

Abstract
Neuronal circuits are defined by synaptic connections between their cellular constituents. Here, I highlight a number of recent studies emphasizing the surprising level of precision exhibited by inhibitory GABAergic synapses within the neocortex and hippocampus. Specifically, GABAergic inputs to dendritic shafts and spines of pyramidal cells play a key role in the localized regulation of neuronal calcium signaling. These findings provide important new insights into the cellular mechanisms underlying the contributions of inhibitory transmission to both normal and abnormal brain activity.
Understanding the precision of neuronal connectivity is a major focus of current neuroscientific research 1. Although most of these efforts focus on excitatory glutamatergic circuits, there is a growing appreciation for the role of GABAergic inhibition in the regulation of cellular and network activity 2. Indeed, the balance between excitation and inhibition is critical for normal brain function, and dysregulation of this balance is implicated in a number of neuropsychiatric disorders, including schizophrenia and autism 3,4. Recent studies have broadened our understanding of the precision by which GABAergic interneurons (INs) innervate and regulate their target cells. That is, different types of INs form connections onto highly specific subregions of their target cell's somatodendritic arbor, enabling fine spatial control of postsynaptic activity. Here, I review several recent findings that support the hypothesis that, rather than serving as a simple brake on action potential output, GABAergic inhibition can sculpt neuronal activity at the subcellular level, exerting complex effects on both electrical and biochemical signaling.
Diversity of GABAergic inhibition in cortical and hippocampal circuits
Neuronal activity in the neocortex and hippocampus is shaped by the interplay of excitatory glutamatergic pyramidal neurons (PNs) and inhibitory GABAergic INs. PNs receive excitatory inputs onto small (∼1 μm) membrane protrusions called dendritic spines, which serve to compartmentalize biochemical and electrical signals 5,6. Activation of AMPA- and NMDA-type glutamate receptors (AMPARs and NMDARs, respectively) causes membrane depolarization and local calcium (Ca2+) influx 7(see Box 1) that contributes to the generation of somatic action potentials and influences long-term changes in synaptic strength. Dendritic integration of these excitatory signals is countered by the actions of GABAergic inhibition, although the subcellular targets and consequences of GABAergic signaling are less well understood.
Box 1. Sources of Dendritic Calcium.
Dendritic Ca2+ sources that are sensitive to GABAergic inhibition include two general categories: glutamate receptors and voltage-gated calcium (Ca2+) channels (VGCCs). The specific contributions made by each of these depend on the brain structure, cell class, and subcellular compartment 7. NMDA-type glutamate receptors contribute a significant fraction of synaptic Ca2+ influx in pyramidal cells of the neocortex and hippocampus 60. The conductance of cations, including Ca2+, through NMDARs is strongly regulated by membrane potential due to pore blockade by extracellular magnesium ions.
Most non-NMDA-type glutamate receptors, including AMPA-type receptors (AMPARs), exhibit minimal Ca2+ permeability in pyramidal neurons. However, AMPARs lacking a GluA2 subunit are Ca2+ permeable and have been primarily described in GABAergic interneurons 61. AMPARs also contribute to Ca2+ signaling by providing membrane depolarization that activates VGCCs and relieves magnesium block from NMDARs 62.
Another significant contributor to dendritic Ca2+ signaling is the diverse group of VGCCs, which comprise a broad class of membrane channels with a wide variety of voltage-dependence, activation, and inactivation properties 63. VGCCs in dendrites and dendritic spines open following strong synaptic depolarization. Indeed, co-activation of many synapses can induce a dendritic spike, a VGCC-dependent regenerative event that causes widespread Ca2+ influx and can influence somatic spike generation 34. Sufficient depolarization for VGCC opening can also be provided by the antidromic propagation of somatically-generated action potentials through at least the proximal portions of the dendritic arbor 34.
Each of these sources is potentially influenced by GABAergic inhibition. First, membrane hyperpolarization (via activation of either GABAA or GABAB receptors) or shunting of synaptic depolarization (predominantly via GABAARs) reduces the open probability of both NMDARs and VGCCs 21,41,42. Second, GABABRs are coupled to biochemical signaling pathways that reduce Ca2+ influx through NMDARs and VGCCs 47,48 (see main text).
