

Abstract
We developed a computational method (ABSOLUTE) that infers tumor purity and malignant cell ploidy directly from analysis of somatic DNA alterations. ABSOLUTE can detect subclonal heterogeneity, somatic homozygosity, and calculate statistical sensitivity to detect specific aberrations. We used ABSOLUTE to analyze ovarian cancer data and identified pervasive subclonal somatic point mutations. In contrast, mutations occurring in key tumor suppressor genes, TP53 and NF1 were predominantly clonal and homozygous, as were mutations in a candidate tumor suppressor gene, CDK12. Analysis of absolute allelic copy-number profiles from 3,155 cancer specimens revealed that genome-doubling events are common in human cancer, and likely occur in already aneuploid cells. By correlating genome-doubling status with mutation data, we found that homozygous mutations in NF1 occurred predominantly in non-doubled samples. This finding suggests that genome doubling influences the pathways of tumor progression, with recessive inactivation being less common after genome doubling.
Introduction
Defining chromosome copy number and allele ratios is fundamental to understanding the structure and history of the cancer genome. Current genomic characterization techniques measure somatic alterations in a cancer sample in units of genomes (DNA mass). The meaning of such measurements is dependent on the tumor's purity and its overall ploidy; they are hence complicated to interpret and compare across samples. Ideally, copy-number should be measured in copies-per-cancer-cell. Such measurements are straightforward to interpret and, for alterations that are fixed in the cancer cell population, are simple integer values. This is considerably more challenging than measuring relative copy-number in units of diploid DNA mass in a tumor-derived sample.
Measuring somatic copy number alterations (SCNAs) on a relative basis is straightforward using microarrays 1,2,3,4,5 or massively parallel sequencing technology 6,7; it has been the standard approach for copy-number analysis since the development of comparative genomic hybridization (CGH) 8.
Inferring absolute copy-number is more difficult because: (i) cancer cells are nearly always intermixed with an unknown fraction of normal cells (tumor purity); (ii) the actual DNA content of the cancer cells (ploidy), resulting from gross numerical and structural chromosomal abnormalities, is unknown 9, 10, 11, 12, 13 and (iii) the cancer cell population may be heterogeneous, perhaps due to ongoing subclonal evolution 14, 15. In principle, one could infer absolute copy-numbers by rescaling relative data based on cytological measurements of DNA mass per cancer cell16, 17, 18, or by single-cell sequencing approaches 15. However, such approaches are not suited to support initial large-scale efforts at comprehensive characterization of the cancer genome19.
We began focusing on this issue several years ago, initially developing ad hoc techniques20, 21. We subsequently developed a fully quantitative method (ABSOLUTE) and applied it to several cancer genome analysis projects, including The Cancer Genome Atlas (TCGA) project. ABSOLUTE provides a foundation for integrative genomic analysis of cancer genome alterations on an absolute (cellular) basis. We used these methods to correlate purity and ploidy estimates with expression subtypes and to develop statistical power calculations and use them to select well-powered samples for whole-genome sequencing in several published 22, 23, 24, and numerous ongoing projects, including breast, prostate, and skin cancer genome characterization. Recently, we extended ABSOLUTE to infer the multiplicity of somatic point-mutations in integer allelic units per cancer-cell.
Our purpose here is to: (i) to present the mathematical inference framework of the ABSOLUTE method, as well as experimental validation of its predictions; (ii) to apply it to analyze a large cancer dataset, enabling novel characterization of the incidence and timing of whole-genome doublings during tumor evolution; and (iii) to describe a novel integrated analysis of point-mutation and copy-number estimates and its application to ovarian carcinoma.
We describe three key mathematical features of ABSOLUTE. First, it jointly estimates tumor purity and ploidy directly from observed relative copy profiles (point-mutations may also be used, if available). Second, because joint estimation may not be fully determined on a single sample, it uses a large and diverse sample collection to help resolve ambiguous cases. Third, the model attempts to account for subclonal copy alterations and point mutations, which are expected in heterogeneous cancer samples.
We then report the first large-scale ‘pan-cancer’ analysis of copy-number alterations on an absolute basis, across 3,155 cancer samples, representing 25 diseases with at least 20 samples. The analysis reveals that whole-genome doubling events occur frequently during tumorigenesis, ultimately resulting in mature cancers descended from doubled cells, bearing complex karyotypes. Despite evidence that genome doublings can result in genetic instability and accelerate oncogenesis 25,13,26 the incidence and timing of such events had not been broadly characterized in human cancer.
We then describe how estimates of tumor purity and absolute copy-number allow us to analyze sequencing data to distinguish clonal and subclonal point-mutations, and to detect macroscopic subclonal structure in an ovarian cancer sample. Clonal events may be classified as homozygous or heterozygous in the cancer cells, guiding interpretation of their function. In addition, the ability to quantify integer multiplicity of point mutations distinguishes events occurring prior to DNA gains including the mutated locus from those occurring later.
Finally, our data allows characterization of somatic cancer evolution with respect to genome doubling, which we demonstrate in ovarian carcinoma and associate with clinicopathological parameters.
Results
Inference of sample purity and ploidy in cancer-derived DNA
A conceptual overview of ABSOLUTE is shown in Figure 1. When DNA is extracted from an admixed population of cancer and normal cells, the information on absolute copy number per cancer cell is lost in the mixing. The purpose of ABSOLUTE is to re-extract these data from the mixed DNA population. This process begins by generation of segmented copy number data, which is input to the ABSOLUTE algorithm together with pre-computed models of recurrent cancer karyotypes and, optionally, allelic fraction values for somatic point mutations. The output of ABSOLUTE then provides re-extracted information on the absolute cellular copy number of local DNA segments and, for point mutations, the number of mutated alleles (Figure 1).
Figure 1. Overview of tumor DNA analysis using ABSOLUTE.

A constant mass of DNA is extracted from a heterogeneous cell population consisting of cancer and normal cells. This DNA is profiled using either microarray or massively parallel sequencing technology, giving a genome-wide profile of DNA concentrations (blue lines). ABSOLUTE uses statistical models of recurrent cancer karyotypes to interpret the DNA concentrations as discrete copy states corresponding to predominantly clonal somatic copy number alterations, although some subclonal alterations are often present. If somatic point mutation data are available (from sequencing of the DNA), then the allelic fractions (fraction of sequencing reads bearing the non-reference allele) of these mutations may be used help to interpret the DNA concentrations. In addition, the allelic fractions may be reinterpreted as integer allelic copies per cancer cell (multiplicity), potentially revealing subclonal point mutations.
We begin by describing the inference framework used in the ABSOLUTE method. Suppose a cancer-tissue sample consists of a mixture of a proportion α of cancer cells (assumed to be monogenomic – that is, with homogenous SCNAs in the cancer cells) and a proportion (1-α) of contaminating normal (diploid) cells. For each locus x in the genome, let q(x) denote the integer copy-number of the locus in the cancer cells. Let τ denote the mean ploidy of the cancer-cell fraction, defined as the average value of q(x) across the genome. In the mixed cancer sample, the average absolute copy number of locus x is αq(x) + 2(1 - α) and the average ploidy is D = ατ+ 2(1 - α), measured in units of haploid genomes.
The relative copy number of locus x is therefore:
| (1) |
Because q(x) takes integer values, R(x) takes discrete values. The smallest possible value is [2(1 - α)/D], which occurs at homozygously deleted loci and corresponds to the fraction of DNA from normal cells. The spacing between values [α/D] corresponds to the concentration ratio of alleles present at one copy per cancer cell and 0 copies per normal cell. Importantly, if a cancer sample is not strictly clonal, copy-number alterations occurring in substantial subclonal fractions will appear as outliers from this pattern (Fig. 1, Supplementary Fig. 1a-c, arrows).
