

Abstract
New chemotherapeutic agents are urgently required to combat the global spread of multi-drug resistant tuberculosis (MDR-TB). The mycobacterial enoyl reductase, InhA, is one of the few clinically-validated targets in tuberculosis drug discovery. Here, we report the identification of a new class of direct InhA inhibitors, the 4-hydroxy-2-pyridones, using phenotypic high-throughput whole-cell screening. This class of orally-active compounds showed potent bactericidal activity against common isoniazid-resistant TB clinical isolates. Biophysical studies revealed that 4-hydroxy-2-pyridones bound specifically to InhA in an NADH-dependent manner and blocked the enoyl-substrate binding pocket. The lead compound NITD-916 directly blocked InhA in a dose-dependent manner and showed in vivo efficacy in acute and established mouse models of infection by Mycobacterium tuberculosis. Collectively, our structural and biochemical data open up new avenues for rational structure-guided optimization of the 4-hydroxy-2-pyridone class of compounds for the treatment of MDR-TB.
Introduction
Tuberculosis (TB) infections caused by Mycobacterium tuberculosis (Mtb) continue to be a major public health threat, particularly in the developing world. Resistance to multiple drugs together with the human immunodeficiency virus (HIV) have created new challenges in the management of TB. In 2012, around 8.6 million people developed TB including ∼400,000 who had multi-drug resistant TB (MDR-TB), with 1.3 million deaths (1). Globally ∼4% of newly diagnosed TB cases and 20% of those previously treated for TB have MDR-TB (1). Hence, there is an immediate need to address the growing problem of clinical drug resistance with new therapeutic entities active against Mtb. Despite some recent successes with several new chemical entities (2), the high attrition rate in drug development and clinical testing requires continued efforts to find better drugs.
Inhibition of the mycobacterial enoyl-reductase InhA is one of the most effective means of killing Mtb, as clinically demonstrated by isoniazid, the most potent TB drug. Unfortunately, both multi-drug and extensively-drug resistant (XDR) Mtb isolates are resistant to isoniazid, predominantly due to mutations in KatG, the catalase-peroxidase involved in the activation of isoniazid (3). This has led to extensive efforts to identify direct InhA inhibitors (4-7). Over the last two decades, these efforts have yielded many potent structurally-diverse direct InhA inhibitors but so far with limited success in achieving an orally active candidate with in vivo efficacy. Here, we report the identification of a new class of small-molecule mycobactericidal agents, the 4-hydroxy-2-pyridones, using phenotypic screening. These compounds blocked the target InhA without requiring bio-activation. The lead candidate, NITD-916, showed in vivo efficacy and was active against common MDR-TB clinical isolates. Our results suggest that the 4-hydroxy-2-pyridones are an attractive candidate for lead optimization in the quest for new drugs to treat TB.
Results
Identification of 4-hydroxy-2-pyridones and microbiological profiling
A whole-cell high-throughput screen of the ∼2.3 million Novartis compound collection against Mtb H37Ra, resulted in 20,000 hits with activity > 50% inhibition at 12.5 μM concentration. Promiscuous pan-active compounds (8), scaffolds of known anti-TB compounds, cytotoxic compounds against mammalian cells (Huh7 or HepG2), compounds containing undesirable functional groups and compounds with MW > 500, clogP < 1 or > 4 were deprioritized, resulting in one of the hits NITD-529, a new anti-TB compound (Fig. 1A). NITD-529, 4-hydroxy-6-isobutyl-3-phenylpyridin-2(1H)-one, is a small and polar molecule with moderate activity against Mtb H37Rv (MIC50 1.5 μM) and good solubility (Table S1). Structure-activity-relationship studies with several 4-hydroxy-2-pyridone analogues (9, 10) revealed the importance of the pyridone core, 4-hydroxy group and R6 lipophilic group (Fig. 1A) for Mtb activity which led to the identification of NITD-564 and NITD-916 (Fig. 1A). NITD-916, a dimethylcyclohexyl derivative at the R6 position, is 30× more potent than the initial screening hit NITD-529. The anti-TB activity of NITD-916 is 5-8 times more potent than isoniazid (MIC50, 0.33 μM) and PA-824 (MIC50, 0.4 μM) (11), and is comparable to bedaquiline (MIC50, 50 nM) (12). 4-hydroxy-2-pyridone analogues showed both concentration- and time-dependent bactericidal activity against in vitro replicating Mtb and were also active against Mtb within macrophages (Fig. 1B and 1C). The in vitro cidal-activity profile of NITD-916 showed rapid killing at concentrations greater than 0.2 μM, similar to isoniazid at 0.5 μM. Viable bacterial counts with isoniazid treatment increased from day 3 to 5, potentially due to the emergence of resistance. However, no such increase in bacterial counts was observed with 4-hydroxy-2-pyridone analogues, possibly suggesting lower mutation frequency. 4-hydroxy-2-pyridones were also shown to be active against both slow-growing (Mtb, M. bovis BCG) and fast-growing (M. smegmatis) mycobacterial species (Table S2). They showed a narrow spectrum of antibacterial activity, with no activity against tested Gram-positive and Gram-negative bacterial species (Table S2). The anti-mycobacterial activity of 4-hydroxy-2-pyridones was restricted to actively-replicating Mtb; they were not active in the Wayne model for non-replicating persistent hypoxic Mtb (Table S2), implying that these molecules block an essential step in active metabolism of Mtb.
Fig. 1. Chemical structures of 4-hydroxy-2pyridones and in vitro and in vivo antimycobacterial activity of these compounds.
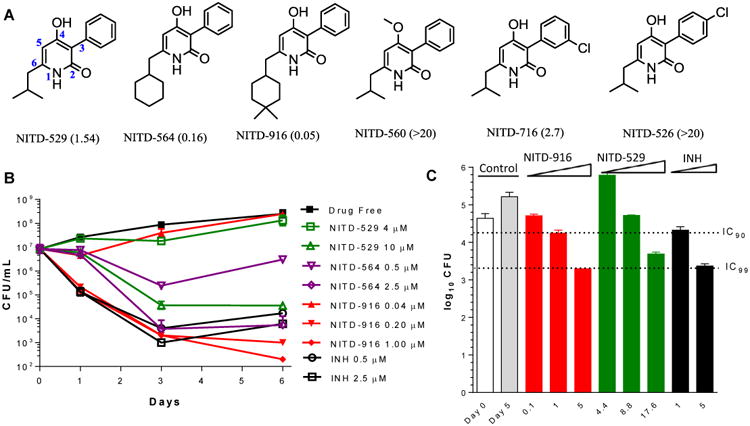
(A) Chemical structures of 4-hydroxy-2-pyridone analogs used in this study. MIC50 values against Mtb are given in parentheses (μM). R1-6 numbering is shown in the NITD-529 structure. (B) The concentration dependent bactericidal kill-kinetics activity of NITD-529 and NITD-916 against in vitro replicating Mtb, and compared with isoniazid. (C) Concentration dependent activity of NITD-916, NITD-529 and isoniazid against Mtb in intracellular activated THP-1 macrophages with five days drug exposure. IC90 and IC99 values are indicated by stippled lines. Both kill kinetic and intra-macrophage analysis were performed in biological replicates (n = 2) and results are shown as mean values with standard errors.