GABAergic inhibition is mediated by two classes of receptors expressed ubiquitously throughout the nervous system (Fig. 1) 8. Type-A receptors (GABAARs) are ionotropic channels, permeable to chloride and bicarbonate, that typically produce minimal direct change in membrane potential but generate a large conductance that shunts the impact of excitatory input-mediated depolarization (see Box 2). Type-B receptors (GABABRs) are G protein-coupled and lead to downregulation of cAMP production, activation of inwardly rectifying potassium channels that hyperpolarize the membrane potential, and inhibition of voltage-gated Ca2+ channels 9. In the cortex and hippocampus, GABA is synthesized and released by inhibitory INs. A major challenge to understanding the contribution of GABAergic signaling to brain activity is the wide diversity of these cells. INs comprise approximately 20-30% of all cortical and hippocampal neurons and can be subdivided into numerous classes with distinct physiology, synaptic specializations, and molecular markers 10-12. Recent work suggests three principal groups: cells expressing (1) the Ca2+ binding protein parvalbumin, (2) the 5HT3a-type serotonin receptor, or (3) the peptide transmitter somatostatin (SOM) 11, each of which plays a distinct role in local circuit function.
Figure 1. GABAergic interneurons and the targets of inhibition in cortical circuits.
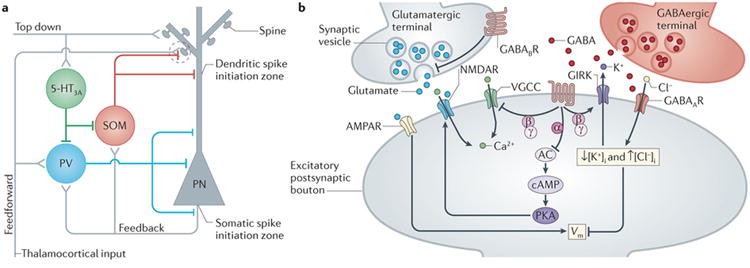
A, Schematic illustration of the three major inhibitory circuits in the neocortex. Excitatory (+) and inhibitory (-) synapses are shown. Perisomatic-targeting interneurons that express parvalbumin (PV) are activated by feedforward and feedback excitation and sharply curtail generation of somatic action potentials in response to afferent inputs. Dendrite-targeting interneurons that express somatostatin (SOM) are strongly engaged by feedback excitation originating from local cortical pyramidal neurons. They form synapses on both dendritic shafts and spines that converge with excitatory inputs (dashed circle) to regulate synaptic integration and dendritic spike initiation. Interneurons expressing the serotonergic 5HT3a receptor target other interneurons and receive excitatory inputs from top-down intracortical projections. B, Schematic of the local actions of GABAergic inhibition in dendritic spines receiving both excitatory and inhibitory inputs. Ionotropic GABAA receptors and metabotropic GABAB receptors influence both spine Ca2+ signals and membrane potential (Vm) through regulation of glutamate receptors (AMPARs and NMDARs), voltage-gated Ca2+ channels (VGCCs), and G protein-coupled potassium channels (GIRKs). Other abbreviations: AC (adenylate cyclase), cAMP (cyclic adenosine monophosphate), Cl- (chloride), K+ (potassium), PKA (cAMP-sensitive protein kinase), Vm (membrane potential).
Box 2. Hyperpolarizing Versus Shunting Inhibition.
An inhibitory synapse is one whose activity reduces the targeted cell's likelihood of crossing the threshold for action potential generation. Within this definition, two non-mutually exclusive modes of inhibition exist: hyperpolarization and shunting. Both types can be mediated by GABAergic signaling, but they produce distinct consequences for the postsynaptic cell.
Hyperpolarizing inhibition is relatively straightforward and is typified by the actions of GABABRs. Activation of GABABRs leads to release of the βγ subunit from the receptor's associated G protein. The βγ subunit then triggers the opening G protein-coupled inwardly rectifying potassium channels (GIRKs). The direction of current flow through any open channel is determined by the relationship between the cell's resting membrane potential (Vm) and the reversal potential of the channel (Vrev) as defined by the Goldman-Hodgkin-Katz equation:
where R is the ideal gas constant, T is temperature, F is Faraday's constant, [C+] and [A-] are the concentrations of cationic and anionic species and PC and PA are their permeabilities, respectively. Synaptic current is then given by the equation:
where ISyn is the current through the channel and GSyn is the channel conductance. In the case of potassium, VRev in pyramidal neurons is approximately -90 mV and is more negative than the resting potential of approximately -70 mV, leading to a positive (outward) synaptic current. Thus, upon GIRK channel opening, potassium flows out of the cell, hyperpolarizing the membrane potential. As this moves Vm further from spike threshold (∼-45 mV), the result is to inhibit the cell's activity.
