

Abstract
Genome-wide association studies (GWAS) have identified 76 variants associated with prostate cancer risk predominantly in populations of European ancestry. To identify additional susceptibility loci for this common cancer, we conducted a meta-analysis of >10 million SNPs in 43,303prostate cancer cases and 43,737 controls from studies in populations of European, African, Japanese and Latino ancestry. Twenty-three novel susceptibility loci were revealed at P<5×10-8; 15 variants were identified among men of European ancestry, 7 from multiethnic analyses and one was associated with early-onset prostate cancer. These 23 variants, in combination with the known prostate cancer risk variants, explain 33% of the familial risk of the disease in European ancestry populations. These findings provide new regions for investigation into the pathogenesis of prostate cancer and demonstrate the utility of combining ancestrally diverse populations to discover risk loci for disease.
Prostate cancer is the most common non-skin cancer in men in the Western world and epidemiological studies have shown strong evidence for genetic predisposition to prostate cancer, based on two of the most important factors, ancestry and family history. Genome-wide association studies (GWAS) have identified 76 common risk loci (reviewed in ref 1); however, over 1,000 additional common SNPs are estimated to contribute prostate cancer risk.2,3 Previous prostate cancer GWAS have been conducted primarily in populations of European ancestry2,4-7, with the majority of risk loci discovered also found to be associated with prostate cancer risk in other racial/ethnic populations.8,9 The generalizability of risk associations for a large fraction of loci suggests that combining GWAS across ancestral populations could increase power to detect risk loci that are shared among diverse populations.
To search for additional genetic risk factors for prostate cancer, we combined data from studies with existing high-density SNP genotyping in prostate cancer GWAS discovery or replication efforts in the following populations: European ancestry[34,379 cases and 33,164 controls from UK/Australia4, Cancer of the Prostate in Sweden (CAPS)10, Breast and Prostate Cancer Cohort Consortium (BPC3)6, PEGASUS, and iCOGS/PRACTICAL2]; African ancestry[5,327 cases and 5,136 controls from the African Ancestry Prostate Cancer GWAS Consortium (AAPC)11and the Ghana Prostate Study12]; Japanese ancestry[2,563 cases and 4,391 controls from a GWAS in Japanese in the Multiethnic Cohort (MEC)8, and Biobank Japan13,14]; and, Latino ancestry[1,034 cases and 1,046 controls from the MEC8]. Imputation was performed in each study using a cosmopolitan reference panel from the 1000 Genomes Project (1KGP; March, 2012). Across the various studies, 5.8-16.8M genotyped and imputed SNPs, as well as insertion/deletion variants ≥1% frequency were examined in association with prostate cancer risk (Online Methods, Supplementary Tables 1-3, Supplementary Information).
We first conducted ethnic-specific meta-analyses, with the large European ancestry sample providing the strongest statistical power for discovery of novel loci, followed by a multiethnic meta-analysis of all populations to identify additional loci with pan-ethnic effects. For these primary analyses we employed a P-value threshold of 5×10-8 to define genome-wide significance. Secondary meta-analyses focused on a) aggressive disease in the large European ancestry sample; b) aggressive disease in the combined multiethnic sample; and c) prostate cancer diagnosed at ≤55 years of age in the European ancestry sample only. Aggressive prostate cancer was defined as a Gleason score ≥8, disease stage as ‘distant’, a prostate-specific antigen (PSA) level >100 ng/ml, or death from prostate cancer. For these two secondary phenotypes, we utilized a more stringent P-value threshold of 5×10-8/2=2.5×10-8 for genome-wide significance. In each study, we tested for gene dosage effects via a 1-d.f. test for trend from logistic regression models adjusted for genetic ancestry (principal components). We observed little evidence of inflation in the test statistics in any single study or population (λ/λ1000: European, 1.14/1.00; African, 1.03/1.01; Japanese, 1.06/1.02; Hispanic, 1.00/1.00) or in the multiethnic analysis (λ=1.08,/λ1000=1.00; Online Methods, Supplementary Table 4, Supplementary Figure 1).
In the meta-analysis of the European ancestry studies,20 novel signals in18 regions ±500 kb outside of previously associated loci were observed to be associated with prostate cancer risk at P<5×10-8 (Figure 1; Supplementary Figure 2 Supplementary Figure 3). The most significant associations in each region were observed with imputed variants and we were able to confirm the imputed genotypes for 15 variants which had high imputation information scores (r2 range, 0.76-1) through direct genotyping or sequencing across multiple studies (Table 1;Online Methods, Supplementary Tables 5-8). Two of the variants were located within 370kb of each other on chromosome Xq13and are independent signals based on conditional analyses (rs6625711, P=6.1×10-10 and rs4844289, P=2.0×10-8; r2<0.01 in EUR 1KGP; Supplementary Table 9). All 15 variants were common, with minor allele frequencies (MAFs) ≥0.09, in the European ancestry population, and all but three (rs80130819/12q13, rs76939039/10q11 and rs17694493/9p21) were also common (MAF≥0.05) in African, Japanese and Latino populations. Evidence of heterogeneity in the per-allele OR was noted with 4 variants (Phet=0.01-8.4×10-6; rs17599629/1q21, rs115306967/6p21, rs17694493/9p21 and rs6625711/Xq13). Four of the 15 variants (rs10009409/4q13, rs4713266/6p24, rs80130819/12q13 and rs2807031/Xp11) had directional effects that were consistent with men of European ancestry and were nominally statistically significant (P<0.05) in at least one other population (Table 1) and for 3 SNPs, combining data across populations strengthened the statistical significance of the association (Table 1). In this large European ancestry sample we also confirmed the reported signal at 22q13 with variant rs58133635 (P=5.8×10-9; r2=0.74 with rs9623117 in 1KGP European ancestry populations (EUR); Supplemental Figure 2; Supplementary Figure 3).15
Figure 1.