4-hydroxy-2-pyridones are also active against six different clinical MDR-TB isolates that are distributed into five prominent clusters representing global populations of Mtb strains (13). The minimum concentration (MIC) required to inhibit 99% growth of the diverse drug-resistant clinical isolates (MDR 1 to 6) by NITD-529, NITD-564 and NITD-916 was in a similar range to that needed to inhibit 99% growth of wild-type Mtb H37Rv (Table 1). The MIC activity of NITD-916 against the MDR-Mtb strains ranged from 0.04 to 0.16 μM, demonstrating the potential of 4-hydroxy-2-pyridones for use against MDR-TB strains.
Table 1.
Activity of 4-hydroxy-2-pyridone analogues against a panel of drug-resistant TB clinical isolates.
| MIC99 (μM) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Strains | Cluster* | Resistance# | Genotype | NITD-529 | NITD-564 | NITD-916 | M | R | I | S |
| MDR1 | I | SIR | - | 2.50 | 0.16 | 0.04 | 0.31 | >5 | >20 | > 5 |
| MDR2 | II | SIR | - | 2.50 | 0.31 | 0.16 | 0.16 | >5 | 10 | > 5 |
| MDR3 | II | SIRM PZA | rpoB (S531L), katG (S315T), rpsL (43AGC), pncA (11ups,A>G) | 3.13 | 0.16 | 0.08 | >5 | >5 | >20 | > 5 |
| MDR4 | IIA | SIR PZA | rpoB (S531L), katG (S315T), pncA (G132R) | 2.50 | 0.31 | 0.04 | 0.16 | >5 | >20 | > 5 |
| MDR5 | III | SI | - | 2.50 | 0.16 | 0.08 | 0.31 | 0.02 | >20 | 2.50 |
| MDR6 | VI | SIR PZA | rpoB (V176F), katG (S315T), pnkΔCCA(P69) | 2.50 | 0.31 | 0.16 | 0.16 | >5 | >20 | > 5 |
| MDR isolate with inhA mutation | - | IR | inhA (I194T) | 20 | 2.5 | 1.25 | > 20 | 0.63 | ||
|
| ||||||||||
| H37Rv | VIII | None (Wild type) | - | 2.50 | 0.31 | 0.08 | 0.16 | 0.02 | 0.62 | 0.31 |
Note:
S, Streptomycin; I, Isoniazid; R, Rifampicin; PZA, Pyrazinamide; M, Moxifloxacin
Clustering is based on Gutacker et al., 2006 (13)
Identification and validation of the molecular target
In an attempt to identify the molecular target of 4-hydroxy-2-pyridones, we isolated and characterized mutants that were spontaneously resistant to NITD-529. The observed frequency of resistance in Mtb H37Rv against a 10×MIC of NITD-529 and NITD-916 was 1 × 10−8, which was nearly two log orders lower than that for isoniazid (Table 2). Five independent Mtb-resistant mutants selected against NITD-529 were at least 25-fold more resistant to NITD-529 than the parental H37Rv strain and also were cross-resistant to both NITD-564 and NITD-916 (Table 2). All of these mutants were equally sensitive to streptomycin. Similarly, the frequency of resistance of M. bovis BCG to NITD-916 was 1 × 10−8 and NITD916-resistant mutants retained full sensitivity to streptomycin (Table S3). Surprisingly, one of the Mtb pyridone-resistant mutants (529-B2) and two from M. bovis BCG (529-2 and 916-B1), showed significant (MIC50 >3 fold) resistance to both isoniazid and ethionamide.
Table 2.
Cross-resistance and whole genome sequencing analysis of 4-hydroxy-2-pyridone resistance mutants of Mtb.
| Strains | inhA genotype | Catalase phenotype* | Compound MIC50 (μM) | |||||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| NITD-529 | NITD-564 | NITD-916 | I | E | S | |||
| Mutation frequency | - | - | 1.2×10-8 | - | 1.0×10-8 | ∼ 1× 10-6 | - | - |
|
| ||||||||
| H37Rv WT | WT | + | 1.54 | 0.16 | 0.05 | 0.25 | 1.66 | 0.08 |
| 529-B2 | tcg to gcg S94A | + | > 40 | 4.04 | 0.78 | 0.86 | 9.74 | 0.06 |
| 529-S3 | gac to ggc D148G | + | > 40 | 1.73 | 0.38 | 0.15 | - | 0.08 |
| 529-B4 | ggg to gtg G96V | + | > 40 | 14.60 | > 5.0 | 0.09 | 1.32 | 0.1 |
| 529-B6 | - | + | > 40 | > 40 | > 5.0 | 0.11 | 1.41 | 0.09 |
| 529-B8 | gac to gaa D148E | + | > 40 | > 40 | > 5.0 | 0.31 | 1.91 | 0.08 |
| H37Rv∷pMV262-InhA | inhA∷inhA | nd | 2.59 (1.7×) | - | 0.108 (2.2×) | 0.51 (2.04×) | - | 0.074 |
| H37Rv∷pMV262-InhA∷D148G | inhA∷inhAD148G | nd | > 60 (> 38×) | - | 3.08 (60×) | - | - | 0.084 |
| H37Rv∷pMV262-InhA∷D148E | inhA∷inhAD148E | nd | > 60 (> 38×) | - | > 6.6 (> 100×) | - | - | 0.079 |
| H37Rv∷pMV262-InhA∷S94A | inhA∷inhAS94A | nd | 28.71 (18 ×) | - | 2.40 (48×) | - | - | 0.075 |
In genetically complemented strains, MIC50 fold shift with the Mtb H37Rv strain is given in parentheses,
+ ve for catalase activity; nd – not determined. I, Isoniazid, E, Ethionamide, S, Streptomycin.
To further elucidate the genetic basis of the action and resistance of 4-hydroxy-2-pyridones, whole-genome sequencing (WGS) of three Mtb-mutant strains was carried-out. Whole-genome sequencing revealed two independent single-nucleotide polymorphisms in the inhA gene encoding the NADH-dependent enoyl-ACP (acyl carrier protein) reductase compared to the parental strain Mtb H37Rv (Table 2). In the 529-B2 mutant, a missense mutation encoding S94A in InhA was observed, whereas 529-S1 and 529-S3 mutants shared a common D148G mutation in InhA. In addition, sequencing of inhA in the remaining Mtb and BCG mutants revealed more missense mutations i.e., G96A, D148E, M161I, M161A and T17A (Table 2 and Table S3). To genetically validate InhA as the molecular target of NITD-916, we overexpressed wild-type or mutated copies of inhA under the control of the hsp60 promoter on a non-integrative plasmid pMV262. Overexpression of mutated copies of InhA (D148G, D148E or S94A) in wild-type Mtb H37Rv resulted in > 10-fold shift in MIC, whereas overexpression of wild-type InhA resulted in a marginal 1.7-2.2-fold shift in MIC for both pyridones and isoniazid (Table 2). Thus, resistance-conferring mutations in InhA seem to exert a dominant effect over the wild-type protein. None of the mutations affected the MIC for streptomycin. Together, these data suggested that a mutation in InhA may be responsible for resistance to the 4-hydroxy-2-pyridones.