Shunting inhibition is somewhat more complex, and is typified by inhibition mediated by GABAARs. GABAARs are primarily permeable to the anions chloride and bicarbonate, and exhibit a reversal potential of approximately -70 mV, very close to the cell's resting potential. Thus, upon GABA binding and channel opening, there is very little net current across the membrane (ISyn ≈ 0). However, the GABAAR-mediated synaptic conductance is quite large, leading to an increase in the overall conductance of the cell's membrane during the time that the channels are open. Imagine another excitatory synaptic channel (e.g., an AMPA-type glutamate receptor) opening at the same time. Inward current through the glutamate receptor would typically depolarize the cell's membrane potential. However, this depolarizing current is short-circuited by the GABAergic conductance rather than charging the membrane. Thus, without changing the resting membrane potential, the GABAAR-mediated shunt has made it less likely that the cell will fire an action potential in response to excitatory input.
The temporal dynamics of these two forms of inhibition are also distinct. In the case of hyperpolarizing inhibition, capacitive filtering of synaptic currents by the cell produces a change in the membrane potential that outlasts the underlying conductance. In contrast, shunting inhibition only occurs for the duration of the inhibitory conductance.
In the neocortex and hippocampus, parvalbumin-expressing basket cells and chandelier cells make strong inhibitory contacts onto the perisomatic regions of their target PNs, including the axon initial segment 13,14 (Fig. 1). These powerful inputs exert well-documented control over the timing and magnitude of neuronal output. For example, feed-forward inhibition mediated by these INs rapidly truncates afferent excitation of the PN, limiting the temporal window during which action potentials can be generated 15-17.
In contrast, INs expressing the 5HT3a receptor were only recently described, and much less is known about their function. They are present throughout the cortex and hippocampus, particularly in more superficial layers 18,19. A subgroup of these neurons that co-expresses vasointestinal peptide (VIP) appears to predominantly contact other INs 12. Recent work suggests that VIP-expressing cells are specifically excited by long-range intracortical projections, such as those projecting from motor to somatosensory cortex 20 (Fig. 1). This long-range circuit may subserve top-down disinhibition of afferent responses, providing a mechanism for sensorimotor integration.
The third group, somatostatin-expressing INs, consists largely of cells whose pial-projecting axons form contacts along PN dendritic arbors, near the sites of glutamatergic inputs 13,21,22 (Fig. 1). The most characteristic of these is the Martinotti cell, whose axon ramifies extensively in layer 1 of the neocortex, contacting the apical tufts of pyramidal neurons 22. Similar cells exist in the hippocampus, where their cell bodies are located in stratum oriens and their projections target distal tufts of CA1 PNs in stratum lacunosum moleculare 23. Somatostatin-expressing INs receive facilitating feedback excitation from neighboring pyramidal cells, making them sensitive to local network activity 24,25.
GABAergic targeting of dendritic spines
Ultrastructural analyses of dye-injected neurons in the cat visual cortex revealed that the majority of GABAergic presynaptic contacts are formed onto dendrites, including dendritic spines where they converge with individual glutamatergic afferents 26,27 (Fig. 1). Immunogold labeling has also identified GABA receptors within dendritic shafts and spines 28-30. However, the precise arrangement of postsynaptic densities and active zones of co-localized glutamatergic and GABAergic synapses remains largely unknown.
Until recently, there was little data on the density and distribution of GABAergic synapses along the length of individual dendritic branches. However, recent efforts utilizing 2-photon imaging have tracked the location of inhibitory synapses in vivo. In two independent studies, the authors expressed a fluorescently tagged version of gephyrin, a scaffolding protein unique to inhibitory synapses, in layer 2/3 pyramidal neurons of the mouse visual cortex 31,32. GABAergic inputs exhibited a density of approximately 0.2 synapses per micron, roughly half that of excitatory contacts estimated by counting dendritic spines 31. Remarkably, of the imaged spines located within 125 microns of the soma, approximately 14% also bore a GABAergic synapse. This fraction was increased two-fold for more distal spines present in the most superficial cortical layers. Each dendritic spine receiving a GABAergic input also received a glutamatergic contact, indicating dual innervation 31. Interestingly, spines receiving inhibitory synapses are targeted by glutamatergic inputs enriched for the thalamocortical synaptic marker Type 2 vesicular glutamate transporter (VGluT2)32,33, suggesting that inhibition to dendritic spines may specifically regulate sensory information arising from ascending thalamic inputs. Both anatomical and physiological studies further indicate that GABAergic inputs to dendritic spines arise, at least in part, from somatostatin-expressing INs 21,22.