Manhattan Plot of genotyped and imputed results from the European ancestry meta-analysis of overall prostate cancer risk. All SNPs within 500kb of known GWAS SNPs are omitted. The green line represents P=5×10-8. This figure shows all new variants with P<5×10-8, regardless of the confirmation results (one signal on chr1, one on chr4, one on chr17, and 2 on chr X were not confirmed). Many of the new signals are in close proximity to one another on the same chromosome (see Supplementary Table 6).
Table 1.
Association results for 23 novel risk variants for prostate cancer.
| SNP ID | Chromosome, positionb |
Nearby Genes | Allelesc | European 35,093 cases 34,599 controls |
African 5,327 cases 5,136 controls |
Japanese 2,563 cases 4,391 controls |
Latino 1,034 cases 1,046 controls |
Multiethnic 44,107 cases 45,172 controls |
PHet valued |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Risk Loci Revealed in European Ancestry Meta-Analysis | OR | P | RAFa | OR | P | RAFa | OR | P | RAFa | OR | P | RAFa | OR | P | ||||
| rs17599629 | 1q21, 150658287 | GOLPH3L | G/A | 1.10 | 5.9×10-11 | 0.22 | 1.09 | 0.13 | 0.08 | 0.97 | 0.48 | 0.18 | 0.92 | 0.23 | 0.26 | 1.08 | 2.6×10-9 | 8.6×10-3 |
| rs9287719 | 2p25, 10710730 | NOL10 | C/T | 1.07 | 1.8×10-8 | 0.46 | 1.00 | 0.98 | 0.26 | 1.07 | 0.06 | 0.42 | 1.00 | 0.99 | 0.45 | 1.06 | 2.8×10-8 | 0.21 |
| rs10009409 | 4q13, 73855253 | COX18 | T/C | 1.09 | 2.1×10-10 | 0.32 | 1.02 | 0.56 | 0.35 | 1.10 | 0.02 | 0.56 | 1.00 | 0.96 | 0.50 | 1.08 | 2.3×10-10 | 0.12 |
| rs4713266 | 6p24, 11219030 | NEDD9 | C/T | 1.07 | 3.9×10-8 | 0.52 | 1.07 | 0.03 | 0.78 | 1.06 | 0.21 | 0.23 | 1.02 | 0.81 | 0.40 | 1.06 | 2.9×10-9 | 0.89 |
| rs115457135 | 6p22, 30073776 | TRIM31 | A/G | 1.08 | 1.9×10-8 | 0.22 | 1.01 | 0.91 | 0.15 | 1.01 | 0.87 | 0.27 | 1.03 | 0.69 | 0.26 | 1.07 | 1.4×10-7 | 0.25 |
| rs115306967 | 6p21, 32400939 | HLA-DRB6 | G/C | 1.08 | 2.7×10-9 | 0.65 | 0.92 | 0.02 | 0.81 | 1.09 | 0.29 | 0.81 | 1.01 | 0.86 | 0.76 | 1.06 | 8.7×10-7 | 5.2×10-4 |
| rs56232506 | 7p12, 47437244 | TNS3 | A/G | 1.07 | 1.8×10-9 | 0.45 | 0.99 | 0.76 | 0.13 | 1.00 | 0.99 | 0.31 | 1.11 | 0.12 | 0.52 | 1.06 | 8.9×10-9 | 0.13 |
| rs17694493 | 9p21, 22041998 | CDKN2B-AS1 | G/C | 1.10 | 4.0×10-8 | 0.14 | 1.00 | 0.97 | 0.11 | 1.04 | 0.78 | 0.02 | 0.78 | 0.04 | 0.08 | 1.08 | 1.1×10-6 | 0.01 |
| rs76934034 | 10q11, 46082985 | MARCH8 | T/C | 1.14 | 4.8×10-9 | 0.91 | 0.98 | 0.88 | 0.98 | -e | 1.06 | 0.64 | 0.92 | 1.13 | 1.1×10-8 | 0.39 | ||
| rs11214775 | 11q23, 113807181 | HTR3B | G/A | 1.08 | 3.0×10-8 | 0.71 | 1.04 | 0.22 | 0.71 | 1.02 | 0.70 | 0.71 | 1.06 | 0.47 | 0.81 | 1.07 | 4.5×10-8 | 0.39 |
| rs80130819 | 12q13, 48419618 | RP1-228P16.4 | A/C | 1.13 | 4.3×10-8 | 0.91 | 1.28 | 0.02 | 0.98 | -e | 1.22 | 0.17 | 0.94 | 1.14 | 2.2×10-9 | 0.44 | ||
| rs8014671 | 14q24, 71092256 | TTC9 | G/A | 1.07 | 1.3×10-8 | 0.59 | 1.00 | 0.85 | 0.46 | 1.03 | 0.40 | 0.36 | 0.98 | 0.75 | 0.60 | 1.06 | 2.5×10-7 | 0.09 |
| rs2807031 | Xp11, 52896949 | XAGE3 | C/T | 1.07 | 8.5×10-10 | 0.18 | 1.06 | 0.02 | 0.22 | 1.17 | 0.16 | 0.05 | 1.02 | 0.82 | 0.09 | 1.07 | 2.7×10-11 | 0.77 |
| rs6625711 | Xq13, 70139850 | SLC7A | A/T | 1.07 | 6.3×10-12 | 0.41 | 0.92 | 0.004 | 0.83 | 0.99 | 0.86 | 0.48 | 0.97 | 0.52 | 0.61 | 1.04 | 6.4×10-7 | 8.4×10-6 |
| rs4844289 | Xq13, 70407983 | NLGN3/BCYRN1 | G/A | 1.05 | 1.3×10-9 | 0.39 | 0.99 | 0.58 | 0.68 | 1.00 | 0.99 | 0.72 | 1.09 | 0.05 | 0.59 | 1.04 | 8.9×10-8 | 0.04 |
| Risk Loci Revealed in Multiethnic Meta-Analysis | ||||||||||||||||||
| rs1775148 | 1q32, 205757824 | SLC41A1 | C/T | 1.06 | 1.0×10-5 | 0.27 | 1.06 | 0.04 | 0.63 | 1.12 | 2.0×10-3 | 0.52 | 1.02 | 0.82 | 0.66 | 1.06 | 3.8×10-8 | 0.40 |