Similar considerations have formed the basis for algorithms to infer purity and ploidy using allelic copy-ratios derived from SNP microarrays27-31, 32. We extend absolute copy-inference to encompass somatic point mutations as follows:
| (2) |
Here, sq represents the multiplicity of the point mutation, in integer values per cancer cell (which cannot exceed q(x)), and Ds = α q(x) + 2(1 - α). The values of F(x) correspond to the expected fraction of sequencing reads that support the mutation, which depend on the sample purity and absolute somatic copy-number at the mutant locus, q(x).
The ABSOLUTE algorithm examines possible mappings from relative to integer copy numbers by jointly optimizing the two parameters α and τ (Supplementary Fig. 1c-d,h-i; Online Methods Eq. 5). In many cases, several such mappings are possible, corresponding to multiple optima.
To help resolve ambiguous cases, we used recurrent cancer-karyotype models based on large data sets of samples (Supplementary Fig. 2; Online Methods Eq. 8) to identify the simplest (that is, most common) karyotype that can adequately explain the data. This method favors simpler solutions, while preserving the flexibility to identify unexpected karyotypes given sufficient evidence from fitting the copy-profile. Indeed, several unusual karyotypes, including near-haploid (<1.2n) and hyper-aneuploid (>6n) genomes, were identified using ABSOLUTE (Supplementary Fig. 2).
Our implementation supports copy-number inference from either total or allelic copy-ratio data, such that array-CGH, SNP microarray, or massively parallel sequencing data may be used. ABSOLUTE is available for download at http://broadinstitute.org/software/ABSOLUTE.
Validation
We validated the purity and ploidy predictions made by ABSOLUTE on Affymetrix SNP microarray data using several approaches: (i) direct measurement of ploidy of 37 TCGA ovarian carcinoma samples by fluorescence-activated cell sorting 33 (Fig. 2a); (ii) Measurement of ploidy for 33 NCI60 cell lines based on spectral karyotyping34 (Fig. 2b,c); and (iii) DNA-mixing experiments, in which cancer cell lines were mixed with paired normal B-lymphocyte-derived DNAs in varying mass proportions (Fig. 2d, Online Methods). We also evaluated a related computational method, ASCAT30, on these data (Fig. 2a-d, Supplementary Note 1). Although the results were broadly concordant with our estimates, ABSOLUTE achieved significantly more accurate results (Fig. 2a-d) on our validation data. Notably, we observed an apparent bias by ASCAT to underestimate cancer cell fraction (Fig. 2 c,d), consistent with previous reports applying ASCAT in similar mixing experiments based on Illumina SNP arrays30, (Figure S4), suggesting that the bias is independent of measurement platform.
Figure 2. ABSOLUTE method validation and comparison a-d Performance of ABSOLUTE and ASCAT on 4 validation assays.

RMSE: root mean squared error. P-values were calculated on the squared errors using the paired one-sided Wilcoxon test (*: P < 0.05, **: P < 0.001). See Supplementary Note 1 for the ASCAT2.1 protocol.
a, FACS-based ploidy measurements vs. inferred ploidy estimates for 37 primary tumor samples.Dashed line indicates y=x.
b, SKY-based ploidy measurements vs. inferred ploidy estimates for 33 cancer cell-lines. Dataare displayed as in a.
c, Estimated purity of the 33 cell lines shown in (b) Dashed horizontal line indicates the truepurity (1.0).
d, Cancer-normal DNA mixing experiment results for two cell lines. DNA from each cancercell line was mixed with DNA from the matched B-lymphocyte in varying proportions (x-axis).(top) predicted vs. true DNA mixing fractions compared to the y=x line (dashed). (bottom)predicted cancer cell-line ploidy vs. mixture purity. The copy-profile of several samples wasmisinterpreted (x's); these points were not included in the RMSE calculations. Ploidy estimateswere generally consistent with previous SKY analysis of these cell lines: http://www.path.cam.ac.uk/∼pawefish/cell%20line%20catalogues/breast-cell-lines.htm.
e, Leukocyte methylation signature enrichment in tumors of histologicaly underestimatedpurity. HGS-OvCa samples are shown grouped according to the indicated histological purityestimates (x-axis)33. Black horizontal lines indicate the median purity of each group, asestimated by ABSOLUTE (y-axis). The color of each point corresponds to the degree to whichthat sample's methylation profile resembled that of purified leukocytes (Online Methods).
Notably, the purity estimates produced by ABSOLUTE appeared to be more accurate for the bulk tumor than those derived from histological examination of frozen tumor sections (Online Methods, Fig. 2e). Estimates of the proportion of contaminating normal cells for 458 ovarian carcinoma samples33 produced by ABSOLUTE were strongly correlated with a molecular signature of genomic methylation (Online Methods) seen in leukocytes (r2 = 0.59, P < 2.2×10-16, Fig. 2e), but only weakly correlated with estimates of contamination from histological examination (r2 =0.1, P = 2.4×10-12; Online Methods; Fig. 2e x-axis scale, Supplementary Fig. 4).
Estimation of tumor purity and ploidy across cancer types
We used ABSOLUTE to analyze allelic copy-ratio profiles derived from SNP arrays from 3,155 cancer samples, comprising 2,791 tissue specimens and 364 cancer cell lines. The samples came from two TCGA pilot studies describing glioblastoma (GBM; 192 samples) 21 and ovarian carcinoma (488 samples) 33, as well as 2,445 profiles incorporated from a previous pan-cancer copy-number analysis 35 (Online Methods). A minority of these samples (519 or 16.4%) could not be analyzed because they lacked clearly identifiable SCNAs, either because they were nearly euploid (“non-aberrant”), or were excessively contaminated with normal cells (“insufficient purity”), (Fig. 3a). Although sequencing data for somatic point mutations may have resolved these cases, such data were not available for the majority of samples in this cohort35.
Figure 3. Pan-cancer application of ABSOLUTE.
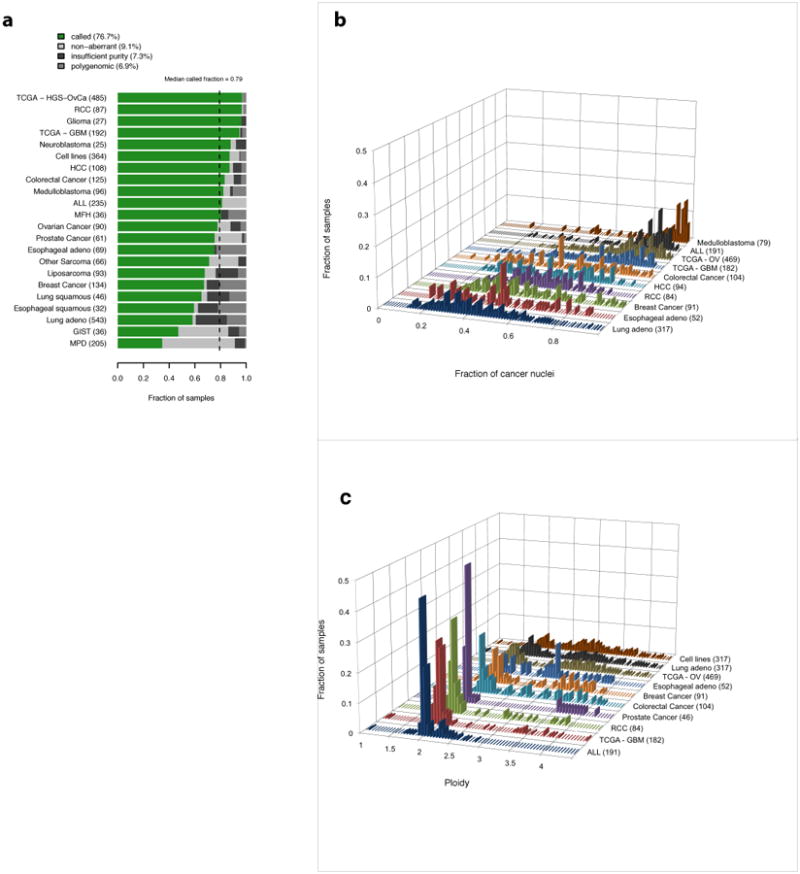
a, ABSOLUTE result types: (i) ‘called’ -- unique purity/ploidy solution; (ii) ‘non-aberrant’ --sample has no detectable somatic copy-number alterations; (iii) ‘insufficient purity’ – insufficient fraction of cancer cells; (iv) ‘polygenomic’ discrete copy-ratio levels could not be determined. See Online Methods and Supplementary Fig. 5 for a description and examples of each result type.