Somatostatin-expressing interneurons regulate dendritic spikes
As a consequence of their target specificity, a critical function of somatostatin-expressing INs appears to be the regulation of dendritic electrical activity in both the neocortex and hippocampus. Several decades of work have now shown that neuronal dendrites do not passively relay synaptic inputs to the cell body. Rather, an array of voltage-gated conductances shape the dynamics of synaptic integration across multiple subcellular compartments within the dendritic tree (recently reviewed in 34). Several studies have emphasized the electrogenic properties of distal apical dendrites of pyramidal neurons in both neocortical layer 5 and hippocampal CA1 35-40. While sodium-based action potentials originate near the cell body in the axon initial segment, a second initiation zone for broad Ca2+-based action potentials called dendritic spikes occurs in these distal dendritic compartments, driven by the activity of voltage-gated calcium (Ca2+) channels (VGCCs) with important contributions from NMDARs. These dendritic spikes can be initiated by spatiotemporally convergent synaptic input and can spread to the soma to evoke bursts of spike output. Evidence from in vivo experiments suggest dendritic spikes shape receptive field properties in somatosensory cortex and participate in feedback control of neuronal activity by long-range intracortical projections 38-40.
Early studies showed that the retrograde invasion of somatic action potentials into distal dendrites, as well as the generation of dendritic spikes, is under control of GABAergic inhibition 35,41,42. More recently, Murayama and colleagues found that the magnitude of dendritic spikes in the neocortex could encode the strength of a somatosensory stimulus in both awake and anesthetized rats, thereby contributing to information representation 37. The slope of the relationship between sensory input and dendritic activity was strongly influenced by the activity of deep layer INs. By using paired recordings in brain slices, they showed that disynaptic inhibition between pyramidal neurons, potentially mediated by somatostatin-expressing Martinotti-type INs, could block initiation of dendritic spikes 37. A similar mechanism appears to regulate the input-output transformation of pyramidal neurons in the CA1 region of the hippocampus. Lovett-Barron and colleagues showed that dendritic inhibition mediated by somatostatin-expressing INs could gate dendritic spike generation and subsequent somatic burst firing, regulating the gain of cellular responsiveness to network activity 43. Together, these findings suggest that dendritic inhibition serves a specific functional role in cortical and hippocampal circuits.
Inhibition by GABAARs regulates Ca2+ signaling in dendritic spines
Until recently, most studies of inhibition have focused on the regulation of postsynaptic spiking, whether initiated in the dendrites or cell body. However, as noted above, GABAergic inputs often converge with individual excitatory synapses on dendritic spines. Synaptic excitation activates both glutamate receptors and voltage-gated Ca2+ channels, producing membrane depolarization and Ca2+ influx that should be sensitive to GABAergic inhibition. However, theoretical studies initially questioned whether dendritic spines are capable of supporting GABAAR-mediated inhibition, as chloride influx into such a small compartment might rapidly diminish the inhibitory synaptic driving force 44. To address this question, we used cell-type specific expression of Channelrhodopsin2 in somatostatin-expressing INs of the mouse prefrontal cortex in combination with 2-photon Ca2+ imaging to monitor the consequences of dendritic inhibition 21. We studied local Ca2+ influx mediated by either VGCCs (opened by retrogradely propagating action potentials) or by NMDARs (opened by synaptic stimulation) and found that activation of GABAARs following optical stimulation of somatostatin-expressing INs could selectively inhibit Ca2+ transients in single spines (Fig. 2) 21. Similar inhibition of action potential-evoked Ca2+ signals was recently described for hippocampal neurons 45. Notably, the magnitude of Ca2+ inhibition was uncorrelated between adjacent spines, suggesting independent control of single excitatory inputs by GABAergic inhibition (Fig. 2) 21. Interestingly, NMDAR-dependent summation of synchronous excitatory inputs to spines directly contacted by a GABAergic synapse was reduced, indicating that synaptic integration depends critically on the precise relationship of excitatory and inhibitory synapses in the dendritic arbor.
Figure 2. Inhibition regulates dendritic spine Ca2+ and synaptic integration.