| rs9443189 | 6q14, 76495882 | MYO6 | A/G | 1.07 | 5.2×10-5 | 0.86 | 1.11 | 4.5×10-4 | 0.47 | 1.07 | 0.08 | 0.68 | 1.01 | 0.93 | 0.86 | 1.08 | 3.9×10-8 | 0.64 |
| rs7153648 | 14q23, 61122526 | SIX1 | C/G | 1.09 | 6.8×10-4 | 0.06 | 1.11 | 8.8×10-4 | 0.34 | 1.17 | 1.4×10-4 | 0.30 | 1.12 | 0.27 | 0.10 | 1.11 | 2.0×10-9 | 0.50 |
| rs12051443 | 16q22, 71691329 | PHLPP2 | A/G | 1.06 | 1.1×10-5 | 0.34 | 1.09 | 0.01 | 0.25 | 1.10 | 0.02 | 0.65 | 1.06 | 0.34 | 0.50 | 1.06 | 3.0×10-8 | 0.69 |
| rs12480328 | 20q13, 49527922 | ADNP | T/C | 1.13 | 1.6×10-7 | 0.93 | 1.14 | 2.3×10-3 | 0.87 | 1.30 | 7.7×10-4 | 0.94 | 0.97 | 0.81 | 0.93 | 1.13 | 4.6×10-11 | 0.18 |
| rs1041449 | 21q22, 42901421 | TMPRSS2 | G/A | 1.06 | 2.6×10-7 | 0.44 | 1.07 | 0.03 | 0.39 | 1.02 | 0.79 | 0.12 | 1.03 | 0.65 | 0.44 | 1.06 | 2.8×10-8 | 0.84 |
| rs2238776 | 22q11, 19757892 | TBX1 | G/A | 1.09 | 1.6×10-7 | 0.80 | 0.98 | 0.81 | 0.95 | 1.08 | 0.03 | 0.60 | 1.09 | 0.22 | 0.73 | 1.08 | 1.8×10-8 | 0.60 |
| Risk Loci Revealed in Early-Onset Meta-Analysisf | ||||||||||||||||||
| rs636291 | 1p35, 10556097 | PEX14 | A/G | 1.18 | 2.1×10-8 | 0.16 | ||||||||||||
Risk Allele Frequency
Genome Build 37
Risk allele/Other allele
P-value for effect heterogeneity across populations.
Minor allele frequency <1%
Analysis limited to European ancestry populations as only small numbers of early onset cases (≤55 years) were available in the other populations.
No novel risk loci were revealed in ethnic-specific analyses within the African, Japanese or Latino ancestry populations possibly due to lack of power (Supplemental Figure 2). However, in combining results across populations in a multiethnic meta-analysis (43,303 cases, 43,737 controls), 11additional variants were identified in association with prostate cancer risk in novel risk regions at P<5×10-8 (Table 1; Supplemental Table 5; Figure 2). We confirmed the imputed genotypes for 7 variants which had high imputation information scores (r2 range, 0.81-1) through additional genotyping and sequencing (Online Methods, Supplementary Tables 6-8). All 7 variants were nominally associated with risk (P<0.05) in at least one of the non-European ancestry populations and the per-allele effects were directionally consistent across all 4 populations for 6 of the 7 variants. All variants had MAFs≥0.05 in all four populations, and no significant evidence of population heterogeneity was noted with any of these 7 variants (Table 1).
Figure 2.

Manhattan Plot of results from the multiethnic meta-analysis of overall prostate cancer risk. All SNPs within 500kb of known GWAS SNPs are omitted. The green line represents P=5×10-8. This figure shows all new variants with P<5×10-8, regardless of the confirmation results, as well as signals that were reported in the European meta-analysis that also reached 5×10-8 in the multiethnic meta-analysis (see Table 1 and Supplementary Table 6).
In secondary GWAS analyses, we detected an association with variant rs636291 at 1p36 (risk allele frequency, 0.16; OR=1.18; P=2.1×10-8; Table 1) and early-onset disease among men of European ancestry (4,147 cases ≤55 years of age and all controls, n=27,212). The association with this variant was weaker for cases diagnosed >55 years of age (23,564 cases versus all controls, n=27,212: OR=1.04; p=0.004; Phet=2.2×10-4; Supplementary Table 10). We did not detect any genome-wide significant associations with aggressive disease in the European population (n=7,903 cases) or in the combined multiethnic sample (n=10,209 cases; Supplemental Figure 4).