InhA is an essential component of the FAS-II (fatty acid synthase-II) complex and is necessary for mycolic acid biosynthesis. Mycolic acids are long-chain fatty acids (C60-80) that are major constituents of a mycobacterial cell wall (14). The mycobacterial FAS-II pathway differs significantly from the mammalian FAS-I pathway, which utilizes a multienzyme complex, in contrast to the distinct enzymes that separately accomplish each step of the bacterial acyl-chain elongation cycle. In the last step, the double bond of the enoyl-ACP is reduced to acyl-ACP by InhA. InhA is a well-known clinically-validated target of the TB drugs isoniazid and ethionamide (6). Isoniazid is a pro-drug activated by the mycobacterial catalase-peroxidase enzyme (KatG) to its acyl-radical form, which reacts with NAD to form an adduct that inhibits InhA (15, 16). Interestingly, all of the tested 4-hydroxy-2-pyridone resistant mutants showed a catalase-positive phenotype and all the MDR-TB clinical isolates tested that contained a mutation in katG were fully sensitive to 4-hydroxy-2-pyridones (Table 1). All five InhA mutations that conferred pyridone resistance were highly conserved across mycobacterial species (Fig. S1), among them only S94 and M161 showed low-level cross-resistance to isoniazid and ethionamide; the other mutations (T17, G96 and D148) remained fully sensitive to both drugs (Table 2 and Table S3). Interestingly, an MDR-TB clinical isolate with a mutation in InhA∷I194T was resistant not only to isoniazid but also to 4-hydroxy-2-pyridones (Table 2).
It is well established that inhibition of Mtb InhA by isoniazid results in the depletion of mycolic acids from the cell wall with a concomitant accumulation of fatty acids (17). [14C] acetate metabolic-labeling experiments revealed that the treatment of mycobacterial cells with NITD-529 and NITD-916 resulted in a dose-dependent inhibition of mycolates, with a concomitant accumulation of fatty acids (Fig. 2A, 2B and fig. S2). However, no significant changes in the lipid profiles were observed with 4-hydroxy-2-pyridone resistant mutants of Mtb (Fig. 2 and fig. S2). Taken together, our data suggests that NITD-916 potentially targets InhA and that its mechanism of action is independent of katG activation.
Fig. 2. Mechanism of action of pyridones.
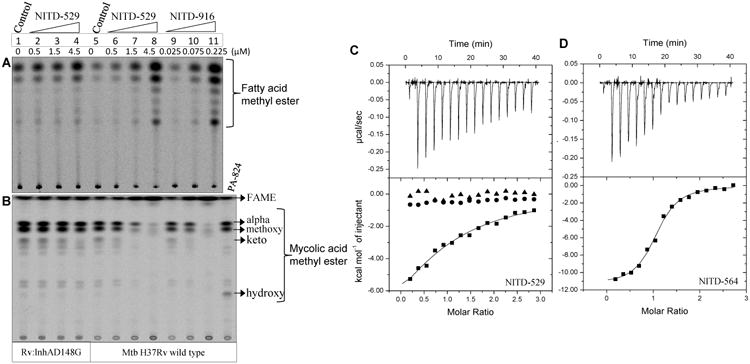
Fatty acid (A) and mycolic acid (B) lipid profiles of Mtb after exposure to 4-hydroxy-2-pyridones. Mycolic acid/fatty acid methyl esters prepared after [14C]acetate metabolic labelling and analyzed by thin layer chromatographic and phosphor-imaging. Isothermal titration calorimetry binding of NITD-529 (C) and NITD-564 (D) to apo-InhA (triangle), the InhA-NAD+ complex (circles), and the InhA-NADH complex (squares).
In vitro inhibition and biophysical interaction of 4-hydroxy-2-pyridones with InhA
To confirm that InhA is the molecular target of 4-hydroxy-2-pyridones, enzyme-inhibition studies in biochemical assays were carried out. In these experiments, NITD-529 (MIC 1.54 μM) and NITD-564 (MIC 0.16 μM) inhibited the InhA enzyme activity with an IC50 of 9.60 μM and 0.59 μM, respectively (Table 3). Although NITD-916 showed ∼3× more cellular potency than NITD-564, no shift in the enzyme IC50 was observed, potentially due to the difference in cell permeability as NITD-916 has more than a log unit higher logP than NITD-564. The 4-methoxy cell-inactive analogue (NITD-560, MIC > 20 μM), was also inactive against the InhA enzyme (Fig. 1A and Table 3). Similarly, a para-chloro substitution on the R3 position (NITD-520, MIC >20 μM) was also inactive against the InhA enzyme, unlike a meta-chloro substitution (NITD-716) (Table 3). Overall, with a limited set of compounds, the InhA enzyme inhibition by 4-hydroxy-2-pyridones correlated with their Mtb MIC. In vitro, the InhA enzyme IC50 of 4-hydroxy-2-pyridone analogues were 4-10 fold higher than Mtb cellular activity. The difference in InhA and cellular IC50 may be due to intracellular accumulation, differential sensitivity of InhA between in vitro and in vivo conditions and also potential direct or indirect effects including secondary molecular targets of 4-hydroxy-2-pyridones inside the cells.
Table 3. Interaction of 4-hydroxy-2-pyridones with InhA.
| Compound | Mtb MIC50 (μM) | InhA IC50 (μM) | Thermaflor assay (Tm °C) | ITC (+NADH) | |||||
|---|---|---|---|---|---|---|---|---|---|
| E-NAD+-I | E-NADH-I | ΔTm | Kd (μM) | ΔH (cal.mol-1) | ΔS (cal.mol-1) | N | |||
| NITD-529 | 1.54 | 9.6±0.7 | 50.3 | 54.7 | 4.4 | 25 ± 7 | 13100 ± 4202 | -22.9 | 0.97 ± 0.23 |
| NITD-564 | 0.16 | 0.59±0.05 | 56.3 | 57.8 | 1.5 | 0.56 ± 0.07 | 11440 ± 250 | -9.8 | 1.04 ± 0.02 |
| NITD-916 | 0.05 | 0.57±0.04 | 55 | 56.3 | 1.3 | - | - | - | - |
| NITD-560 | > 20 | > 20 | - | - | - | - | - | - | - |
| NITD-716 | 2.67 | 4.3±0.5 | - | - | - | - | - | - | - |
| NITD-526 | >20 | > 20 | - | - | - | - | - | - | - |
| PT70a | 11a | <0.05a | 66.7 | 60.1 | -6.6 | - | - | - | - |
| PT166b | 10.5b | <0.05b | 52.3 | 71.5 | 19.2 | - | - | - | - |
Isothermal titration calorimetry (ITC) revealed that NITD-529 bound to InhA only in the presence of NADH and no binding was observed in the presence of NAD or without any cofactor (Fig. 2C, D). NITD-529 and NITD-564 bound to the InhA-NADH complex with a Kd of 25 and 0.56 μM, respectively (Fig. 2, Table 3). Compound binding to NADH fits well with one binding site per monomer. Differential scanning fluorimetry also confirmed that 4-hydroxy-2-pyridones preferentially bound to the InhA-NADH complex similar to PT166 (18) and in contrast to PT70 (19), a diphenyl ether derivative which forms a stable ternary complex with NAD+ (Table 3). NITD-529 binding to the InhA-NADH complex led to a modest increase (ΔTm + 4.4 °C) in thermal stability compared to binding to the InhA-NAD+ complex; in contrast, the thermal stability of the diphenyl ether was higher with NAD+ (ΔTm -6.6 °C) (Table 3). Similar NADH-dependent binding has recently been shown for methyl-thiazoles (7). Collectively, the enzymology and biophysical-binding data demonstrate that 4-hydroxy-2-pyridones are direct InhA inhibitors and that they preferentially bind to the InhA-NADH complex.
