degree of uncertainty as far as site localization is concerned. For this reason, the conventional target-decoy framework often leads to underestimation of the error rates for site localization of PTMs (Fermin et al., 2013). Hence, an additional procedure to control the number of false site localizations is necessary when reporting PTM localization analysis results at the proteome scale.
2 Functional features
2.1 Improved implementation
To address this issue, we recently published a computational method called LuciPHOr for site localization analysis of phosphorylation events using MS/MS data (Fermin et al., 2013). LuciPHOr was first implemented with a novel target-decoy framework specifically designed for phospho-site localization. Here, we have converted this tool into a general PTM localization program (LuciPHOr2) for estimating false localization rate (FLR) for any PTMs of a fixed mass. The new implementation provides enhancements over the previous version: (i) it is written entirely in Java, making it operating system independent; (ii) the software now processes search results from any proteomics search engine.
2.2. Key parameters for site localization
In LuciPHOr2, the user is expected to specify key parameters for statistical modeling, which determine inclusion/exclusion of MS/MS peaks into the scoring algorithm. LuciPHOr2 constructs a probability model of peak intensity and mass accuracy across all spectra for correct and incorrect localizations respectively, and computes the likelihood ratio as the localization score for each candidate site. This requires an appropriate choice of low/high mass accuracy model (ALGORITHM parameter), fragment mass tolerance (MS2_TOL) and the criteria for selecting high confidence localizations to train the probability model (SELECTION_METHOD).
Two additional parameters are worth mentioning in this new version: the target modification and the mass shift of the PTM of interest. Since LuciPHOr2 was designed to score generic PTMs, the user must specify what amino acids should be treated as potential modification sites along with their respective mass shifts. The mass shift is required for the program to place decoy PTMs on arbitrary sites (non-specific residues) and accurately estimate the FLR.
To facilitate the optimal configuration, LuciPHOr2 generates a template input file that the user can edit for their specific needs. However, we note that the default parameters generated in this file are optimized for phosphorylation analysis.
3 Application to generic PTM analysis
We demonstrate the program through the analysis of a published dataset for lysine acetylation in 12 human skeletal muscle biopsies (Lundby et al., 2012). The RAW files were converted into the open-source format mzML using MSConvert from the ProteoWizard package (Kessner et al., 2008), and searched with X!Tandem (Fenyo and Beavis, 2003). The database search was performed as described by Lundby et al. with two exceptions: the precursor tolerance was increased to 20 ppm and up to five missed cleavages were allowed. The peptide search results were post-processed using PeptideProphet of the TPP (Deutsch et al., 2010; Keller et al., 2002). All peptide-to-spectrum matches (PSMs) with a lysine acetylation event were reanalyzed with LuciPHOr2 configured for lysine-acetylations (K+42.01056).
A total of 35 482 PSMs were reported with acetylation by X!Tandem/TPP. To filter high-quality spectra, 30 602 PSMs within the 1% FDR threshold (PeptideProphet probability 0.809) were selected, and among these 20 754 PSMs had site ambiguity (68%). As expected for a non-labile modification, X!Tandem/TPP and LuciPHOr2 showed concordant site localization on the majority of ambiguous cases (19 557/20 754, 94%). For a non-negligible number of PSMs (673/19 557, 3.4%), however, LuciPHOr2 delta score was <3 between the top two localizations, indicating that those sites were localized based on merely a single fragment ion or less credible low intensity fragments, implying considerable ambiguity.
On the other hand, LuciPHOr2 and X!Tandem/TPP disagreed on 1197 site localizations, consisting of two major types. The first type were 987 PSMs with the ambiguity on two consecutive lysines at the end of the peptide sequences (e.g. VLTLELYKK), whereas the second type were 210 PSMs with the ambiguity in the middle of the sequences (e.g. DNKKPFIKEAAMAK). In nearly all of the latter cases, there was no major site discriminating fragment ion. This is illustrated in Figure 1a, where only a few minor peaks support both candidate sites (Supplemental Table S1).
Fig. 1.