b, Distribution of estimated tumor purity for several datasets. The number of called tumor samples in each group is shown in parentheses. We note that, because heavily contaminated tumors are difficult to call using ABSOLUTE, several of these distributions are biased towards higher purity samples.
c, The number of called tumor samples in each group is shown in parentheses. Because tumors without SCNAs cannot be called using ABSOLUTE, these distributions do not incorporate the prevalence of such samples.
For the 2,636 samples with detectable SCNAs, ABSOLUTE provided purity and ploidy calls for 92% of cases, and designated the remaining samples as “polygenomic” (genomically heterogeneous) (Fig. 3a), (Online Methods; Supplementary Fig. 5). The fraction of called samples varied by disease type, from 34.6% (myeloproliferative disease; mostly non-aberrant genomes) to 96.7% (ovarian carcinoma, 100% aberrant genomes), with a median call-rate of 79.2% (Fig. 3a).
The distributions of estimated purity varied among cancer types, with the tested lung, esophageal, and breast cancer samples being the least pure on average in our dataset (Fig. 3b). The effect of contamination was readily visible in the copy ratios of impure tumor types (Supplementary Fig. 6). Distributions of estimated ploidy (Fig. 3c) were qualitatively consistent with those derived from previously obtained cytological data for each tumor type 13.
A table giving the characteristics of each tumor sample, as well as predicted purity / ploidy values, is available as Supplementary Table 1. An additional table detailing the segmented absolute allelic copy number of each tumor is given as Supplementary Table 2.
Power for detection of somatic point-mutations by sequencing
Both tumor purity and ploidy will affect the local depth of sequencing necessary to detect point mutations. For example, suppose that a region is present at 6 copies with only 1 copy carrying a mutation, in a sample that has 50% contamination with normal cells. In this case, only 1 of 8 alleles at this locus (6 from the cancer cells and 2 from the normal cells) carry the mutation (Supplementary Fig. 7a). We therefore expect that the mutation will be observed in only 12.5% of reads. Given this allelic fraction, local sequence coverage of 33-fold is required to detect the mutation with 80% sensitivity, assuming a sequencing error rate of 10-3 per base and a false positive rate controlled at <5×10-7, (Online Methods, Eq. 9, Supplementary Fig. 7b).
Using ABSOLUTE's estimates of purity and genome-wide integer copy-numbers, we can calculate the required coverage for powered detection of mutations present at specified allelic multiplicity per cancer cell. Similar considerations apply to detecting subclonal mutations, present in a fraction of cancer cells, by using fractional multiplicities (Supplementary Fig. 7c). We note that consideration of tumor purity in units of cells, rather than DNA fraction, is preferred for devising power calculations for sequencing experiments, since many somatic alterations of interest are expected to occur at a single copy per cancer cell.
We analyzed the distribution of purity and ploidy values in cancer samples analyzed for allelic copy number 35, 21, 33 to determine an appropriate depth of sequencing coverage needed to detect clonal mutations with power 0.8 in each sample. For this purpose, we calculated the number of reads needed to detect a mutation present in one copy at a locus present at the average copy number, given the sample's purity. (One could alternatively choose a particular percentile on the copy-number distribution.) For such a locus, we found that 30× local coverage would suffice for most samples (Supplementary Fig. 7d). By contrast, a locus of average copy number with a mutation carried in a subclone at 20% frequency would require coverage of ∼ 100-fold to allow detection in about half of the samples (Supplementary Fig. 7e). Using these calculations and the distribution of local coverage along the genome (which depends on the specific sequencing technology), one can determine the average coverage necessary to obtain sufficient power in a predefined fraction of the genome (e.g. >80% power in >80% of the genome).
We then examined whole-exome sequencing data (∼150× average coverage) from 214 TCGA ovarian carcinoma samples33 to determine whether detection power was related to the number of mutations actually observed. For each sample, we calculated the proportion of loci for which the local coverage provided at least 80% power to detect mutations present at single copy in a subclone present at 5%. Those samples with the lowest proportion of such well-powered loci tended to be 2 those in which the least mutations were detected (r2 = 0.24, P = 2.7×10-13; Supplementary Fig. 7f), suggesting that the failure to find such mutations was due to the lack of power. This result also demonstrates the importance of power calculations for characterization of the subclonal frequency spectrum.
Multiplicity analysis of somatic point mutations
We next used ABSOLUTE to convert the allelic-fraction of mutations to cellular multiplicities. For this purpose, we examined 29,268 somatic mutations identified in whole-exome hybrid capture Illumina sequencing36 data from 214 ovarian carcinoma tumor-normal pairs33 (Online Methods, Fig. 4a). Tumor purity, ploidy, and absolute copy-number values were obtained from Affymetrix SNP6.0 hybridization data on the same DNA aliquot that was sequenced, allowing the rescaling of allelic-fractions to units of multiplicity (Fig. 4a,b; Online Methods, Eq. 12).
Figure 4. Characterization of subclonal evolution in ovarian cancer by integrative analysis of SNP array and whole-exome sequencing data.

a, Histogram of allelic fraction (alternate/total read-count) values for 29,628 somatic point-mutations detected in 214 primary HGS-OvCa samples33.
b, Allelic fractions for the mutations shown in (a) were converted to point estimates of integer allele-counts per cancer cell (cellular multiplicity; x-axis) by correcting for sample purity and local copy-numbers. Subclonal mutations were identified using the model defined in equation 10.
c, The fraction of each of the 6 distinguishable nucleotide substitutions for clonal vs. subclonal point-mutations. The solid grey line indicates y=x. RMSE: root mean squared error.
d-f, Analysis of distinct subclonal populations in HGS-OvCa sample TCGA-24-1603 (purity=0.96, ploidy= 1.75).
d, Tumor SCNA profile with modeled absolute copy-numbers, as in Supplementary Fig. 1c,h. Regions of normal homologous copy-number = 1 are grayed out, clonal SCNAs are brown.Subclonal SCNAs (light blue) appear in several clusters (arrows).
e, Point mutation allelic-fraction profile. Each solid curve corresponds to a single mutation, withthe density according to the posterior (Beta) distribution implied by the observed allelic fractionand local read depth (Online Methods, Eq. 12). Color indicates degree of classification as clonalor subclonal, as in (b). Dashed curves indicate summed density of individual posteriors.
f, SCNAs from (d) and point mutations from (e) were rescaled to units of cancer cell fraction. Subclonal cancer cell fractions of ∼0.2, 0.3, and 0.6 are supported both by SCNAs and point mutations (purple, blue, and orange arrows, respectively; see corresponding copy ratios in d).