Crystal structure of cofactor-bound InhA with NITD-564 and NITD-916
To further understand the molecular interaction of 4-hydroxy-2-pyridones with InhA, co-crystal structures of ternary complexes with NADH-NITD5-64 and NADH-NITD-916 were solved at a resolution of 2.9 and 3.2 Å, respectively (Table S4 and Fig. 3). The refined structures are similar, with an r.m.s. deviation for all Cα atoms of 0.199 Å, and the ligands NITD-564 and NITD-916 were bound in the same position and orientation (Fig. 3A). Consistent with the ITC and thermal-shift data, both NITD-916 and NITD-564 bound to the E-NADH complex (Fig. 3B). The co-crystal structure revealed five key interactions of 4-hydroxy-2-pyridones with InhA and the NADH cofactor: (i) the aromatic pyridone ring of 4-hydroxy-2-pyridones π-stacks against the pyridine ring of the cofactor NADH, (ii) the oxygen at the 4-hydroxy group of NITD-916 hydrogen bonds with the 2′-hydroxyl moiety of the nicotinamide ribose sugar and the hydroxyl of Y158 of InhA, a conserved residue of the enoyl-reductase active-site triad, (iii) the N- of the pyridone core interacts with the S of M199 (Fig. 3B, 3C), (iv) the R3 phenyl ring is exposed to a narrow pocket of the enzyme and (v) the cyclohexyl or di-methyl cyclohexyl moiety of NITD-564 and NITD-916, respectively, occupies the large hydrophobic pocket comprising the side chains of F149, M155, Y158, M199, G192, P193, I215, L218 and W222 (Fig. 3B). Consequently, at the R6 position, when the isopropyl group in NITD-529 was replaced by cyclohexyl (NITD-564), it provided a better interaction between the ligand and these hydrophobic side chains, leading to a 10-fold increase in anti-Mtb potency as well as in the potency against the InhA enzyme (Table 3). Replacing the cyclohexyl with the more hydrophobic dimethyl-cyclohexyl (NITD-916) further enhanced Mtb activity by 10 fold. NITD-916 binding to InhA led to ordering of the substrate-binding loop encompassing residues 196 to 211 (Fig. 3A). All five pyridone-resistant mutations mapped in InhA (T17, S94, G96, D148 and M161) were within 6Å of the NADH binding site (Fig. 3C).
Fig. 3. Structural analysis of the 2-pyridone binding site in the InhA-NADH complex.
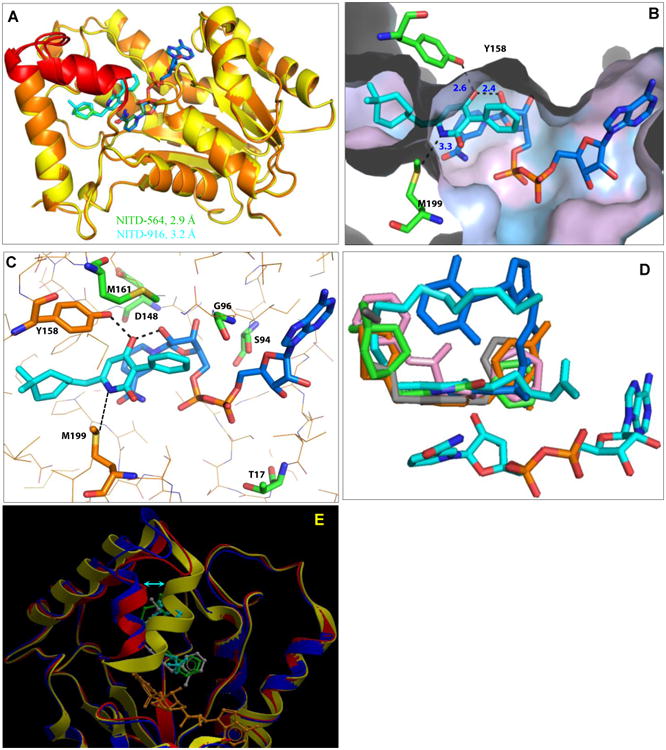
(A) Superimposed crystal structures of InhA-NADH-NITD564 (yellow) and InhA-NADH-NITD916 (orange); respective 2-pyridone ligands are shown in green and cyan. Substrate binding loop encompassing residues 196 to 211 is shown in red. (B) Close-up of NITD-916 (cyan) binding pocket in InhA-NADH complex, with protein polar (cyan) and hydrophobic (grey) surfaces shown. The side chains of Y158 and M199 residues are shown. The distance (in Å) between the ligand and side chains of with Y158, M199 and 2′-OH on the ribose sugar of NADH is highlighted by dotted lines. (C) Hydrogen bonding interactions of NITD-916 with critical residues in the active site of InhA. Side chains of amino acid residues responsible for NITD-916 resistance (T17, S94, G96, D148 and M161) are shown. (D) The InhA-NITD916 (green) structure overlaid with the fatty acyl substrate (cyan, 1BVR), along with other direct InhA inhibitors namely, triclosan derivative (orange, 3FNG), alkyl diphenyl ether (grey, 2×23), pyrrolidine carboxamides (pink, 2H7I) and methyl-thiazoles (blue, 4BQP). (E) The InhA-NITD916 structure (red) overlaid with co-crystal structures of fatty acyl substrate (blue, 1BVR) and alkyl diphenyl ether (yellow, 2×23). NITD-916, fatty acyl substrate and alkyl diphenyl ether ligands are colored in green, cyan and grey respectively. The shift in the conformation of the substrate binding loop is shown by an arrow.
Superimposition of the InhA-NITD-916 structure with the enoyl substrate bound form (20) revealed that NITD-916 partly occupies the fatty acyl-substrate binding pocket and the dimethyl cyclohexyl group forms hydrophobic interactions with the substrate-binding loop (Fig. 3D). Thus, it is likely that the binding of the NITD-916 to InhA-NADH complex blocks enoyl substrate access to its binding site on the enzyme. Earlier efforts to identify direct InhA inhibitors to overcome KatG-mediated resistance yielded many inhibitors that blocked the lipid-binding site (4-7, 19) (Fig. S3). Overlaying a few of these inhibitors (7, 21-23) along with the fatty acyl substrate in the NITD-916 structure revealed that these inhibitors all occupied the enoyl-substrate binding site (Fig. 3D). Recently, the natural product, pyridomycin has also been shown to be a direct InhA inhibitor occupying both NADH and enoyl-substrate binding sites (5, 24). The InhA-NITD-916 structure attained an open substrate binding loop conformation similar to the enoyl-substrate bound structure (20), unlike PT-70 (23), which has a closed conformation (Fig. 3E). Thus, the co-crystal structures confirmed the NADH-dependent binding of 4-hydroxy-2-pyridones, and showed that these ligands partly occupy the enoyl-substrate binding pocket.
Pharmacology of 4-hydroxy-2-pyridones
Considering the lengthy treatment period for MDR-TB (more than 18 months), cytotoxicity, cardiotoxicity or mutagenicity of compounds would impede the progression to clinical testing (25). Hence, it is important to identify the potential safety liabilities of 4-hydroxy-2-pyridones. In a battery of in vitro assays these compounds were found to have an adequate cellular selectivity, displayed no mutagenic or cardiotoxicity potential, and showed no in vitro safety pharmacological liabilities (Table S1). Any new TB drug has to be clinically administered in combination with other antitubercular or anti-retroviral medications, so the absence of drug-drug interactions is critical for clinical development (25). NITD-916 and other pyridone analogues did not inhibit the major CYP450 isoenzyme 3A4 in either reversible or time-dependent inhibition assays and also did not induce hPXR (human pregnane X receptor) activation (Table S1).