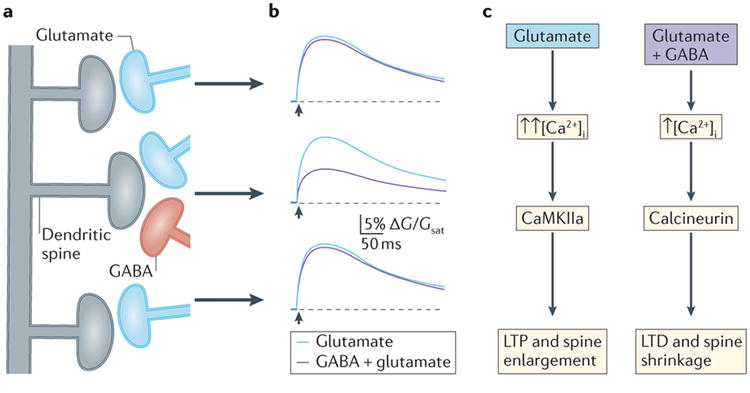
A, Inhibition of responses evoked by 2-photon uncaging of glutamate (yellow arrowheads) using one-photon GABA uncaging (large blue circle). Scale bar, 1 μm. B, Ca2+ transients and excitatory postsynaptic potentials (EPSP) for glutamatergic responses alone (black) or paired with preceding GABAergic stimulation (red), for spines indicated in (A). Scale bars, 4% ΔF/Fmax, 50 ms (top) or 0.1 mV, 5 ms (bottom). C, Integration of responses evoked by 2-photon glutamate uncaging (yellow arrowheads). Scale bar, 1 μm. D, Average EPSPs (±SEM) for glutamatergic response alone (black) and when paired with preceding inhibition (red) evoked on three neighboring spines, recorded in control saline or with NMDARs blocked by CPP. Scale bars, 0.25 mV, 20 ms. E, Relative summation of responses recorded in control saline or with CPP. *P<0.05 (Wilcoxon matched pairs test); n.s., not significant. Modified with permission from 21.
Pharmacological and computational studies showed that this local inhibition was mediated by a highly compartmentalized shunting conductance that reduces the membrane depolarization necessary for opening VGCCs and NMDARs 21. Notably, the high electrical resistance of the spine neck isolates the shunt in one spine from its neighbors. This conclusion was supported by recent theoretical work from Gidon and Segev 46 who showed that shunting inhibition can spread across large-caliber dendrites but is restricted by high resistance structures such as fine dendritic branches and dendritic spine necks. Thus, the compartmentalization of GABAAR-mediated inhibition within the dendritic arbor is heavily dependent on the structural elements innervated by presynaptic GABAergic INs.
GABA-B receptors also regulate dendritic Ca2+ signaling
Dendritic inhibition is not limited to ionotropic signaling, as two recent studies by Chalifoux and colleagues demonstrated that GABABRs regulate Ca2+ influx into dendritic spines by multiple parallel mechanisms 47,48 (see Fig. 1). First, they showed that presynaptic GABABRs decrease glutamate release from presynaptic terminals in the mouse prefrontal cortex by reducing the number of vesicles released per action potential. Second, they found that postsynaptic GABABRs decrease Ca2+ influx through NMDARs into single dendritic spines. Activation of the GABABR and its associated G protein αi subunit inhibits adenylate cyclase, leading to reduced cAMP production and downregulation of cAMP-dependent kinase (PKA). PKA-mediated phosphorylation of the NR2B subunit normally enhances Ca2+ permeability through the NMDAR 49,50, a process reversed by activation of GABABRs and other Gαi-coupled receptors 47,51. In a subsequent study, the authors showed that GABABRs directly inhibit spine Ca2+ influx through VGCCs via a PKA-independent mechanism that may involve direct channel modulation by the G protein βγ subunit 48. In addition to direct modulation of dendritic Ca2+ sources, GABABRs can also produce hyperpolarizing inhibition of PNs via their activation of G protein-coupled inwardly rectifying potassium channels (GIRKs) 52 that may deactivate voltage-dependent NMDARs and VGCCs. Thus, in combination with the above data on GABAAR-mediated Ca2+ inhibition, these results suggest independent but parallel GABAergic control over electrical and biochemical signaling in dendritic spines.
GABAergic inhibition regulates synaptic plasticity
One clear hypothesis emerging from these findings is that GABAergic inhibition likely regulates the strength and direction of Ca2+-dependent synaptic plasticity. Indeed, multiple studies have suggested that long-term potentiation (LTP) of excitatory synaptic strength in both the cortex and hippocampus is modified by GABAergic activity 53-56. In a recent report, when a LTP induction protocol that normally caused dendritic spine enlargement (a structural correlate of glutamatergic synaptic strengthening) was paired with local GABA uncaging, spine shrinkage corresponding to long-term depression occurred 55. This result appeared to be a consequence of a reduction in bulk Ca2+ mediated by GABAergic inhibition. Residual Ca2+ influx through NMDARs was still necessary for plasticity induction, indicating that dendritic inhibition does not “veto” individual synaptic contacts, but instead more subtly modulates transmission 21,55. Moreover, the effect of GABA uncaging on plasticity was limited to within 15 μm of the targeted spine, further demonstrating that functional consequences of inhibition are highly localized in the dendritic arbor 55.