For the 23 novel risk variants (15 in European, 7 in multiethnic and 1 in the early onset analysis), the per-allele effects ranged from 1.06-1.14 and were consistent with log-additive effects (Supplemental Table 11). The association of each variant was noted for both aggressive and non-aggressive prostate cancer (Supplemental Table 12); for only one variant, rs7153648 at 14q23, there was suggestive evidence of a difference by disease severity (OR=1.17 for aggressive and OR=1.09 for non-aggressive disease; Phet=0.03). These results confirm what has been observed in prostate cancer GWAS to date; risk loci appear to confer risk for prostate cancer overall and not discriminate between the aggressive and indolent disease. In analyses stratified by age, 17 of the 23 variants demonstrated larger effects at younger ages (≤55 versus >55 years), although only 6 had evidence of a significant difference (p<0.05) (Supplemental Table 9). Only two of the 23 variants was modestly associated with PSA levels among controls (rs9287719 at 2p25, P=0.03 and rs115306967 at 6p21, P=0.05; Supplemental Table 13).
Of the 23 novel risk variants, 13 are located in intronic regions of genes and 2 are correlated with non-synonymous variants in adjacent genes (rs12051443/16q22, r2=0.98 with rs4788821/E60Kin MARVELD3; rs2238776/22q11, r2=0.67 with rs72646967/N397HinTBX1). Based on functional annotations of transcription factor (TF) occupancy, response element disruption, histone marks and DNaseI sensitive regions in prostate cancer cell lines (Online Methods), 12 of the risk variants are either directly located within putative functional elements or are correlated (at r2>0.9 in 1KGP EUR) with such variants (Supplementary Table 14). Using gene expression data for 145 prostate cancer tumor samples from The Cancer Genome Atlas (TCGA) (Online Methods) we also examined the cis-associations between the index SNP and expression of gene transcripts within a 1Mbregion. Among the 23 loci, 5 cis-associations were observed, albeit the associations were modest (Supplemental Table 14;Online Methods).
A number of the novel susceptibility regions are located in close proximity to genes which have either an established role, or have been directly implicated, in cancer (Table 1). The most notable is rs1041449 on chromosome 21q22, which is situated 20kb 5′ of theTMPRSS2 gene which encodes a member of a serine protease family.16 Expression of TMPRSS2 is highly specific to prostate tissue and chromosomal translocation resulting in fusion of the TMPRSS2 promoter/enhancer region with the ETS transcription factors ERG and ETV1 are frequently observed in prostate cancer.17 In analyzing data of 552 tumors characterized for the TMPRSS2-ERG fusion (46% positive) (Online Methods), we found no evidence of an association between the risk allele and fusion status (p=0.53; Supplementary Table 15). The variant risk rs1041449 is located within a number of histone marks and TF occupancy sites in the predicted enhancer region of TMPRSS2 (Figure 3) however we found little evidence that this variant influences TMPRSS2 expression in prostate tumors (n=244, P=0.60), or in normal prostate tissue (n=87, P=0.62) (Online Methods).
Figure 3.
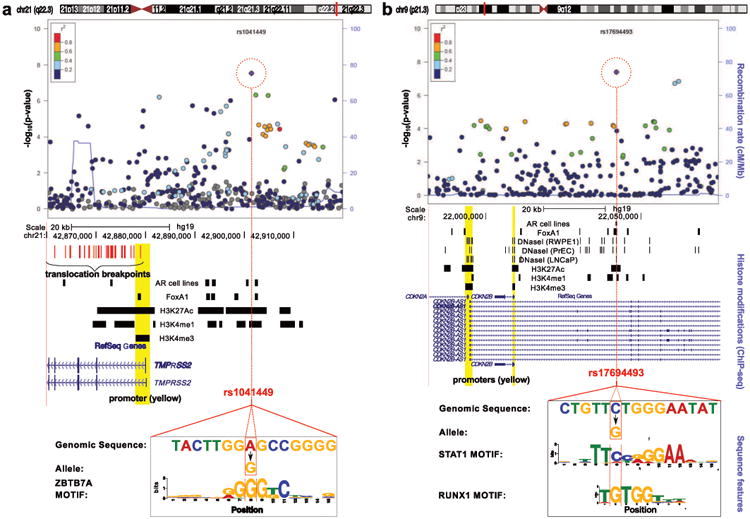
Regional plots of two novel genome-wide significant loci associated with prostate cancer risk. rs1041449/21q22 (TMPRSS2 region, left) and rs17694493/9p21 (CDKN2B-AS1region, right). Top: SNPs are plotted by their position 500kb on either side of the index SNP (purple diamond) on the chromosome against their association (-log10 P) with prostate cancer from the multiethnic meta-analysis (rs1041449) and European meta-analysis (rs17694493). SNPs surrounding the index SNP are colored to indicate the local LD structure using pairwise r2 data from the EUR panel of the 1000 Genomes (March 2012). MIDDLE: Significant peaks from TF and histone modification ChIP-seq experiments in the same genomic window (see Online Methods). All ChIP-seq in LNCaP unless otherwise indicated. BOTTOM: Genomic sequence (enclosed in black box) surrounding the SNP (red box) aligned to a LOGO graphic representing the proposed motif disruption.