NITD-916 has low aqueous solubility and high permeability in vitro with a low to moderate metabolic clearance in mouse and human hepatic microsomes (Table S1). Considering the highly lipophilic nature of the compound, a special lipid-based microemulsion preconcentrate (MEPC) formulation was required for oral pharmacokinetic (PK) studies (26). Upon intra-venous administration, the compound showed a low total systemic clearance and low volume of distribution (Vd 0.54 L/kg) but good oral bioavailability (66% at 25 mg/kg) in rodents (Table S5). The plasma concentration of NITD-916 in vivo (Table S5) was above the in vitro MIC against Mtb, justifying mouse-efficacy studies. Nonetheless it is worth noting that the compound distribution in the lungs was lower than in plasma [at 25 mg/kg dose the ratio was 0.2 for Cmax and 0.4 for area under the curve (AUC)]. In the acute and established murine-efficacy models, all animals tolerated 1 month of daily dosing of NITD-916 in MEPC formulation. In the acute model, NITD-916 showed a dose-dependent in vivo activity in both lungs and spleen (Fig. 4A). Treatment of mice with NITD-916 for one month at a dose of 100 mg/kg resulted in 1.92 and 2.82-log CFU reduction in lung and spleen tissues, respectively, compared to untreated control animals. The efficacy of NITD-916 (100 mg/kg) observed in the acute-infection model is comparable to rifampicin (10 mg/kg) and ethambutol (100 mg/kg), but inferior to isoniazid (25 mg/kg). In an established-infection mouse model, four weeks of treatment with NITD-916 (100 mg/kg) reduced the bacterial burden by 0.95 log in the lungs (Fig. 4B). The in vivo efficacy of NITD-916 in the established model was comparable to the first-line TB drug ethambutol (100 mg/kg) and a potential drug candidate, PA-824 (25 mg/kg). However, under similar conditions rifampicin and isoniazid were significantly more potent than NITD-916. Despite its very high plasma protein binding, low volume of distribution and low lung-to-plasma ratio, the lead candidate NITD-916 showed dose-dependent in vivo efficacy in the lungs of infected mice when given orally.
Fig. 4. In vivo efficacy of NITD-916 in Mtb infected mouse models.

BALB/c mice were infected intranasally with 103 H37Rv strain of Mtb, and animals were orally treated for 4 weeks following 1 or 4 weeks of infection in acute (A) and established (B) efficacy models, respectively. Efficacy of test compounds was measured by Δlog CFUs compared to untreated controls independently for lung and spleen. Statistical evaluation was done using one way ANOVA and the data was analyzed using Tukey's multiple comparison test. Statistical significance was accepted with P values <0.05. All bars labeled with the same letter of the alphabet do not differ significantly. INH, isoniazid; RIF, rifampicin and EMB, Ethambutol.
Discussion
Isoniazid, which primarily targets InhA, is one of the key pillars of TB chemotherapy. Considering the frequent emergence of KatG-mediated isoniazid resistance in patients, several groups have identified structurally-diverse direct InhA inhibitors (6, 27). However, translating the potent enzyme inhibitors into compounds with anti-Mtb activity and the physicochemical properties required for achieving optimal bioavailability and in vivo efficacy remains a significant challenge. Here, using unbiased phenotypic screening, we report the identification of a new class of anti-mycobactericidal agents, the 4-hydroxy-2-pyridones, that act as potent and direct InhA inhibitors. Importantly, these compounds do not require katG-mediated activation; isoniazid-resistant MDR/XDR-TB clinical isolates with katG mutations were fully susceptible to the lead compound NITD-916. Moreover, the 4-hydroxy-2-pyridones have appropriate physicochemical properties and a mode of binding that translated into potent anti-TB activity and in vivo efficacy in both acute and established mouse infection models.
InhA is highly conserved in eubacteria with 27-33% amino-acid sequence identity (Fig. S1). Sequence alignment of enoyl-reductase homologues across multiple bacterial species revealed that the five NITD-916-resistant InhA mutations are highly conserved in the genus mycobacteria, but not in other Gram-positive and Gram-negative bacteria, conceivably explaining the narrow spectrum of activity (Table S2). Moreover, other pathogenic mycobacteria such as M. leprae and M. ulcerans, which cause leprosy and buruli ulcer respectively, also share a nearly identical InhA sequence and thus are expected to be susceptible to 4-hydroxy-2-pyridones. It has been shown previously that InhA S94A and D148G mutants have 7-14 fold less affinity for NADH than the wildtype enzyme (24, 28). This suggests that the resistance to NITD-916 arises due to remodeling of the cofactor-binding site in InhA. Recently, resistance to other direct InhA inhibitors, the methyl-thiazoles, has been mapped to G96 and M103, other conserved residues in the vicinity of the NITD-916 binding pocket (7). Likewise, resistance to pyridomycin, a natural product known to block both the NADH and enoyl-substrate-binding pockets of InhA, has been mapped to residue D148 (5, 24). Thus, the mutation data offers strong genetic evidence for molecular target engagement by 4-hydroxy-2-pyridones. The in vitro frequency of spontaneous 4-hydroxy-2-pyridone resistance mutants of Mtb is 100 times lower than for isoniazid (table 2), suggesting a lower risk for developing drug resistance.
Unlike most other ligands that are known to bind to the InhA-NAD product complex (e.g. triclosan, alkyl diphenyl ethers, pyrrolidine carboxamides), NITD-916 preferentially binds to the InhA-NADH complex [like methylthiazole (7) and pyridone inhibitors of other FabIs (18)] and occupies part of the lipid-substrate binding site. Interestingly, the affinity (Km) of InhA enzyme for NADH is ∼two log orders higher than NAD+ (7), therefore, ligands that bind to the E-NADH complex are likely to be more efficient than those that bind to the E-NAD complex. Structural data indicate that NITD-916 binds to the InhA-NADH complex with multiple hydrogen bonds, π-stacking and hydrophobic interactions. Thus, we propose that binding of 4-hydroxy-2-pyridones to the InhA-NADH complex inhibits the fatty-acid elongation step resulting in blocking of the biosynthesis of mycolic acids, weakening of the cell-wall mycolyl-arabinogalactan-peptidoglycan complex and ultimately lysis of Mtb (Fig. S4).
NITD-916 is a promising small molecule lead candidate that is not mutagenic, lacks cardiotoxicity and does not show CYP-mediated drug-drug-interactions. Addressing the solubility and lipophilicity concerns of 4-hydroxy-2-pyridones will be critical to move away from lipid-based formulation and also to de-risk the drug during clinical development. Phosphate ester pro-drugs are typically designed to enhance their aqueous solubility to allow a more favorable oral administration and are generally rapidly hydrolysed by intestinal alkaline phosphatases (29). As a proof of concept, we have synthesized NITD-113, a 4-hydroxy methyl phosphate ester pro-drug of NITD-916, which improved the aqueous solubility by two log orders (Fig. S5A). The oral mouse PK of NITD-113 in simple 0.5% carboxy-methyl cellulose (CMC) aqueous formulation resulted in good bioconversion to NITD-916 and the drug exposure achieved with CMC formulation was similar to NITD-916 in MEPC formulation (Fig. S5B). In addition, preliminary analysis of the binding mechanism of NITD-916 suggested replacing R3 phenyl with suitable ring systems could potentially address solubility and increase basicity, leading to a higher volume of distribution and lower binding to plasma proteins. Thus, the co-crystal structure of NITD-916 with the InhA-NADH complex has opened up avenues for structure-based rational lead optimization. The real value of identifying the molecular target of a phenotypic hit has yet to be exploited.