Interactions between excitatory and inhibitory synapses also appear to influence plasticity in vivo. By analyzing the appearance and disappearance of dendritic spines and GFP-tagged GABAergic synapses, two recent studies estimated the stability of excitatory and inhibitory synapses. The dynamics of GABAergic inputs were found to depend on their location, with inhibitory spine contacts showing significantly greater turnover rates than those on dendritic shafts 31,32. Inhibitory synapses were also sensitive to sensory experience. Within the first four days of monocular visual deprivation, there was a dramatic loss of GABAergic contacts to both dendritic shafts and spines that did not recover after re-opening the deprived eye 32. Moreover, the turnover of excitatory and inhibitory synapses was spatially clustered, as dynamic (versus stable) excitatory and inhibitory inputs were likely to occur within 10 μm of each other, suggesting mechanistic links between excitatory and inhibitory plasticity 31. This hypothesis is supported by a recent study showing that GABAergic synaptic plasticity in a single mouse visual cortex neuron modifies induction of glutamatergic plasticity in the same cell 56.
Conclusions and open questions
In recent years, a remarkable combination of methodological approaches has yielded data supporting a novel hypothesis for the role of GABAergic function in cortical circuits. The consequences of GABAergic transmission, like glutamatergic excitation, depend critically on the precise subcellular targeting of inhibitory synapses. Specifically, multiple studies demonstrate that GABAergic inhibition can be localized to small dendritic compartments, down to the level of individual dendritic spines. This localized inhibition has critical consequences for both electrical and biochemical (e.g., Ca2+) activity in postsynaptic neurons, and may regulate both action potential generation and excitatory synaptic plasticity.
Importantly, these observations raise several questions regarding the mechanisms that control targeting of GABAergic synapses to pyramidal neuron dendrites. One possibility is that the recruitment of inhibitory contacts is activity dependent. Ca2+ influx through glutamate receptors is coupled to both potentiation and depression of GABAergic synapses depending on the relative activity of CaMKIIa and calcineurin 57,58. Thus, active excitatory inputs may specifically attract (or repel) an inhibitory bouton. A second possibility is that GABAergic inputs are recruited by the presence of specific glutamatergic afferents. As noted above, spines receiving a GABAergic synapse appear to be targeted by excitatory terminals expressing the thalamocortical synaptic marker VGlut2 32,33. The mechanisms underlying this convergent targeting remain unknown, though studies in the cerebellum have begun to uncover molecular signals, such as ankyrin and neurofascin, that govern the subcellular localization of GABAergic synapses 59.
Finally, the growing links between GABAergic dysfunction and neuropsychiatric disorders 3,4 suggest that inhibitory control of Ca2+ may be a key factor in the maintenance of synaptic connections in the brain. Future studies must begin to address how alterations in GABAergic interneurons and inhibitory synapses may lead to widespread perturbations of neuronal circuits and behavioral deficits.
References
- 1.Alivisatos AP, et al. Nanotools for neuroscience and brain activity mapping. ACS nano. 2013;7:1850–1866. doi: 10.1021/nn4012847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–243. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gogolla N, et al. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord. 2009;1:172–181. doi: 10.1007/s11689-009-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis DA, Hashimoto T. Deciphering the disease process of schizophrenia: the contribution of cortical GABA neurons. Int Rev Neurobiol. 2007;78:109–131. doi: 10.1016/S0074-7742(06)78004-7. [DOI] [PubMed] [Google Scholar]
- 5.Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annual review of neuroscience. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- 6.Yuste R. Electrical compartmentalization in dendritic spines. Annual review of neuroscience. 2013;36:429–449. doi: 10.1146/annurev-neuro-062111-150455. [DOI] [PubMed] [Google Scholar]
- 7.Higley MJ, Sabatini BL. Calcium signaling in dendrites and spines: practical and functional considerations. Neuron. 2008;59:902–913. doi: 10.1016/j.neuron.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 8.Sivilotti L, Nistri A. GABA receptor mechanisms in the central nervous system. Progress in neurobiology. 1991;36:35–92. doi: 10.1016/0301-0082(91)90036-z. [DOI] [PubMed] [Google Scholar]
- 9.Chalifoux JR, Carter AG. GABAB receptor modulation of synaptic function. Current opinion in neurobiology. 2011;21:339–344. doi: 10.1016/j.conb.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ascoli GA, et al. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci. 2008;9:557–568. doi: 10.1038/nrn2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. 2011;71:45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M. Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nature neuroscience. 2013;16:1068–1076. doi: 10.1038/nn.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Cristo G, et al. Subcellular domain-restricted GABAergic innervation in primary visual cortex in the absence of sensory and thalamic inputs. Nature neuroscience. 2004;7:1184–1186. doi: 10.1038/nn1334. [DOI] [PubMed] [Google Scholar]