This procedure identified pervasive subclonal point-mutations in ovarian carcinoma samples. While many of the mutations were clustered around integer multiplicities, a substantial fraction occurred at multiplicities substantially less than 1 copy per average cancer cell, consistent with subclonal multiplicity (Fig. 4b).
Several lines of evidence support the validity of these subclonal mutations, including Illumina resequencing of an independent whole genome amplification aliquot, which confirmed both their presence (Supplementary Fig. 8a,b), and that their allelic fractions corresponded to subclonal multiplicity values (Supplementary Fig. 8c,d). In addition, the mutation spectrum seen for clonal and subclonal mutations was similar (RMSE = 0.02, Fig. 4c), consistent with a common mechanism of origin. Power calculations showed that these samples were at least 80% powered for detection of subclonal mutations occurring in cancer-cell fractions ranging from 10% to 53%, with a median of 19% (Supplementary Fig. 7e).
The distribution of multiplicities of the subclonal mutations was similar in the majority of samples (Fig. 4b) -- it rapidly increased at the sample-specific detection limit and then decreased in a manner approximated by an exponential decay in the multiplicity range of 0.05 to 0.5 when pooling across all samples (not shown). In contrast, the HGS-OvCa sample TCGA-24-1603 (Fig. 4d-f) showed evidence for discrete “ macroscopic subclones”. Rescaling of subclonal SCNAs (Fig. 4d) and point mutations (Fig. 4e) to units of cancer cell-fraction (Fig. 4f) revealed discrete clusters near fractions 0.2, 0.3, and 0.6 (Fig. 4f), implying the alterations within each cluster likely co-occurred in the same cells. We note that this combination of cell fractions sums to more than 1, implying that at least one of the detected subclones was nested inside another.
We next used ABSOLUTE to analyze the multiplicity of both the reference and alternate alleles, to classify point mutations as either heterozygous or homozygous in the affected cell-fraction (Fig. 5a-c). We considered 15 genes with mutations recently identified in these data 33, including 5 known tumor suppressor genes (TSGs) and 5 oncogenes (Fig. 5d). The frequency of homozygous mutations in known TSGs and oncogenes were significantly different, with a significantly elevated fraction of homozygous mutations in the TSGs (P = 0.006, Fig. 5d) and no homozygous mutations in the oncogenes: (P = 0.012, Fig. 5d). This result provides evidence supporting CDK12 as a candidate TSG in ovarian carcinoma 33, since 7 of 12 CDK12 mutations were homozygous (P= 6.5×10-5; Fig. 5d).
Figure 5. Classification of somatic mutations by multiplicity analysis in 214 primary HGS-OvCa tumor samples.

a, Empirical density estimate of allelic concentration-ratios, which are obtained by multiplication of the allelic fraction by the copy-ratio at that locus.
b, Density estimate of allelic multiplicity estimates, as in Fig. 4b, for reference vs. mutant allele.Mutations were classified into the four indicated categories according to their mutant andreference allele multiplicity.
c, The density estimates of allelic concentration-ratios are shown for each of the four mutation classes in b are shown superimposed.
d, Mutation classification profiles of genes identified as significantly recurrent in HGS-OvCa33, aswell as several COSMIC genes with previously observed mutations in these data. Note that onlyindividual point mutations were considered here; the possibility of recessive inactivation viamultiple events (compound heterozygosity) was not considered. Histograms of gene classification fractions for 1412 genes having at least 5 recurrent mutations. Dashed vertical lines denote the 5th (top) and 95th (other) percentiles of each distribution. No mutations occurring at multiplicity > 1were observed in NF1 (not shown).
Overall, TP53 had among the greatest fraction of clonal, homozygous, and multiplicity > 1 mutations of any gene in the coding exome (Fig. 5e), demonstrating the clear identification of a key initiating event in HGS-OvCa carcinogenesis 37 directly from genomic data and independently of statistical recurrence analysis.
Whole genome doubling occurs frequently in human cancer
For many cancer types, the distribution of total copy number (ploidy) was markedly bi-modal (Fig. 3c), consistent with chromosome-count profiles derived from SKY10, 13. Although these results are consistent with whole-genome doubling during their somatic evolution, it has been difficult to rule out the alternative hypothesis that evolution of high-ploidy karyotypes result from a process of successive partial amplifications 12.
To study genome doublings, we used homologous copy-number information – that is, the copy numbers, bi and ci, of the two homologous chromosome segments at each locus. By looking at the distribution of bi, ci across the genome, we could draw inferences regarding genome doubling. Immediately following genome doubling, both, bi and ci would be even numbers. Following the loss of a single copy of a region, the larger of bi and ci will remain even, but the smaller would become odd. In fact, when we looked at high-ploidy samples, we discerned that the higher of bi and ci was usually even throughout the genome, consistent with their having arisen by doubling of the entire genome (Supplementary Fig. 9). Using simulations, we found that the observed profiles were unlikely to arise due to SCNAs occurring in serial fashion at multiple independent chromosomes (P < 1e-3; Online Methods).
Using such information, we could classify samples into three groups, which we interpreted as corresponding to 0, 1 and > 1 genome doubling events in the clonal evolution of the cancer. These three groups had modal ploidy values of 1.75, 2.75, and 4.0, respectively (Fig. 6a), and also segregated into three clusters by ploidy and mean homologous copy-number imbalance (Fig. 6b). We interpreted this as evidence of SNCAs occurring with net losses, interspersed with the genome doublings. This process resulted in intermediate ploidy values for the doubled clones (2.2 – 3.4N), with pervasive imbalance of homologous chromosomes (Fig. 6b).
Figure 6. Incidence and timing of whole genome doubling events in primary cancers.

a, b, Ploidy estimates were obtained from ABSOLUTE. Mean homologue imbalance was calculated as the average difference in the homologous copy numbers at every position in the genome. Genome doubling status was inferred from the homologous copy numbers (Online Methods, Supplementary Fig. 9).
c, MPD – myeloproliferative disease, ALL – acute lymphoblastic leukemia, GBM - Glioblastomamultiforme, RCC - renal cell carcinoma, HCC - hepatocellular carcinoma, HGS-OvCa - high-gradeserous ovarian carcinoma.
d, LOH (loss of heterozygosity) was defined as 0 allelic copies. Amplification was defined as > 1allelic copy for samples with 0 genome doublings, and as > 2 allelic copies for those with 1genome doubling. Calls were made based on the modal allelic copy numbers of eachchromosome arm. Dashed lines indicate y=x.
e, SCNAs, defined as regions differing from the modal absolute copy number of each sample,were binned at adaptive resolution to maintain 200 SCNAs per bin, and renormalized by binlength. The value in each bin was further divided by the number of tumor samples in eachgenome doubling class, indicated by color as in a. The black line indicates slope = −1. Linearregression models were fit independently for each class using SCNAs 0.5 < x < 20 Mb. Thisresulted in fitted slope values of -1.05, -0.96, and -0.88 for 0, 1, and > 1 genome doublings,respectively (not shown).
The frequency of genome doubling varied across tumor types (Fig. 6c), reflecting differences in disease specific biology and clinical progression status. Hematopoietic neoplasms (MPD, ALL) had nearly no doubling events, whereas GBM, RCC, prostate cancer, various sarcomas, HCC, and medulloblastoma all had ∼25% incidence of doubling. Genome doubling was more common in epithelial cancers, with colorectal, breast, lung, ovarian, and esophageal cancers all having > 50% incidence of doubling (Fig. 6c). Esophageal adenocarcinoma had the greatest doubling incidence, consistent with previous reports of frequent “4N” populations at various stages of Barrett's esophagus progression 38,39.