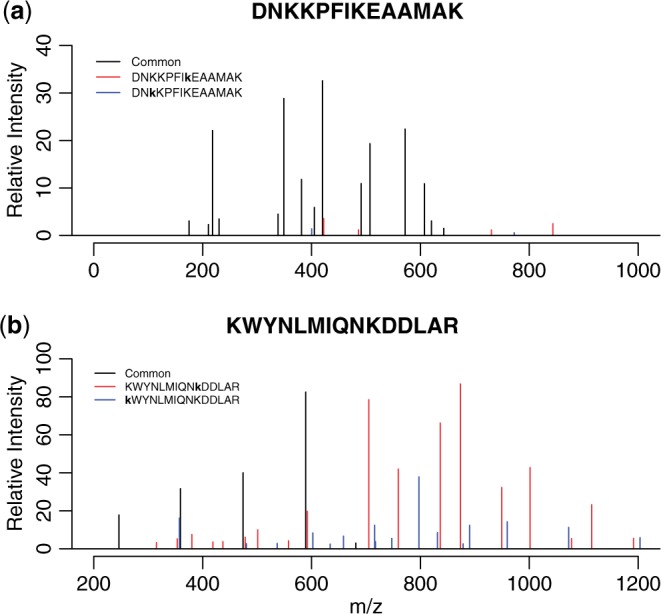
MS/MS spectra with site determining ions for top two site localizations (matched peaks only). Black peaks are shard ions, while red and blue ones are the site determining ions for each site localization. The two site localizations are shown in the text in the upper left corner of each panel (lower case indicates PTM sites). (a) Peptide with a low score due to lack of clear site determining ions. (b) Peptide with a high score with evidence for both localizations, indicating the possibility of co-eluting positional isomers
Another advantage of LuciPHOr2 is that it produces a peak-by-peak score report for the two best site localizations, which can assist identification of positional isomers. The term ‘positional isomers’ refers to two co-eluting species of the same peptide with different site localizations (Courcelles et al., 2012), often captured in the same MS/MS spectrum. To find these, the peak score report can be queried for the peptides with high likelihood scores (e.g. >30 for low mass accuracy data, >50 for high mass accuracy data) with a sufficient number of site determining ions for each localization. Figure 1b shows the spectrum for KWYNLMIQNKDDLAR, which has 18 and 16 site determining ions for the two lysines (Supplemental Table S1). This example was easily queried with the following criteria: the top two localizations score above 50 on the same spectrum and have more than five site determining ions.
4 Conclusions
In sum, the post hoc analysis of site localization of PTMs using the database search output is an essential step toward the control of type I errors in large-scale PTM analysis. The LuciPHOr2 package facilitates such analysis using the peptide identification from various database search engines, and is expected to contribute to more robust analysis of proteome-level PTM analysis in a wide range of applications.
Funding
This work was supported by Singapore MOE [grant R-608-000-088-112 to H.C.] and NIH [R01-GM-094231 to A.I.N.].
Conflict of Interest: none declared.
Supplementary Material
References
- Courcelles M., et al. (2012) Occurrence and detection of phosphopeptide isomers in large-scale phosphoproteomics experiments. J. Proteome Res., 6, 3753–3765. [DOI] [PubMed] [Google Scholar]
- Deutsch E.W., et al. (2010) A guided tour of the trans-proteomic pipeline. Proteomics, 10, 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenyo D., Beavis R.C. (2003) A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem., 75, 768–774. [DOI] [PubMed] [Google Scholar]
- Fermin D., et al. (2013) LuciPHOr: algorithm for phosphorylation site localization with false localization rate estimation using modified target-decoy approach. Mol. Cell. Proteomics, 12, 3409–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman M.D., et al. (2008) Current approaches for global post-translational modification discovery and mass spectrometric analysis. Anal. Chim. Acta, 627, 50–61. [DOI] [PubMed] [Google Scholar]
- Keller A., et al. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem., 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Kessner D., et al. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics, 24, 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A., et al. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep., 2, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A.M., et al. (2013) Post-translational modifications and mass spectrometry detection. Free Radical Biol. Med., 65, 925–941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.