The molecular target of 4-hydroxy-2-pyridones, InhA, is a well-recognized clinically validated target. High throughput phenotypic screening approach and strategies used for target deconvolution of NITD-916 by whole genome-sequencing of spontaneous resistant mutants, is now a well-established strategy to elucidate the mechanisms of action in infectious diseases. And also other groups have previously discovered direct inhibitors of InhA have helped to characterize the ligand-binding pocket of InhA.
Nonetheless, the “re-discovery” of InhA as the molecular target of a phenotypic screening hit with an NADH-dependent mode of binding and in vitro and in vivo potency, emphasizes the power of combining cell–based screening with target deconvolution approaches. Given that the lead compound NITD-916 has a low volume of distribution and reduced lung-to-plasma ratio, further medicinal chemistry optimization will be required to achieve the necessary drug exposure in the lung cavities of TB patients. The biochemical and structural characterization of NITD-916 bound to InhA reported here should facilitate further structure-guided rational approaches to identify better preclinical candidates.
Materials and Methods
Strains and growth conditions
M. tuberculosis H37Rv (ATCC 27294), M. tuberculosis H37Ra, M. bovis BCG Pasteur (ATCC 35734), M. smegmatis MC2 155, and derivative strains were maintained in Middlebrook 7H9 broth medium supplemented with 0.05 % Tween 80 and 10 % ADS supplement.
High-throughput screening, MIC determination, kill kinetics analysis and catalase assay
High-through put screening of Novartis compounds collection was carried out using Mtb H37Ra strain, MIC50 against Mtb H37Rv and kill kinetic analysis was determined as previously (26). For determining growth inhibition against diverse MDR Mtb clinical isolates (13), pellet formation method was used (26). MIC is defined as the minimum concentration of the drug required to inhibit 50% of H37Rv growth or 99% of growth in MDR clinical isolates after 5 days or 10 days incubation respectively. Catalase activity was assayed using a mixture of H2O2 (15 %) and Tween-80 (10 %) in water as described earlier (30).
Cytotoxicity determination and intra-macrophage activity
Cytotoxicity against HepG2 and THP-1 cell lines was determined as previously described (26). For intra-macrophage activity 5×105 THP-1 cells differentiated using 50 nM PMA for 48hrs. The differentiated cells were activated with 10 μM IFN-γ for 4 hrs and were infected with Mtb at MOI of 1:1. After one hour of post-infection extracellular mycobacteria were removed by washing twice with warm PBS and replaced with fresh medium with or without compounds. The number of viable intracellular mycobacteria was determined by plating after lysing the macrophages with 500 μl of 0.1% triton X-100 at 1 hour, 5 days post-infection. Inhibitory concentration 90 (IC90) and inhibitory concentration 99 (IC99) were defined as the lowest concentration of drug resulting in 90% and in 99% reduction in CFU, respectively (26).
Efficacy studies in mouse models of acute and established infection
Acute and established in vivo efficacy studies were carried out as described previously (26). Briefly, BALB/c mice were infected intranasally with 103 Mtb H37Rv, and animals were orally treated for 4 weeks following 1 or 4 weeks of infection for acute and established efficacy model, respectively. Ethambutol (100mg/kg), Rifampicin (10 mg/kg), Isoniazid (25 mg/kg) and PA-824 (25 mg/kg) were used as control. Bacterial load in lung and spleen (mean ± SD from 6 mice per group and per time point) were analyzed at 4 weeks post-treatment by enumerating CFUs. Statistical evaluation was done using one way ANOVA and the data analyzed using Tukey's multiple comparison test. Statistical significance was accepted with P values <0.05.
Generation of pyridone-resistant mutants and whole genome sequencing
Mtb H37Rv strain was plated on 7H11 agar plates containing 10× and 20× MIC50 of the test compounds. After 3 weeks of incubation at 37°C single isolated colonies were propagated in drug free 7H9 broth. The resistance phenotype to the pyridones was confirmed by testing MIC50 values against NITD-529, 564 and 916. Whole genome sequencing and single-nucleotide polymorphism analysis of the spontaneous resistant mutants was carried out as described earlier (26, 31).
Lipid labeling, extraction, and analysis
Radiolabeling of Mtb lipids with 14C-acetate was carried out by adding 1 μ Ci/ml of [1,2-14C]acetic acid sodium salt to a 5 ml mid log phase (A600 nm OD = 0.3) H37Rv cells treated with the indicated concentrations of drugs / inhibitors for 2 hours. After one hour 14C-acetate labelling cultures were centrifuged, and the cell pellet was used directly to analyze total cellular lipids using the methyl esterification protocol for preparing mycolic acid methyl esters (MAMEs) and fatty acid methyl esters (FAMEs) as described earlier (32).
Recombinant expression and genetic complementation of Mtb InhA
Mtb inhA gene (Rv1484) was amplified by PCR (Pfx polymerase; Invitrogen) from H37Rv genomic DNA using forward (5-CTTTAAGAAGGAGATATCATATGACAGGACTGCTGGACGGC-3) and reverse (5′-GACGCCGGATCCTAGAGCAATTGGGTGTGCGC-3) primers. PCR amplified fragments were cloned into NdeI and BamHI sites of pET15(b) vector to obtain pET15b-inhA (33). Mtb InhA in pET15b vector encoding C-terminal His tag was transformed into BL21 (DE3) cells and protein expression was induced at 0.6 OD with 0.1 mM IPTG for 16 h. Soluble recombinant His6-InhA protein was purified on a nickel affinity binding column in buffer containing 20 mM Tris-HCl (pH 8.0) and 500 mM NaCl. Subsequently the protein was subjected to gel filtration on Superdex-200 in 20 mM HEPES (pH 7.0), 150 mM NaCl, and 1 mM TCEP buffer. The purified protein was concentrated to ∼ 10 mg/mL in 20 mM HEPES (pH 7.0), 150 mM NaCl, 1 mM TCEP is used for ITC binding and X-ray crystallographic studies. For genetic complementation, Mtb inhA gene was amplified from the genomic DNA isolated from wild type or mutants using forward (5- TAGGATCCATGACAGGACTGCTGGACGG -3) and reverse (5′-CGGAATTCCTAGAGCAATTGGGTGTGCGCG-3) primers. PCR amplified fragments were cloned into BamHI and EcoRI sites of pMV262 vector, transformed into Mtb H37Rv and selected for kanamycin resistant recombinant clones.
Binding assay
Isothermal titration calorimetry experiments were performed using an iTC200 (GE Healthcare) at 25°C, with 10-20 μM InhA in 20 mM Hepes pH 7.0, 150 mM NaCl and 2 mM TCEP in the sample cell. The compound at 125-500 μM was diluted, from a 10 mM stock in DMSO, into the same buffer and titrated into the InhA sample supplemented with the same concentration of DMSO. NADH from a 5 mM stock dissolved in water was added to both the cell and syringe at a final concentration of 50 μM. Typically, 15 injections of 2.6 μl of compound were injected into the sample cell at 2.5 min intervals. The data were fitted to a single-site binding equation using Origin. The Thermofluor stability assay was performed on a Bio-Rad CFX96 Real-Time System, using 30 mM PIPES buffer (pH 6.8) containing 150 mM NaCl and 1.0 mM EDTA. Briefly, a 20 μL solution of 7.4μM InhA, 2.5mM cofactor (NADH or NAD+) and 25μM inhibitors were incubated on a 96-well plate at room temperature for 2 hours before adding 5× Sypro Orange protein gel stain. A thermo cycle was run from 25°C to 90°C. At the end of each 0.20°C increment for 10 min, the fluorescence intensity of the plate was read after 10 min. Diphenyl ether was used as a positive control for compounds binding to NAD+ and 4-pyridone analogue was used as a positive control for compounds binding to NADH.