Specific aneuploidies precede genome doubling
We then used ABSOLUTE to infer the temporal order of genome doubling in tumorigenesis, relative to SNCAs involving specific chromosome arms. In many cancer types, the fixation of arm-level SCNAs was inferred to occur prior to genome doubling, since both doubled and non-doubled samples had similar frequencies of specific arm-level SNCAs (Fig. 6d, Supplementary Fig. 10).
In GBM samples, LOH involving chromosomes 9 and 10, and gains of chromosome 7 occurred at equivalent frequencies (Fig. 6d), demonstrating that the most common broad SCNAs in GBM occur prior to genome doubling. Gain of chromosomes 19 and 20 was nearly exclusive to non-doubled samples, and several arms had greater LOH frequency in doubled samples (Fig. 6d), suggesting that additional biological differences underlie these samples.
Because ABSOLUTE could not distinguish between ploidy 2N and 4N in cases with no observed SCNAs, we discarded such non-aberrant samples from our analysis (Fig. 3a). For many tumor types, such cases were rare, due to the tendency for chromosomal losses following doubling (Fig. 3c, Fig. 6a,b, Supplementary Fig. 9). The representation of specific cancer subtypes may be biased by differences in ascertainment, however.
In contrast to broad chromosomal alterations, focal SCNA events occurred at greater frequency in doubled genomes (Fig. 6e). Consistent with previous reports 35, 40, 41, the observed frequency of focal SCNAs as a function of their length (L) followed power-law scaling: P(L) ∝ L−α, for L > 0.5 Mb (Fig. 6e). Genome doubling was associated with a larger overall number of SCNAs, however we obtained estimates of α near 1 for each group (Fig. 6e), suggesting that the mechanism(s) by which they were generated did not greatly depend on ploidy.
Genome doubling influences progression of ovarian carcinoma
We next sought to correlate whole-genome doubling occurrence in ovarian carcinoma with other genetic and clinical features. Genome-doubled samples showed a higher incidence of heterozygous mutations, but correcting for sample ploidy removed this effect (Fig. 7a), suggesting that the per-base mutation rates are equivalent. Clonal mutations at multiplicity > 1 were approximately ten-fold more prevalent in doubled samples; many of these events likely occurred prior to the doubling event. Genome-doubled samples had a lower frequency of homozygous deletions (Fig. 7b) and a two-fold lower rate of clonal homozygous mutations (P = 1.55×10-8, Fig. 7c). We expect that many of the observed homozygous alterations in the doubled samples were fixed prior to genome doubling.
Figure 7. Genetic and clinical associations with genome doubling in primary HGS-OvCa samples.

a-e, Colors correspond to putative genome doubling status, as indicated. Significance codes: **– P < 10-5, * – P < 0.05, NS – P > 0.05.
a-c, Number of mutations in indicated classes as a function of genome doublings. P-values were calculated with the two-sided Wilcoxin rank-sum test comparing samples with 0 and 1 genome doublings. Error bars indicate standard errors of the means.
d, P-values were calculated with the two-sided Wilcoxin rank-sum test.
e, P-values were calculated using the log-rank test.
The lower incidence of homozygous mutations in genome-doubled samples may reflect the fact that more events are required to render a mutation homozygous in a genome-doubled sample (although the effect may be partially offset, however, by a possible increase in genetic instability following doubling, e.g., by centrosome duplication 42). These considerations suggest that genome-doubled samples evolve via distinct trajectories, because inactivation of tumor suppressors may occur less frequently following doubling.
We note that 13 of the 15 detected point mutations in the tumor suppressor NF1 occurred in the 93 ovarian samples that had not undergone genome doubling (P = 0.002; Fisher's exact test), and these mutations were uniformly homozygous (not shown). This is consistent with selection for recessive inactivation of NF1, a typical pattern for a tumor suppressor gene. It also suggests that non-genome-doubled ovarian carcinoma samples evolved via a distinct trajectory, rather than being precursors to doubled samples. If not, many NF1 mutations would be homozygous with multiplicity > 1 in doubled samples, as is seen for TP53.
Finally, we noted that genome-doubled samples were associated with a significant increase in the age at pathological diagnosis (Fig. 7d) and with a significantly greater incidence of cancer recurrence (Fig. 7e).
Discussion
We here report the development of a reliable high-throughput method to infer absolute homologous copy-numbers from tumor-derived DNA samples, as well as multiplicity values of point mutations (ABSOLUTE). It may be possible to extend ABSOLUTE to other types of genomic alterations, such as structural rearrangements and small insertions/deletions, although this may require longer sequence reads in order to ensure accurate sequence alignment.
ABSOLUTE analysis of SCNAs demonstrated that many of the copy-number alterations analyzed were fixed in the cancer lineage represented in the sample (Fig. 3). This was recapitulated in ovarian cancer by somatic point-mutations, many of which were fixed at integer multiplicity (Fig. 4b). Classification of point mutations based on their multiplicities may help distinguish tumor suppressors and on cogenes (Fig. 5d). Knowledge of discrete tumor copy-states, subclonal structure, and genome doubling status provides a foundation for further reconstruction of the phylogenetic relationships within a cancer and the temporal sequence by which a given cancer genome arose43, 44, 45.
ABSOLUTE provides a framework for the design of studies using genomic sequencing to detect variant alleles in cancer tissue samples, based on calculation of sensitivity to detect mutations as a function of sample purity, local copy-number and sequencing depth (Supplementary Fig. 7). The high accuracy of tumor purity and ploidy estimates produced by ABSOLUTE based on SNP microarray data (Fig. 2) make it possible to determine the sequencing depth required for a given sample or to select suitable samples given a fixed sequencing depth. Such considerations are vital to the interpretation of subclonal point-mutations (Supplementary Fig. 7f, 10).
Analysis of the predicted absolute allelic copy-number profiles across human cancers produced by ABSOLUTE shed new light on cancer genome evolution. The observed SCNA profiles (Supplementary Fig. 9) were consistent with a common trajectory consisting of an early period of chromosomal instability followed by the emergence of a stable aneuploid clone, as previously described 11. Our data further indicate that genome doublings occur in a subset of cancer cells already harboring arm-level SCNAs characteristic of the corresponding tumor type. The genomes of these cancers were therefore shaped by selection at chromosomal arm-level resolution prior to doubling and further clonal outgrowth (Fig. 6d, Supplementary Fig. 10).
These findings are broadly consistent with an earlier interpretation of primary breast cancer FACS/SKY profiles 46, and has recently been recapitulated in studies of macro-dissected and ploidy-sorted cell populations 14, and single cell sequencing15 of primary breast tumors. We note that this model represents a departure from the idea that tetraploidization is an initiating event 47,13,26,48,49. In addition, the association of genome lineage (Fig. 6c) and with age at diagnosis in ovarian carcinoma (Fig. 7d) is consistent with a recently described mechanism linking telomere crisis, DNA damage response, and genome doubling in cultured mouse embryonic fibroblasts 48.
The analysis of clonality presented in this work offers a path forward for clinical sequencing of cancer, and provides means to address recently reported concerns regarding intra-tumor heterogeneity50, 14, 44, 15, 45, 51,52. Analysis using ABSOLUTE can identify alterations present in all cancer cells contributing to the DNA aliquot (Fig. 1), even if such clonal alterations correspond to the minority of observed mutations. Such alterations are candidate founding oncogenic drivers for a given cancer, which may be the preferred therapeutic targets. Further characterization of subclonal somatic alterations in cancer may become important for understanding variable response to targeted therapeutics, with the clonalilty of targeted mutations potentially affecting response level.