- 14.Taniguchi H, Lu J, Huang ZJ. The spatial and temporal origin of chandelier cells in mouse neocortex. Science. 2013;339:70–74. doi: 10.1126/science.1227622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Higley MJ, Contreras D. Balanced excitation and inhibition determine spike timing during frequency adaptation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:448–457. doi: 10.1523/JNEUROSCI.3506-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- 17.Kruglikov I, Rudy B. Perisomatic GABA release and thalamocortical integration onto neocortical excitatory cells are regulated by neuromodulators. Neuron. 2008;58:911–924. doi: 10.1016/j.neuron.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wierenga CJ, et al. Molecular and electrophysiological characterization of GFP-expressing CA1 interneurons in GAD65-GFP mice. PloS one. 2010;5:e15915. doi: 10.1371/journal.pone.0015915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, Roby KD, Callaway EM. Immunochemical characterization of inhibitory mouse cortical neurons: three chemically distinct classes of inhibitory cells. The Journal of comparative neurology. 2010;518:389–404. doi: 10.1002/cne.22229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S, Kruglikov I, Huang ZJ, Fishell G, Rudy B. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nature neuroscience. 2013;16:1662–1670. doi: 10.1038/nn.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu CQ, et al. Compartmentalization of GABAergic inhibition by dendritic spines. Science. 2013;340:759–762. doi: 10.1126/science.1234274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, et al. Anatomical, physiological and molecular properties of Martinotti cells in the somatosensory cortex of the juvenile rat. J Physiol. 2004;561:65–90. doi: 10.1113/jphysiol.2004.073353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leao RN, et al. OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nature neuroscience. 2012;15:1524–1530. doi: 10.1038/nn.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapfer C, Glickfeld LL, Atallah BV, Scanziani M. Supralinear increase of recurrent inhibition during sparse activity in the somatosensory cortex. Nature neuroscience. 2007;10:743–753. doi: 10.1038/nn1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silberberg G, Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron. 2007;53:735–746. doi: 10.1016/j.neuron.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Tamas G, Buhl EH, Somogyi P. Fast IPSPs elicited via multiple synaptic release sites by different types of GABAergic neurone in the cat visual cortex. J Physiol. 1997;500(Pt 3):715–738. doi: 10.1113/jphysiol.1997.sp022054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Somogyi P, Tamas G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev. 1998;26:113–135. doi: 10.1016/s0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- 28.Lopez-Bendito G, et al. Distribution of metabotropic GABA receptor subunits GABAB1a/b and GABAB2 in the rat hippocampus during prenatal and postnatal development. Hippocampus. 2004;14:836–848. doi: 10.1002/hipo.10221. [DOI] [PubMed] [Google Scholar]
- 29.Sabaliauskas N, Shen H, Homanics GE, Smith SS, Aoki C. Knockout of the gamma-aminobutyric acid receptor subunit alpha4 reduces functional delta-containing extrasynaptic receptors in hippocampal pyramidal cells at the onset of puberty. Brain research. 2012;1450:11–23. doi: 10.1016/j.brainres.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serwanski DR, et al. Synaptic and nonsynaptic localization of GABAA receptors containing the alpha5 subunit in the rat brain. The Journal of comparative neurology. 2006;499:458–470. doi: 10.1002/cne.21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen JL, et al. Clustered dynamics of inhibitory synapses and dendritic spines in the adult neocortex. Neuron. 2012;74:361–373. doi: 10.1016/j.neuron.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Versendaal D, et al. Elimination of inhibitory synapses is a major component of adult ocular dominance plasticity. Neuron. 2012;74:374–383. doi: 10.1016/j.neuron.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 33.Kubota Y, Hatada S, Kondo S, Karube F, Kawaguchi Y. Neocortical inhibitory terminals innervate dendritic spines targeted by thalamocortical afferents. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:1139–1150. doi: 10.1523/JNEUROSCI.3846-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Major G, Larkum ME, Schiller J. Active properties of neocortical pyramidal neuron dendrites. Annual review of neuroscience. 2013;36:1–24. doi: 10.1146/annurev-neuro-062111-150343. [DOI] [PubMed] [Google Scholar]
- 35.Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999;398:338–341. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- 36.Losonczy A, Magee JC. Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:291–307. doi: 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 37.Murayama M, et al. Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature. 2009;457:1137–1141. doi: 10.1038/nature07663. [DOI] [PubMed] [Google Scholar]