Another region of notable importance is on chromosome 9p21. The risk variant, rs17694493, is intronic in CDKN2B-AS1, which encodes a long non-coding RNA – ANRIL, and is part of the CDKN2B-CDKN2A gene cluster (Figure 3). The region contains highly penetrant alleles for familial melanoma and common susceptibility alleles for melanoma, breast cancer, basal cell carcinoma, lung cancer and glioma.18-24 The index SNP, rs17694493, falls within chromatin bio features and is predicted to disrupt two TF motifs (STAT1 and RUNX1) suggesting that it may have a functional effect on the regulation of the CDKN2B-AS1 or CDNK genes (Figure 3, Supplementary Table 14), however, the variant was not found to be strongly associated with expression of either CDKN2A (P=0.19) or CDKN2B (P=0.40) in the 145 TCGA prostate tumors.
Variant rs4713266 at chromosome 6p25, is located in intron 1 of NEDD9, a gene that participates in cell adhesion, motility, the cell cycle and apoptosis, and has been implicated in progression and metastasis of several cancer types.25 Variant rs9443189 on chromosome 6q14 is intronic in MYO6, a modulator of androgen-dependent gene expression which has been found to be overexpressed in prostate cancer tumors and enhance prostate tumor growth and metastasis.26-28 Variant rs636291 on chromosome 1p36, which we found in association with early-onset prostate cancer, is located in intron 2 of PEX14 and is correlated with rs616488 (r2=0.66 in 1000 Genomes Project, EUR population), a variant reported in a GWAS of breast cancer.29
The identification of novel risk loci for prostate cancer through a multiethnic analysis demonstrates the value of combining genetic data across populations to increase statistical power for discovery. As further support for conducting multiethnic analyses, we examined the genome-wide evidence for consistency in the direction of the allelic associations between populations. Excluding SNPs ± 500kb of index signals at known loci (n=77), we defined independent signals (r2<0.2) for the European ancestry population of nominal significance at various P-value thresholds between <10-2-10-5. For the sets of SNPs defined for men of European ancestry, 53-64% had ORs that are directionally concordant for African (p=0.04-0.003, dependent on the p-value threshold bin), Asian (p=0.31-0.02) or Hispanic men (p=0.04-0.002) with the ORs in Europeans. This same observation remained once we removed the 23 risk loci identified by the current study (Supplementary Figure 5). The excess of directionally consistent associations between populations implies that additional common risk loci for prostate may be revealed through discovery efforts in multiethnic studies.
These 23 novel loci (includingrs58133635 at 22q13)15 bring the total number of susceptibility variants for prostate cancer to 100 (Supplementary Table 16). In total, we estimate these 100 risk loci account for ∼33% of the familial risk of prostate cancer in populations of European ancestry, with these additional 23 loci, with effect sizes ranging from 1.06 to 1.14, explaining ∼3.1% of the familial risk (Online Methods). Based on a polygenic risk score comprising these 100 variants for men of European ancestry (Online Methods), the top 10% of men in the highest risk stratum have a 2.9 fold (95% CI 2.8-3.1) relative risk of prostate cancer and the top 1% of men have a 5.7 fold (95% CI 4.8-6.6) relative risk compared with the population average (Supplemental Table 17). The top 10% is at a RR compared with the average of the population where it will be important to examine whether targeted screening based on family history genetic risk may reduce the over-diagnosis of indolent disease, which is a main limitation of PSA screening. Our findings demonstrate the importance of conducting large-scale genetic studies in diverse populations for the discovery of novel risk loci which continue to provide novel insights into disease mechanisms for complex traits.
Online Methods
Primary genotype data were used from four prostate cancer GWAS in men of European ancestry (UK/Australia Stages 1 and 2; CAPS 1 and 2; BPC3 and Pegasus), and a ∼200K custom replication array (iCOGS), two GWAS in men of African ancestry (AAPC and Ghana Prostate Study), two GWAS in Japanese men (JAPC and BBJ) and a single scan in Latinos (LABC).2,4-8,10-14 (Supplementary Tables 1-3; Supplementary Information). Genotypes in all scans were imputed for ∼17 M SNPs/indels using the 1000 Genome Project (March 2012 release) as a reference panel. UK/Australia stages 1 and 2, CAPS 1 and 2, Pegasus, iCOGS, AAPC, Ghana Prostate Study, LABC and JAPC were imputed using IMPUTE V2.30 BPC3, BBJ and Pegasus were imputed using Minimac. Betas and standard errors for each SNP were estimated stratified by study adjusting for principal components. In addition to analyses of overall prostate cancer risk, we performed secondary analyses of aggressive and early onset disease (age at diagnosis ≤55). Aggressive prostate cancer was defined as a Gleason score ≥8, disease stage as ‘distant’, a prostate-specific antigen (PSA) level >100 ng/ml, or death from prostate cancer. We included imputed data for SNPs with quality information scores >0.3 (IMPUTE V2) or with estimated correlation between the genotype scores and the true genotypes (r2)>0.3 (Minimac). We limited the analysis to SNPs/indels on chromosomes 1-22 as well as the X with minor allele frequency greater than 1%, except in iCOGS and Pegasus, which utilized arrays with coverage of less common alleles, where the MAF threshold was reduced to 0.5%.