Online Methods
Inference of purity, ploidy, and absolute somatic copy-numbers
populations presents a challenge to such analysis, both cytological 10 and genomic data 11 have supported this assumption, as have reports of similar SCNA profiles obtained from paired primary and metastatic lesions Homologue-specific copy ratios (HSCRs; copy-ratio estimates of both homologous chromosomes), are preferred for analysis with ABSOLUTE, and were used for all analyses in this study Although ABSOLUTE can be run on total copy-ratio data (e.g. from array CGH or low-pass sequencing data), we do not present such results here. The use of HSCRs reduces the ambiguity of copy profiles. For example, the total copy-ratio profile of a sample without SCNAs would be equivalent for ploidy values of 1,2,3, etc., however the HSCR profile would rule out odd ploidy values, since these would not be consistent with equal homologous copy-numbers. In addition, since subclonal SCNAs will generally affect only one of the two HSCR values in a given genomic segment, the ratio of clonal to subclonal SCNAs genome-wide is generally higher when considering HSCRs rather than only total copy-numbers.
HSCRs were derived from segmental estimates of phased multipoint allelic copy-ratios at heterozygous loci using the program HAPSEG 53 on data from Affymetrix SNP arrays. As part of this procedure, haplotype panels from population linkage analysis HAPMAP3 54 were used in conjunction with statistical phasing software BEAGLE 55 in order to estimate the phased germline genotypes at SNP markers in each cancer sample. This increased our sensitivity for resolving these genotypes, since it naturally exploits the local statistical dependencies between SNPs 53. In addition, this allowed greater resolution of small differences between homologous copy-ratios, since phase information from allelic imbalance of heterozygous markers due to SCNA could be combined with the statistical phasing from the haplotype panels 53.
Identification and evaluation of candidate tumor purity and ploidy values
We describe identification of candidate tumor purity and ploidy values and calculation of their SCNA-fit log-likelihood scores using a probabilistic model. This is accomplished by fitting the input HSCR estimates with a Gaussian mixture model, with components centered at the discrete concentration-ratios implied by eq. 1. The model also supports a small fraction of subclonal events which are not restricted to the discrete levels. Candidate solutions are identified by searching for local optima of this likelihood over a large range of purity and ploidy values. This results in a discrete set of candidate solutions with corresponding SCNA-fit likelihoods (eq. 1, Supplementary Fig. 1d,h).
These scores quantify the evidence for each solution contributed by explanation of the observed HSCRs as integer SCNAs. These computations are independent for each sample. The input data consist of N HSCRs xi, i ∈ {1, …, N}. Each of these is observed with standard error σi, and corresponds to a genomic fraction denoted wi. Each of the xi is assumed to have arisen from either one of Q integer copy-number states: Q = {0,1, …, Q − 1}, or an additional state Z corresponding to subclonal copy-number. We refer to the collection of possible copy-states as S = Q ∪ Z. We define Q + 1 indicators s for the copy-state of each segment, with p(si) representing the probability of segment i having been generated from state s ∈ S. The integer copy-states of S are indexed by q ∈ Q; the non-integer state is denoted by z.
The expected copy-ratio corresponding to each integer copy-number q(x) in a tumor sample is given by equation 1. Note that when homologous copy-ratios are used, this equation becomes:
| (3) |
since HSCRs are measured relative to haploid concentrations, as opposed to the diploid values assumed by eqn. 1. D is related to tumor purity and ploidy (α and τ) (eq. 1, Supplementary Fig. 1). The observed xi are modeled with a mixture of Q Gaussian components located at μ = {μq∈Q} representing integer copy-states Q and an additional uniform component Z. The mixture Z allows segments to be assigned non-integer copy values so that occasional subclonal alterations or artifacts do not dramatically impact the likelihood.
| (4) |
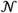 and
and
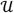 denote the normal and uniform densities, respectively. The free parameter σH represents sample-level noise in excess of the HSCR standard-error σi, which might represent a moderate number of related clones in the malignant cell population, ongoing genomic instability, or excessive noise due to variable experimental conditions. The mixture weights θ = {θs∈S} specify the expected genomic fraction allocated to each copy-state. The parameter d represents the domain of the uniform density, corresponding to the range of plausible copy-ratio values (we used d = 7).
denote the normal and uniform densities, respectively. The free parameter σH represents sample-level noise in excess of the HSCR standard-error σi, which might represent a moderate number of related clones in the malignant cell population, ongoing genomic instability, or excessive noise due to variable experimental conditions. The mixture weights θ = {θs∈S} specify the expected genomic fraction allocated to each copy-state. The parameter d represents the domain of the uniform density, corresponding to the range of plausible copy-ratio values (we used d = 7).
Some complication arises due to the fact that the data consist of copy-ratios calculated from a segmentation of the genome. For consistent interpretation, the mixture weights (P(si|wi, θ)) must be calculated for each segment separately, taking into account the variable genomic fraction wi. This is accomplished by constraining the canonical averages of genomic mass allocated to each copy-state to match those specified by θ:
where 〈·〉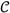 denotes the average over all configurations {si}, weighted by the function
= P(si|wi,λ) This density corresponds to the maximum entropy distribution over s subject to these constraints:
denotes the average over all configurations {si}, weighted by the function
= P(si|wi,λ) This density corresponds to the maximum entropy distribution over s subject to these constraints:
where s# indicates the order of state s in a sequence of copy-states, beginning with 0. Values of the Q Lagrange multipliers λ are determined via Nelder-Mead optimization of L2 loss:
This approximation allows for robustness of the SCNA-fit score to over-segmentation of the data. The likelihood of a given segment i is then calculated as:
and the full log-likelihood of the data is then:
| (5) |
We define the parameterization b = 2(1 − α), δτ = α/D, which determines μ via eq. (3). Candidate purity and ploidy solutions for a tumor sample are identified by optimization of eq. (5) with respect to b and δτ. Calculation of eq. (5) requires estimates of θ and σH, which are not known a priori. We make an approximation (scale-separation) assuming that locations of the modes of eq. (5) are invariant to moderate fluctuations in these parameters. A provisional likelihood for each xi may then be calculated by
Candidate purity and ploidy solutions are then identified by optimization of
initiated from all points in a regular lattice spanning the domain of b and δτ. The parameter σP was set to 0.01 for this study. We verified that the above approximation identified modes equivalent to those obtained through a full Metropolis-Hastings Markov chain Monte Carlo (MCMC) simulation (data not shown). The approximation allows for much simpler computations to be used.
The SCNA-fit score for each solution is calculated after optimization of σH:
with the elements of θ calculated for each value of σH by:
| (6) |
The final calculation of the SCNA-fit log-likelihood for each mode is obtained by inserting μ̂, θ ^, and σ̂H into eq. (5). Estimates of the copy-state indicators for each segment are calculated as:
Note that each q̂i is a vector representing the posterior probability of each Q ∈ Q integer copy-states, corresponding to the copy-ratios (locations) μ.
Genome-wide absolute copy-profiles are over-determined with respect to DNA ploidy estimates. An alternate estimate of ploidy may be calculated as the expected absolute copy-number over the genome:
| (7) |
By definition, this quantity (τ̂g) is an alternate estimate of cancer ploidy (note an additional factor of 2 is added when HSCRs are used). Because (τ̂g) is a weighted average over discrete states in the modeled data, it is expected to be more robust to experimental fluctuations that shift or scale the copy-profile slightly. Note that, for this computation, the q̂ij were calculated with θ̂z = 0, so that the above expectation is over integer states only.
We verified that these estimates were generally close to the values of (τ̂) obtained by optimization of the SCNA-fit likelihood (RMSE=0.26, Supplementary Fig. 12a). However, we noted a relationship between the level of discordance between ploidy estimators and the mean of the calibrated data (Supplementary Fig. 12b). Noting that correctly calibrated copy-ratio data always has mean = 1, we examined whether the miscalibration was due to scaling bias in the data. We found that this model explained nearly two thirds of the discordance between the two estimates (corrected RMSE=0.09, Supplementary Fig. 12c), by which we inferred that scaling biases dominated our miscalibration. This is significant, as such biases do not effect the estimation of tumor purity (Supplementary Fig. 12).