- 38.Xu NL, et al. Nonlinear dendritic integration of sensory and motor input during an active sensing task. Nature. 2012;492:247–251. doi: 10.1038/nature11601. [DOI] [PubMed] [Google Scholar]
- 39.Lavzin M, Rapoport S, Polsky A, Garion L, Schiller J. Nonlinear dendritic processing determines angular tuning of barrel cortex neurons in vivo. Nature. 2012;490:397–401. doi: 10.1038/nature11451. [DOI] [PubMed] [Google Scholar]
- 40.Palmer LM, et al. NMDA spikes enhance action potential generation during sensory input. Nature neuroscience. 2014;17:383–390. doi: 10.1038/nn.3646. [DOI] [PubMed] [Google Scholar]
- 41.Miles R, Toth K, Gulyas AI, Hajos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/s0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- 42.Tsubokawa H, Ross WN. IPSPs modulate spike backpropagation and associated [Ca2+]i changes in the dendrites of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:2896–2906. doi: 10.1152/jn.1996.76.5.2896. [DOI] [PubMed] [Google Scholar]
- 43.Lovett-Barron M, et al. Regulation of neuronal input transformations by tunable dendritic inhibition. Nature neuroscience. 2012;15:423–430. S421–423. doi: 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]
- 44.Qian N, Sejnowski TJ. When is an inhibitory synapse effective? Proceedings of the National Academy of Sciences of the United States of America. 1990;87:8145–8149. doi: 10.1073/pnas.87.20.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanemoto Y, et al. Spatial distributions of GABA receptors and local inhibition of Ca2+ transients studied with GABA uncaging in the dendrites of CA1 pyramidal neurons. PloS one. 2011;6:e22652. doi: 10.1371/journal.pone.0022652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gidon A, Segev I. Principles governing the operation of synaptic inhibition in dendrites. Neuron. 2012;75:330–341. doi: 10.1016/j.neuron.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 47.Chalifoux JR, Carter AG. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron. 2010;66:101–113. doi: 10.1016/j.neuron.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chalifoux JR, Carter AG. GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:4221–4232. doi: 10.1523/JNEUROSCI.4561-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy JA, et al. Phosphorylation of Ser1166 on GluN2B by PKA is critical to synaptic NMDA receptor function and Ca2+ signaling in spines. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:869–879. doi: 10.1523/JNEUROSCI.4538-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skeberdis VA, et al. Protein kinase A regulates calcium permeability of NMDA receptors. Nature neuroscience. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- 51.Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca influx by D2 dopamine and A2A adenosine receptors. Nature neuroscience. 2010 doi: 10.1038/nn.2592. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Padgett CL, Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Advances in pharmacology. 2010;58:123–147. doi: 10.1016/S1054-3589(10)58006-2. [DOI] [PubMed] [Google Scholar]
- 53.Chen YJ, et al. ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21818–21823. doi: 10.1073/pnas.1010669107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meredith RM, Floyer-Lea AM, Paulsen O. Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:11142–11146. doi: 10.1523/JNEUROSCI.23-35-11142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayama T, et al. GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nature neuroscience. 2013;16:1409–1416. doi: 10.1038/nn.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang L, Maffei A. Inhibitory plasticity dictates the sign of plasticity at excitatory synapses. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:1083–1093. doi: 10.1523/JNEUROSCI.4711-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marsden KC, Beattie JB, Friedenthal J, Carroll RC. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABA(A) receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:14326–14337. doi: 10.1523/JNEUROSCI.4433-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muir J, et al. NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the gamma2 subunit. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16679–16684. doi: 10.1073/pnas.1000589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang ZJ. Subcellular organization of GABAergic synapses: role of ankyrins and L1 cell adhesion molecules. Nature neuroscience. 2006;9:163–166. doi: 10.1038/nn1638. [DOI] [PubMed] [Google Scholar]
- 60.Bloodgood BL, Sabatini BL. NMDA Receptor-Mediated Calcium Transients in Dendritic Spines. In: Van Dongen AM, editor. Biology of the NMDA Receptor. Boca Raton (FL): 2009. [PubMed] [Google Scholar]
- 61.Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Current opinion in neurobiology. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 62.Denk W, Sugimori M, Llinas R. Two types of calcium response limited to single spines in cerebellar Purkinje cells. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8279–8282. doi: 10.1073/pnas.92.18.8279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hille B. Ion channels of excitable membranes. Sinauer Associates; Sunderland, MA: 2001. [Google Scholar]