Tests of homogeneity of the ORs across populations and study were assessed using likelihood ratio tests. Risk heterogeneity by disease aggressiveness and age was assessed using a case-only analysis. The associations between SNP genotypes and PSA level were assessed using linear regression, after log-transformation of PSA level to correct for skewness. Analyses were performed using SNPTEST, ProbABEL31, PLINK, Stata and an in-house C++ program (Supplementary Table 2). METAL was used to perform fixed effect ethnic-specific and multi-ethnic meta-analyses for overall prostate cancer, as well as secondary meta-analyses of aggressive and early-onset disease.32
Inflation
We excluded SNPs with ± 500kb distance of any previously known prostate cancer risk locus and estimated the inflation for each study based on the 45th percentile of the test statistic. The inflation was estimated to be 1.00 in the Latino, 1.03 in the African, 1.06 in the Japanese and 1.14 in the European ancestry studies, and, 1.07 in the European ancestry studies when SNPs at known risk loci and the iCOGS and UK2 studies were removed (see Supplementary Table 4). The inflation was converted to an equivalent inflation for a study with 1000 cases and 1000 controls (λ1000) by adjusting by effective study size, namely
where nk and mk were the number of cases and controls, respectively, for study k. Following the conversion the study-specific lambdas ranged from 0.995-1.083.
Genotyping and Concordance
The most significant associations in the meta-analyses were observed with imputed SNPs. To validate the accuracy of the imputed genotypes we genotyped each variant in ≥1847 samples (except rs9443189 and rs12051443 which were sequenced in 183 and 265 samples, respectively) that were included in the meta-analysis, and estimated the correlation between imputed and genotyped alleles. A correlation of ≥0.75 was used as the confidence threshold for imputation quality (Supplemental Table 6).
Functional Annotation
We used a number of publicly available prostate epithelia and prostate cancer ENCODE datasets of chromatin features to identify putative enhancer/regulatory regions at each risk locus.33,34 The integration of chromatin bio feature annotations with the index SNPs and correlated markers (r2>0.9) from 1KGP EUR populations was performed using FunciSNP.35 These datasets included LNCaP and RWPEI DnaseI HS sites (GSE32970) ENCODE; PrEC DNaseI HS sites (GSE29692) ENCODE; LNCaP CTCF ChIP-seq peaks (GSE33213) ENCODE; LNCaP H3K27ac and TCF7L2 (GSE51621)33, H3K4me3 and H3K4me1 histone modification ChIP-seq peaks GSE2782336; FoxA1 ChIP-seq peaks (GSE28264)37; Androgen Receptor (AR) ChIP-seq peaks38 and AR binding sites (GSE28219)39; NKX3-1 ChIP-seq peaks (GSE28264).37 We also used the highly conserved set of predicted targets of microRNA targeting (miRcode 11, June 2012 release)40. To determine whether any of the putative functional SNPs potentially affect the binding of known transcription factors, position-specific frequency matrices were employed from Factorbook.33,41
cis-eQTL analysis
Each risk locus is represented by an index SNP. For each index SNP, we retrieved all the correlated (r2≥0.9) variants EUR populations from 1KGP. The genotypes of the correlated variants in 145 prostate tumor samples and 33 normal tissue samples were downloaded from TCGA database (Feb 2013). If a variant was not represented in the TCGA data, the genotypes were imputed using IMPUTE2.30 A cis-eQTL analysis was performed for these variants and any transcript within a 1 Mb interval (500 kb on either side). Gene expression values were adjusted for somatic copy number and CpG methylation as previously described (ref. 42). Each risk variant was corrected for the number of transcripts in the interval. Significant associations were defined as a nominal p-value <0.05 and a false discovery rate <0.05 based on Benjamini-Hochberg method.
For the TMPRSS2 locus, we also used gene expression data generated from formalin-fixed paraffin embedded (FFPE) tissue in the Physicians' Health Study cohort.43 RNA was extracted with the Agencourt Form a Pure FFPE kit (Beckman Coulter, Indianapolis, IN) and amplified using the WT-Ovation FFPE System V2 (NuGEN, San Carlos, CA). cDNA was hybridization on the GeneChip Human Exon 1.0 ST microarray (Affymetrics, Santa Clara, CA). The residuals were shifted to have the original mean expression values and normalized using the RMA method.44,45 The SNP (rs1041449) was available in the BPC3 GWAS samples6; 99 participants had both tumor expression and genotype data; 54 had both normal prostate expression and genotype data.
Determination of TMPRSS2-ERG fusion status
The TMPRSS2-ERG fusion was assessed in a subset of 552 cases from study samples of FHCRC, UKGPCS, TAMPERE, ULM and IPO-PORTO. The majority of cases were typed for TMPRSS2-ERG rearrangements on FFPE tumor materials using FISH techniques according to Summersgill, et al.46 (for UKGPCS and FHCRC), Perner, et al.47 (for ULM), or Saramaki, et al.48 (for TAMPERE). The IPO-PORTO group applied qRT-PCR on RNA from fresh-frozen tumor tissues using a TaqMan gene expression assay (Hs03063375_ft, Life Technologies, Carlsbad, CA) for the fusion transcript T1G4, which is present in approximately 90% of all TMPRSS2-ERG positive prostate cancer.
Comparison of Number of Associated Loci among populations
We used the meta-analysis results from each population to evaluate the excess fraction of directionally consistent effect estimates (ORs) across populations, as evidence for additional shared susceptibility loci. We excluded the previously known prostate cancer risk regions as well as those identified in the current study (±500kb of index SNP) and compared the direction of association of SNPs defined in the European ancestry population with the other populations for several p-value thresholds. The p-values provided are based on a Chi-square binomial test for comparing proportions versus 50% chance to be in the same direction for each p-value cut-off.