Two additional transformations of the copy-state locations μ are used when copy-ratios are measured using microarrays. The first of these accounts for the effect of attenuation with an isothermal adsorbtion model 7:
where the value ϕ̂ parameterizes the attenuation response in a given sample, and is estimated via HAPSEG. The second transformation is a variance stabilization for microarray data adapted from 56:
where ση and σε represent multiplicative and additive noise scales for each microarray, estimated by HAPSEG. This transformation is applied to the marker-level data during estimation of the xi values, after which their distribution is approximately normal. The normal mixture component specified in (4) then becomes h(xi) = h(g(μq))+εi, and the corresponding likelihood calculations are performed under these transformations.
Karyotype models
Additional information is generally required in order to reliably select the correct tumor purity and ploidy solution from the set of candidates identified by fitting the model in (4). In a given tumor sample, several combinations of theoretically possible purity, ploidy, and copy number values may map to equivalent copy ratios (Supplementary Fig. 1c,h). Furthermore, the presence of subclonal SCNAs may result in a spuriously high ploidy solution with an implausible karyotype receiving a greater SCNA-fit likelihood by over-discretizing the copy profile, allowing their assignment to integer copy-levels (Supplementary Fig. 1h-j).
ABSOLUTE models common cancer karyotypes by grouping tumor sets according to similarities in their absolute homologous copy-number profiles (Supplementary Fig. 2). These models are constructed directly from the tumor data in a ‘boot-strapping’ fashion, whereby a subset of tumors with relatively unambiguous profiles (e.g., due to high purity values) is used initialize the models, iteratively allowing more tumors to be called, etc. Previous cytogenetic characterizations of human cancer were used to guide this process 13. These models enable calculation of a karyotype likelihood, for each candidate purity/ploidy solution, reflecting the similarity of the corresponding karyotype to models associated with the specified disease of the input tumor sample (8). Integration of the SCNA-fit and karyotype likelihoods favors robust and unambiguous identification of the correct purity and ploidy values in many tumor samples (Supplementary Fig. 1d,h).The selection of a solution implying a less common karyotype requires greater evidence from the SCNA-fit of the copy profile.
Prior knowledge of karyotypes characteristic of a particular disease is summarized as a mixture of K multivariate multinomial distributions over the integer homologous copy-states Q = [0,7] of each chromosome arm. For a given candidate purity and ploidy solution, the corresponding segmental copy-state indicators for each segment i, q̂ij, are summarized into estimates of the J arm-level homologous copy-numbers, denoted Ĉ. The karyotype log-likelihood score is calculated as:
| (8) |
where wi denotes the weight of each mixture component. The karyotype models Ki are J × Q SCNA probability matrices obtained by clustering arm-level homologous copy-states of modeled copy-profiles using the standard expectation-maximization (EM) algorithm 57 for multinomial mixtures. This calculation identifies groups of disease subtypes with similar genomic copy profiles (Supplementary Fig. 2). Note that copy-states for both homologues of each arm are modeled (J = 78). Karyotype scores for samples with only total copy-ratio data are calculated using convolution of the multinomial probabilities for the two homologous chromosomes.
The number of clusters K for each disease was chosen by minimizing the Bayesian information criterion (BIC) complexity penalty: −2L̂k + KJ log (N), where L̂k indicates the sum of ℒK values over the N input samples, computed using K clusters. In order to avoid local minima, the EM algorithm was run 25 times for each value of K ∈ [2,8] with randomized starting points and the best model was retained.
The models were constructed in a semi-automated fashion by seeding with relatively unambiguous copy-profiles. As tumors were added, the use of recurrent karyotypes clearly identified the correct solutions of additional samples, etc. For example, LOH of chr17 occurs in nearly 100% of ovarian carcinoma samples 33, allowing the model to learn that solutions implying LOH of chr17 are likely to be correct. In total, models for 14 disease types were created. Diseases with fewer than 40 samples called by ABSOLUTE were omitted from this procedure. In addition, a “master” model was created by combining called primary cancer profiles. This model was used for diseases with no specific karyotype model.
Limitations of joint purity/ploidy inference from copy-profile data
Accurate calibration of both the SCNA-fit and karyotype models to the true level of certainty implied by the data would allow for assignment of probabilities to each candidate solution; we do not believe that the models we have presented here sufficiently capture the complexity of cancer genomes to allow for such interpretations. Even with manual review, analysis with ABSOLUTE may occasionally result in incorrect interpretations, for example genome-doubling without subsequent detectable gains or losses would result in a solution implying half the true ploidy value, which in some cases may correspond to a plausible karyotype model. Alternately, when multiple subclonal SCNAs appear close to the midpoints between adjacent clonal peaks, a solution implying twice the true ploidy may be chosen. We note that samples with no reliably detected SCNAs could not be called in our framework (ploidy 2N or 4N; purity undetermined). Such samples were therefore excluded from downstream analysis (see below). Estimation of inference error-rate requires independent measurement of sample ploidy. Further validation experiments in diverse tumor types will help to clarify any disease specific caveats.
We note that the use of somatic mutation allelic-fractions, combined with the SCNA copy-ratios, generally allows for increased sensitivity for samples with few SCNAs. In addition, the mutation data helps distinguish genome-doubling ambiguity in purity/ploidy estimation, although they do not inform ambiguities of the type b′ = b + 2(1-α)/D (Supplementary Fig. 1d,i, eq. 1). Thus, combined analysis generally facilitates obtaining higher call-rates using ABSOLUTE (not shown).
Fortunately, many samples in our pan-cancer SNP array dataset were consistent with frequent SCNA both before and after genome doubling, enabling unambiguous inference for many samples without use of somatic point-mutation data. This aspect of cancer genome evolution was noted previously in breast cancer cytogenetic data 46. We note that manual review of ABSOLUTE results was performed prior to generation of the FACS validation data or analysis of the NCI60 cell-line ploidy estimates (Fig. 2a,b).
Identification of samples refractory to purity/ploidy inference
In order to facilitate rapid analysis of many cancer samples used in this study, ABSOLUTE was programed to automatically identify copy profiles that cannot be reliably called and to classify them into informative failure categories (Fig. 3a), which were defined by the following criteria. Define m̂ as the sorted vector of posterior genome-wide copy-state allocations (θ̂), so that m̂1 represents the greatest element of θ̂ (the modal copy-state). This vector was constructed with θ0 replaced by 0 if θ0 < 0.01 and b < 0.15, so that germline copy-number variants (CNVs) or regions of inherited homozygosity are not confused with small SCNAs implying very pure samples. The categories are then:
non-aberrant: m̂3 < 0.001, m̂2 < 0.005, σ̂H < 0.02
insufficient purity: m̂3 < 0.001, m̂2 < 0.005, σ̂H ≥ 0.02
polygenomic: θ̂z > 0.2.
These criteria were applied to the top-ranked mode for each sample (combined SCNA-fit and karyotype scores). Several examples of each outcome are shown in Supplementary Fig. 5. The above designations led to reasonably good concordance of automated calls with those obtained after manual review. We note that the use of somatic point-mutation data increases the calling sensitivity within these sample categories.