InhA enzyme assay
Trans-2-Dodecenoyl-CoA (DD-CoA) was synthesized from trans-2-dodcenoic acid. IC50 was performed with 25μM trans-2-Dodecenoyl-CoA, 100nM InhA, 0-40 μM inhibitor in 30mM PIPES pH 6.8 buffer containing 150mM NaCl and 1mM EDTA as described earlier (34). Initial velocity (vi) was measured for the first 10% of the reaction. Percentage activity was plotted as a function of inhibitor concentration into the following equation:
Crystallization and structure determination
InhA in 20 mM Hepes pH 7.0, 150 mM NaCl and 2 mM TCEP at 10 mg/ml was mixed with 1 mM NADH (from a 20 mM stock dissolved in water) and 500 μM compound (from a 10 mM stock in DMSO) prior to setting up hanging-drop crystallisation trials. This is approximately a 1:3:1.5 ratio of protein to NADH to compound. Crystals grew in 10 % PEG 4000, 0.1 M Hepes pH 7.0 and 0.2 M ammonium acetate. Crystals appeared within a week and were transferred to a cryoprotectant containing the same solution supplemented with 25% DMSO before rapid cooling in liquid nitrogen. X-ray diffraction data were collected at beamline ×10SA of the Swiss Light Source, integrated using iMOSFLM (35) and scaled using SCALA (36), part of the CCP4 suite (Winn 2011). The structures were solved by molecular replacement using the structure of InhA (PDB ID 3OEW) (37) as a search model. Model-building was done with COOT (38) and the structures were refined using REFMAC5 (39), using NCS restraints.
Accessible Summary.
“Re-discovering” InhA for the treatment of MDR-TB
Isoniazid, a key component of the drugs combination currently used to treat tuberculosis, inhibits the Mycobacterium tuberculosis InhA enzyme. Unfortunately, isoniazid was rendered increasingly obsolete with the spread of multi-drug resistant tuberculosis (MDR-TB). Through phenotypic screening and subsequent target identification, we discovered 4-hydroxy-2-pyridones, a new class of InhA inhibitors. Their direct mode of binding to InhA circumvents the main mechanisms of isoniazid resistance and these compounds show activity against MDR-TB clinical isolates. Preliminary medicinal chemistry efforts yielded a lead compound NITD-916, which displays potent oral activity in tuberculosis mouse models. Our structural data provide a path for further optimization of 4-hydroxy-2-pyridones through rational design.
Acknowledgments
We would like to thank Barry Kreisworth for TB clinical isolates; Gianfranco De Pascale, Meena Sachdeva and Jennifer Leeds for broad-spectrum antibacterial testing and Shahul Nilar and David McNeeley for feedback on the manuscript. We would like to thank Vivian Lim, Zhong Chen, Boon Heng Lee, Pamela Thayalan, Mahesh N., Sindhu Ravindran and other colleagues from Novartis Institute for Tropical Diseases (NITD) and Genomics Institute of the Novartis Research Foundation (GNF) for their support.
Funding: This work was supported by NITD and GNF. PT and WY are supported in part by NIH grant GM102864. We thank the beam-line scientists at the Swiss Light Source for assistance with data collection.
Footnotes
Author contributions: UM, SPSR, LC, BT, SN, KK, DB Performed / analyzed mycobacterial cell based assays; UM, SR, BT, SWB, JW Performed / analyzed whole genome sequencing; UM, BT, SN Designed / performed genetic complementation studies; UM, SR, Performed / analyzed lipid profiling; PT, WY, Performed / analyzed InhA thermofluor binding and enzyme inhibition studies; UM, CN, BT, SN, Protein expression, crystallography and ITC binding studies; SL, FB Performed / analyzed animal pharmacokinetic studies; UM, SR, MH, performed / analyzed mouse efficacy studies; RK, PN, NM, PS did the cheminformatic analysis, and designed and synthesized analogues; UM, SR, RK, LC, FB, DB, RG, PS, TTD supervised and directed the work; UM, SR, CN, TD wrote the paper; All authors discussed the results and commented on the manuscript.
Competing interests: UHM, SR, RK, and NM are named as inventors on the patent application WO2014/093606A1 titled “Pyridone derivatives and uses thereof in the treatment of tuberculosis”. The other authors declare no competing interests.
Data and materials availability: Atomic coordinates and structure factors for InhA-NADH-NITD564 and InhA-NADH-NITD916 complex structures have been deposited in PDB under accession codes 4R9R and 4R9S respectively. All requests for compounds are subject to a Material Transfer Agreement.
References
- 1.World Health Organization (WHO) WHO Press; Geneva: 2012. Global Tuberculosis Report. http://apps.who.int/iris/bitstream/10665/75938/1/9789241564502_eng.pdf. [Google Scholar]
- 2.Zumla A, Nahid P, Cole ST. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov. 2013;12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 3.Ramaswamy SV, Reich R, Dou SJ, Jasperse L, Pan X, Wanger A, Quitugua T, Graviss EA. Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2003;47:1241–1250. doi: 10.1128/AAC.47.4.1241-1250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Encinas L, O'Keefe H, Neu M, Remuinan MJ, Patel AM, Guardia A, Davie CP, Perez-Macias N, Yang H, Convery MA, Messer JA, Perez-Herran E, Centrella PA, Alvarez-Gomez D, Clark MA, Huss S, O'Donovan GK, Ortega-Muro F, McDowell W, Castaneda P, Arico-Muendel CC, Pajk S, Rullas J, Angulo-Barturen I, Alvarez-Ruiz E, Mendoza-Losana A, Ballell Pages L, Castro-Pichel J, Evindar G. Encoded library technology as a source of hits for the discovery and lead optimization of a potent and selective class of bactericidal direct inhibitors of Mycobacterium tuberculosis InhA. J Med Chem. 2014;57:1276–1288. doi: 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- 5.Hartkoorn RC, Pojer F, Read JA, Gingell H, Neres J, Horlacher OP, Altmann KH, Cole ST. Pyridomycin bridges the NADH- and substrate-binding pockets of the enoyl reductase InhA. Nat Chem Biol. 2014;10:96–98. doi: 10.1038/nchembio.1405. [DOI] [PubMed] [Google Scholar]
- 6.Pan P, Tonge PJ. Targeting InhA, the FASII enoyl-ACP reductase: SAR studies on novel inhibitor scaffolds. Curr Top Med Chem. 2012;12:672–693. doi: 10.2174/156802612799984535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirude PS, Madhavapeddi P, Naik M, Murugan K, Shinde V, Nandishaiah R, Bhat J, Kumar A, Hameed S, Holdgate G, Davies G, McMiken H, Hegde N, Ambady A, Venkatraman J, Panda M, Bandodkar B, Sambandamurthy VK, Read JA. Methyl-Thiazoles: A novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J Med Chem. 2013;56:8533–8542. doi: 10.1021/jm4012033. [DOI] [PubMed] [Google Scholar]
- 8.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 9.Kondreddi RR, Manjunatha UH, Ngai LM, Peukert S, Rao SPS. Pyridone Derivatives and uses thereof in the treatment of tuberculosis. 2014 WO2014/093606 A1. [Google Scholar]
- 10.Ng P, Manjunatha UH, Rao SPS, Camacho LR, Ma NL, Herve M, Noble CG, Goh A, Peukert S, Diagana TT, Smith PW, Kondreddi RR. Structure activity relationships of 4-hydroxy-2-pyridones: a novel anti-tuberculosis agents. doi: 10.1016/j.ejmech.2015.10.008. unpublished material. [DOI] [PubMed] [Google Scholar]