Contribution to Familial Risk and Risk Stratification
The contribution of the known SNPs to the familial risk of prostate cancer, under a multiplicative model, was computed using the formula
where λ0 is the observed familial risk to first degree relatives of prostate cancercases, assumed to be 2, and λk is the familial relative risk due to locus k, given by:
where pk is the frequency of the risk allele for locus k, qk =1 − xpk and rk is the estimated per-allele odds ratio.2
Based on the assumption of a log-additive model, we constructed a polygenic risk score (PRS) from the summed genotypes weighted by the per-allele log-odds ratios.3 Thus for each individual j we derived:
Where:
N: Number of SNPs
gij: Allele dose at SNP i (0, 1, 2) for individual j
βi: Per-allele log-odds ratio of SNP i
The risk of prostate cancer was estimated for percentiles of the distribution of the PRS (<1%, 1-10%, 10-25%, 25-57%, 75-90%, 90-99%, >99%). We used effect sizes obtained from the meta-analysis of the European ancestry population and used the data from the iCOGS study for this estimation.
Supplementary Material
Acknowledgments
A full listing of acknowledgements is detailed in the Supplementary Note.
Footnotes
Author Contributions: C.A.H, R.A.E., Z.K.-J., D.F.E., B.E.H., S.J.C., S.I.B., P.K., F.W., H.N. and M.B.C. designed the study. C.A.H., Z.K.-J., A.A.A.O. and R.A.E. wrote the manuscript. A.A.A.O., F.S., Y.H., Z.W., P.W., C.C., E.S., D.L., T.D. S.J.L. performed the statistical analysis. D.O.S. and D.C. provided statistical support. D.J.H., A.S., K.P., X.S., G.A.C, Q.L., M.L.F. provided bioinformatic support as well as functional annotation and QTL data. L.C.P, K.P., L.X., L.B., M.T. conducted the genotyping and sequencing. S.B., C.G. and M.A. managed the PRACTICAL and COGS database. K.G., M.G managed the UKGPCS database. The following authors provided samples and data to the study and commented on the manuscript. L.N.K, L.L.M. and B.E.H. are principal investigators of the MEC. J.X. and S.L.Z are principal investigators of NCPCS. R.T., T.K., A.S., F.C. are EPIC investigators. E.R. is the principal investigator of EPIC. A.T., M.K. and H.N. are principal investigators of BBJ. J.L.S. is the principal investigator of KCPCS; S.K. coordinated data collection. V.L.S and R.W.D. are investigators and S.M.G. is the principal investigator of CPSII. S.S.S. and C.P. are principal investigators of the MDA prostate cancer studies. S.L., D.J.Hunter, P.K., L.M., E.L.G., J.M., M.S. are co-investigators of the Harvard cohorts and BPC3. H.G. is principal investigator of CAPS and STHLM1. W.B.I is the principal investigator of the IPCG study. A.S.K. is the principal investigator of WUGS. E.M.J. is the principal investigator of SFPCS. S.A.I. is the principal investigator of LAAPC. R.A.K. and A.B.M. are investigators of the DCPC. W.B., L.B.S. and W.Z. are principal investigators of SCCS. D.A. and J.V. are principal investigators and S.W. is study coordinator of ATBC. B.N., J.C., C.L. S.-Y. W. and A.H. are principal investigators of PCBP. B.A.R. and C.N.-D. are principal investigators of GECAP. J.W. and G.C. are principal investigators of CaP Genes. D.S. is the program officer of GAME-ON. P.G., E.A.K., A.H and L.C are investigators of SELECT. F.C.H, J.L.D. and D.E.N. are principal investigators of ProtecT. E.D.Y., Y.T., R.B.B., A.A.A., E.T., A.T., S.N., are investigators of the Ghana Prostate Study. S.J.C., S.I.B., R.N.H., M.M., M.Y., C.C.C., A.H. and K.Y. are investigators of PLCO. M.R.T. is the principal investigator and P.P. and S.M are investigators of IPO-Porto. J.B., J.C., A.S. are principal investigator of QLD. R.K. and C.S are the principal investigators, and V.M is an investigator of PCMUS. J.P. and T.S. and H.-Y.L. are the investigators of the MOFFITT study. L.C.-A. is the principal investigator of the Utah study. C.C. is the principal investigator of the Poland study. S.T. is the principal investigator of the Mayo study. P.P. and N.P. are investigators of SEARCH. C.M is the principal investigator of ULM; M.L., K.H. and A.E.R. are investigators of ULM. M.W., S.F.N., B.G.N., C.K., A.R. and P.I. are the principal investigators of CPCS1 and CPCS2. T.W, A.A. and T.T are investigators and J.S. is the principal investigator of TAMPERE. K.M. is a UKGPCS investigator. H.B. is the principal investigator, A.K.D prepared the data and C.S. coordinated the data collection of the ESTHER study. G.G.G. and G.S. are the principal investigators of MCCS; M.S. is an investigator. H.P, A.M and A.K are principal investigators of the PPF-UNIS study.
References
- 1.Eeles R, et al. The genetic epidemiology of prostate cancer and its clinical implications. Nat Rev Urol. 2014;11:18–31. doi: 10.1038/nrurol.2013.266. [DOI] [PubMed] [Google Scholar]
- 2.Eeles RA, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet. 2013;45:385–91. 391e1–2. doi: 10.1038/ng.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park JH, et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–5. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eeles RA, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–21. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 5.Gudmundsson J, et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet. 2009;41:1122–6. doi: 10.1038/ng.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schumacher FR, et al. Genome-wide association study identifies new prostate cancer susceptibility loci. Hum Mol Genet. 2011;20:3867–75. doi: 10.1093/hmg/ddr295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas G, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–5. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 8.Cheng I, et al. Evaluating genetic risk for prostate cancer among Japanese and Latinos. Cancer Epidemiol Biomarkers Prev. 2012;21:2048–58. doi: 10.1158/1055-9965.EPI-12-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haiman CA, et al. Characterizing genetic risk at known prostate cancer susceptibility loci in African Americans. PLoS Genet. 2011;7:e1001387. doi: 10.1371/journal.pgen.1001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duggan D, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst. 2007;99:1836–44. doi: 10.1093/jnci/djm250. [DOI] [PubMed] [Google Scholar]
- 11.Haiman CA, et al. Genome-wide association study of prostate cancer in men of African ancestry identifies a susceptibility locus at 17q21. Nat Genet. 2011;43:570–3. doi: 10.1038/ng.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cook MB, et al. A genome-wide association study of prostate cancer in West African men. Hum Genet. 2013 doi: 10.1007/s00439-013-1387-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akamatsu S, et al. Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nat Genet. 2012;44:426–9. S1. doi: 10.1038/ng.1104. [DOI] [PubMed] [Google Scholar]
- 14.Takata R, et al. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat Genet. 2010;42:751–4. doi: 10.1038/ng.635. [DOI] [PubMed] [Google Scholar]
- 15.Sun J, et al. Sequence variants at 22q13 are associated with prostate cancer risk. Cancer Res. 2009;69:10–5. doi: 10.1158/0008-5472.CAN-08-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–24. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 17.Morris DS, Tomlins SA, Montie JE, Chinnaiyan AM. The discovery and application of gene fusions in prostate cancer. BJU Int. 2008;102:276–82. doi: 10.1111/j.1464-410X.2008.07665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falchi M, et al. Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat Genet. 2009;41:915–9. doi: 10.1038/ng.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussussian CJ, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 20.Shete S, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stacey SN, et al. New common variants affecting susceptibility to basal cell carcinoma. Nat Genet. 2009;41:909–14. doi: 10.1038/ng.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turnbull C, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42:504–7. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wrensch M, et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet. 2009;41:905–8. doi: 10.1038/ng.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timofeeva MN, et al. Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Hum Mol Genet. 2012;21:4980–95. doi: 10.1093/hmg/dds334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tikhmyanova N, Little JL, Golemis EA. CAS proteins in normal and pathological cell growth control. Cell Mol Life Sci. 2010;67:1025–48. doi: 10.1007/s00018-009-0213-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loikkanen I, et al. Myosin VI is a modulator of androgen-dependent gene expression. Oncol Rep. 2009;22:991–5. doi: 10.3892/or_00000526. [DOI] [PubMed] [Google Scholar]
- 27.Puri C, et al. Overexpression of myosin VI in prostate cancer cells enhances PSA and VEGF secretion, but has no effect on endocytosis. Oncogene. 2010;29:188–200. doi: 10.1038/onc.2009.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei S, Dunn TA, Isaacs WB, De Marzo AM, Luo J. GOLPH2 and MYO6: putative prostate cancer markers localized to the Golgi apparatus. Prostate. 2008;68:1387–95. doi: 10.1002/pros.20806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michailidou K, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45:353–61. 361e1–2. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aulchenko YS, Struchalin MV, van Duijn CM. ProbABEL package for genome-wide association analysis of imputed data. BMC Bioinformatics. 2010;11:134. doi: 10.1186/1471-2105-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hazelett DJ, et al. Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet. 2014;10:e1004102. doi: 10.1371/journal.pgen.1004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coetzee SG, Rhie SK, Berman BP, Coetzee GA, Noushmehr H. FunciSNP: an R/bioconductor tool integrating functional non-coding data sets with genetic association studies to identify candidate regulatory SNPs. Nucleic Acids Res. 2012;40:e139. doi: 10.1093/nar/gks542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang D, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–4. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan PY, et al. Integration of regulatory networks by NKX3-1 promotes androgen-dependent prostate cancer survival. Mol Cell Biol. 2012;32:399–414. doi: 10.1128/MCB.05958-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andreu-Vieyra C, et al. Dynamic nucleosome-depleted regions at androgen receptor enhancers in the absence of ligand in prostate cancer cells. Mol Cell Biol. 2011;31:4648–62. doi: 10.1128/MCB.05934-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma NL, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23:35–47. doi: 10.1016/j.ccr.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 40.Jeggari A, Marks DS, Larsson E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics. 2012;28:2062–3. doi: 10.1093/bioinformatics/bts344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Q, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152:633–41. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Final report on the aspirin component of the ongoing Physicians' Health Study. Steering Committee of the Physicians' Health Study Research Group. N Engl J Med. 1989;321:129–35. doi: 10.1056/NEJM198907203210301. [DOI] [PubMed] [Google Scholar]
- 44.Irizarry RA, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 46.Summersgill B, Clark J, Shipley J. Fluorescence and chromogenic in situ hybridization to detect genetic aberrations in formalin-fixed paraffin embedded material, including tissue microarrays. Nat Protoc. 2008;3:220–34. doi: 10.1038/nprot.2007.534. [DOI] [PubMed] [Google Scholar]
- 47.Perner S, et al. TMPRSS2-ERG fusion prostate cancer: an early molecular event associated with invasion. Am J Surg Pathol. 2007;31:882–8. doi: 10.1097/01.pas.0000213424.38503.aa. [DOI] [PubMed] [Google Scholar]
- 48.Saramaki OR, et al. TMPRSS2:ERG fusion identifies a subgroup of prostate cancers with a favorable prognosis. Clin Cancer Res. 2008;14:3395–400. doi: 10.1158/1078-0432.CCR-07-2051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.