- 11.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE., 3rd Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:431–436. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 13.Gutacker MM, Mathema B, Soini H, Shashkina E, Kreiswirth BN, Graviss EA, Musser JM. Single-nucleotide polymorphism-based population genetic analysis of Mycobacterium tuberculosis strains from 4 geographic sites. J Infect Dis. 2006;193:121–128. doi: 10.1086/498574. [DOI] [PubMed] [Google Scholar]
- 14.Barry CE, 3rd, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. Mycolic acids: structure, biosynthesis and physiological functions. Progr Lipid Res. 1998;37:143–179. doi: 10.1016/s0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- 15.Vilcheze C, Jacobs WR., Jr The mechanism of isoniazid killing: clarity through the scope of genetics. Annual Rev Microbiol. 2007;61:35–50. doi: 10.1146/annurev.micro.61.111606.122346. [DOI] [PubMed] [Google Scholar]
- 16.Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci USA. 2003;100:13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vilcheze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, Weisbrod TR, Alland D, Sacchettini JC, Jacobs WR., Jr Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat Med. 2006;12:1027–1029. doi: 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- 18.Schiebel J, Chang A, Shah S, Lu Y, Liu L, Pan P, Hirschbeck MW, Tareilus M, Eltschkner S, Yu W, Cummings JE, Knudson SE, Bommineni GR, Walker SG, Slayden RA, Sotriffer CA, Tonge PJ, Kisker C. Rational design of broad-spectrum antibacterial activity based on a clinically relevant enoyl-ACP reductase inhibitor. J Biol Chem. 2014;289:15987–16005. doi: 10.1074/jbc.M113.532804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan P, Knudson SE, Bommineni GR, Li HJ, Lai CT, Liu N, Garcia-Diaz M, Simmerling C, Patil SS, Slayden RA, Tonge PJ. Time-dependent diaryl ether inhibitors of InhA: structure-activity relationship studies of enzyme inhibition, antibacterial activity, and in vivo efficacy. ChemMedChem. 2014;9:776–791. doi: 10.1002/cmdc.201300429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rozwarski DA, Vilcheze C, Sugantino M, Bittman R, Sacchettini JC. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J Biol Chem. 1999;274:15582–15589. doi: 10.1074/jbc.274.22.15582. [DOI] [PubMed] [Google Scholar]
- 21.Freundlich JS, Wang F, Vilcheze C, Gulten G, Langley R, Schiehser GA, Jacobus DP, Jacobs WR, Jr, Sacchettini JC. Triclosan derivatives: towards potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. ChemMedChem. 2009;4:241–248. doi: 10.1002/cmdc.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He X, Alian A, Stroud R, Ortiz de Montellano PR. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J Med Chem. 2006;49:6308–6323. doi: 10.1021/jm060715y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li HJ, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem Biol. 2006;1:43–53. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- 24.Hartkoorn RC, Sala C, Neres J, Pojer F, Magnet S, Mukherjee R, Uplekar S, Boy-Rottger S, Altmann KH, Cole ST. Towards a new tuberculosis drug: pyridomycin - nature's isoniazid. EMBO Mol Med. 2012;4:1032–1042. doi: 10.1002/emmm.201201689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Food and Drug Administration Guidance for Industry- Pulmonary Tuberculosis: Developing Drugs for Treatment. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM373580.pdf.
- 26.Rao SP, Lakshminarayana SB, Kondreddi RR, Herve M, Camacho LR, Bifani P, Kalapala SK, Jiricek J, Ma NL, Tan BH, Ng SH, Nanjundappa M, Ravindran S, Seah PG, Thayalan P, Lim SH, Lee BH, Goh A, Barnes WS, Chen Z, Gagaring K, Chatterjee AK, Pethe K, Kuhen K, Walker J, Feng G, Babu S, Zhang L, Blasco F, Beer D, Weaver M, Dartois V, Glynne R, Dick T, Smith PW, Diagana TT, Manjunatha UH. Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci Transl Med. 2013;5:214ra168. doi: 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- 27.Tonge PJ, Kisker C, Slayden RA. Development of modern InhA inhibitors to combat drug resistant strains of Mycobacterium tuberculosis. Curr Top Med Chem. 2007;7:489–498. doi: 10.2174/156802607780059781. [DOI] [PubMed] [Google Scholar]
- 28.Quemard A, Sacchettini JC, Dessen A, Vilcheze C, Bittman R, Jacobs WR, Jr, Blanchard JS. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochem. 1995;34:8235–8241. doi: 10.1021/bi00026a004. [DOI] [PubMed] [Google Scholar]
- 29.Heimbach T, Oh DM, Li LY, Forsberg M, Savolainen J, Leppanen J, Matsunaga Y, Flynn G, Fleisher D. Absorption rate limit considerations for oral phosphate prodrugs. Pharmaceut Res. 2003;20:848–856. doi: 10.1023/a:1023827017224. [DOI] [PubMed] [Google Scholar]
- 30.Guo H, Seet Q, Denkin S, Parsons L, Zhang Y. Molecular characterization of isoniazid-resistant clinical isolates of Mycobacterium tuberculosis from the USA. J Med Microbiol. 2006;55:1527–1531. doi: 10.1099/jmm.0.46718-0. [DOI] [PubMed] [Google Scholar]
- 31.Leeds JA, Sachdeva M, Mullin S, Barnes SW, Ruzin A. In vitro selection, via serial passage, of Clostridium difficile mutants with reduced susceptibility to fidaxomicin or vancomycin. J Antimicrob Chemother. 2014;69:41–44. doi: 10.1093/jac/dkt302. [DOI] [PubMed] [Google Scholar]
- 32.Slayden RA, Barry CE., 3rd Analysis of the lipids of Mycobacterium tuberculosis. Methods Mol Med. 2001;54:229–245. doi: 10.1385/1-59259-147-7:229. [DOI] [PubMed] [Google Scholar]
- 33.Parikh S, Moynihan DP, Xiao G, Tonge PJ. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochem. 1999;38:13623–13634. doi: 10.1021/bi990529c. [DOI] [PubMed] [Google Scholar]
- 34.Parikh SL, Xiao G, Tonge PJ. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochem. 2000;39:7645–7650. doi: 10.1021/bi0008940. [DOI] [PubMed] [Google Scholar]
- 35.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta crystallogr D Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans P. Scaling and assessment of data quality. Acta crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 37.Molle V, Gulten G, Vilcheze C, Veyron-Churlet R, Zanella-Cleon I, Sacchettini JC, Jacobs WR, Jr, Kremer L. Phosphorylation of InhA inhibits mycolic acid biosynthesis and growth of Mycobacterium tuberculosis. Mol Microbiol. 2010;78:1591–1605. doi: 10.1111/j.1365-2958.2010.07446.x. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faller B, Wang J, Zimmerlin A, Bell L, Hamon J, Whitebread S, Azzaoui K, Bojanic D, Urban L. High-throughput in vitro profiling assays: lessons learnt from experiences at Novartis. Expert Opin Drug Metab Toxicol. 2006;2:823–833. doi: 10.1517/17425255.2.6.823. [DOI] [PubMed] [Google Scholar]
- 41.Faller B. Artificial membrane assays to assess permeability. Curr Drug Metabol. 2008;9:886–892. doi: 10.2174/138920008786485227. [DOI] [PubMed] [Google Scholar]