Cancer cell-line DNA mixing experiment
DNA extracted from two cancer cell lines (HCC38, HCC1143) was mixed with DNA from matched B-lymphocyte cell lines (HCC38BL, HCC1143BL) in various proportions, and hybridized to Affymetrix 250 K Sty SNP arrays. Stock DNA aliquots were created for each cell-line by normalization of DNA concentration to 50ng/μl. Mixing of cancer and matched B-lymphocyte DNA to each required mixing fraction was done by volume.
FACS analysis of primary tumor samples
Formalin-fixed and paraffin-embedded blocks from ovarian serous carcinoma cases were available from tumor-sections corresponding to the frozen blocks from which DNA-aliquots were obtained for SNP-array hybridization. Multiple curls containing at least 70% tumor cell nuclei were cut to an aggregate thickness of 150 microns. Sections were disaggregated and labeled with propidium iodide (DNA stain). FACS was performed to determine ploidy.
Determination of tumor purity via pathology review
Frozen ovarian serous cystadenocarcinoma specimens were collected from multiple hospital tissue banks and maintained frozen in liquid nitrogen vapors. A tissue portion was created with two flanking H&E slides (arbitrarily named top and bottom) as follows: tissues were mounted in optimal cutting temperature media (OCT) and brought to -20°C. A 4μm frozen section (top slide) was cut with a cryostat (Leica Microsystems, Wetzlar, Germany). A specimen for molecular extraction was created by shaving 100 mg of tumor tissue from the tissue face with a scalpel, then a second 4μm frozen section was cut (bottom slide). An H&E stain was conducted on both slide tissue sections using an Autostainer XL with integrated coverslipper (Leica). Digital images of slides were created at 20× resolution using a Scanscope XT (Aperio, Vista, CA, USA). Board-certified pathologists conducted the pathology review remotely via ImageScope software (Aperio). Pathologists initially reviewed each slide at low magnification to determine low power microscopic morphology, then increased magnification to 20× and reviewed 10 representative high power fields on each slide. Diagnosis of ovarian serous cystadenocarcinoma was verified, and tumor purity was determined as the proportion of tumor nuclei present compared to the total nuclei present on the slide. The tumor purity of the extracted specimen was calculated as the average purity score of the top and bottom slides. Quality control included a random review of 10% of slides by a second pathologist to verify consistency of reads.
Leukocyte methylation signature
DNA methylation data for 489 high stage, high grade serous ovarian tumors and eight normal fallopian tube samples was obtained from http://tcga.cancer.gov/dataportal/. In addition, buffy coat samples from two female individuals were obtained. All data were generated with Illumina Infinium HumanMethylation 27 arrays, which interrogate 27,578 CpG sites located in proximity to the transcription start sites of 14,475 consensus coding sequencing in the NCBI Database (Genome Build 36). The level of DNA methylation at each probe was summarized with beta values ranging from 0 (unmethylated) to 1 (methylated) 58.
The leukocyte methylation signature was derived as follows. Each probe was ranked by the difference in mean beta value in buffy coat and fallopian tube samples. We retained the 100 probes with the largest positive difference and the 100 with the largest negative difference between mean DNA methylation in normal fallopian tube tissues and peripheral blood leukocytes, designated BC and FT, (buffy coat and fallopian tube enriched, respectively). Let Tik denote the beta value for probe k in tumor sample i. Let Bk denote the average beta value of buffy coat samples for each probe. Let Tk denote the minimum observed beta value across all tumor samples for the BC probes and the maximum for the FT probes. Denote by fB the fraction of buffy coat components in the sample, then we have the following equation for each probe: Tik = BkfB + Tk(l−fB). Solving this equation for fB gives: fB = (Tik − Tk)/(Bk − Tk). The values of fB for each of the 200 probes in the signature were calculated and a kernel density estimate was obtained. The leukocyte signature was then calculated as the mode of this density estimate.
Selection of datasets
We analyzed 2445 Affymetrix 250 K Sty SNP samples from a previous pan cancer survey 35 containing 3131 cancer samples. Because our processing of the data required use of the Birdseed algorithm 59, external datasets lacking diploid PCR controls could not be used. In addition, cancer types with fewer than 20 samples were excluded. In addition, 680 Affymetrix SNP6.0 samples were taken from the TCGA GBM 21 and HGS-OvCa 33 studies, as well as 30 cell-line samples, bringing to total sample count to 3155. The complete table of cancer samples analyzed is available as Supplementary file 1. The complete table of ABSOLUTE results is available as Supplementary file 2.
Power calculation for somatic mutation detection in cancer tissue samples
We develop a framework for calculation of statistical power for the detection of mutations. Power to detect a variant depends on the allelic fraction f and local depth of coverage n. To calculate power, we model the idealized scenario in which random sequencing errors occur uniformly with rate ε. We calculate a minimum number of supporting reads k such that the probability of observing k or more identical non-reference reads due to sequencing error is less than a defined false-positiverate (FPR):
Where
Variants with ≥ k supporting reads are then considered detected. We specified the sequencing error rate ε = 1 × 10−3 and FPR = 5× 10−7 for all computations in this study. Power is then calculated as:
| (9) |
Where
We consider the case of detecting clonal somatic variants present at a single copy per cancer cell in cancer-tissue derived DNA samples. Given estimates of purity (α) and local absolute copy-number (qt), the allelic fraction of such variants is:
| (10) |
Power is calculated in such cases as Pow(n, δ).
In order to simplify the relationship between power and tumor purity/ploidy for presentation in Supplementary Fig. 7, we considered the detection power of the expected locus, over the genome-wide copy average. Power as such is determined by the sample allelic index δτ = α/D, which is solely a function of tumor purity/ploidy (eqn. 1). Expected power is obtained by using allelic fraction f =δτ in eq. (9). This calculation differs only in the substitution of expected genomic copy-number, i.e. ploidy (τ)for the local copy-number qt in eq: (10).
Power for expected subclonal variants present in fraction sf of cancer cells is given by Pow(n, sfδτ) This calculation was used for Supplementary Fig. 7c,e. Local copy calculations using Pow(n, sfδ) were used for Supplementary Figs. 7f, 11.
Detection of somatic point mutations in ovarian carcinoma
We analyzed whole-exome hybrid capture Illumina sequencing (WES) 36 data from 214 ovarian carcinoma tumor-normal pairs previously analyzed by the TCGA consortium 33. We used the program muTect (K. Cibulskis, et al., in preparation.) We have used a newer version of the program muTect than used in previous analysis of this data 33. The primary improvement in the new version is a reduction in the prior that somatic mutations be at an allelic fraction of 0.5, allowing greater sensitive at low allelic-fraction mutations, such as clonal events in impure samples, or to subclonal mutations. This procedure resulted in 29,268 somatic mutations.
Inference of point mutation multiplicity
We develop a probabilistic model for inference of the integer multiplicities for both germline and somatic variants, based on knowledge of tumor purity and genome-wide absolute copy-numbers. Denote the absolute homologous copy-numbers at a mutant locus as q1 and q2, with q1 ≥ q2. The possible multiplicities of germline variants are then:
where qt = q1 + q2. Under the assumption that all somatic point-mutations arise uniquely on a single haplotype, the possible multiplicities are:
Note that when only total copy-ratio data are available, q2 above is unknown, and qt is used instead.
Germline mutations are generally present in both the cancer and normal cell populations, with somatic copy-number alterations affecting the allelic fraction. A heterozygous variant in the germline, with multiplicity gq in the cancer genome, has allelic fraction:
| (11) |
Where as the allelic fraction of homozygous germline variants is 1 regardless of α. For somatic point mutations, the expected allelic fraction at multiplicity sq is fsq = sqδ, with δ as in eq. (10).
Consider an observed somatic point-mutation of unknown copy sq ∈ sq, observed allelic fraction f̂, and with n total reads covering the locus. The complete likelihood of f̂ may represented as a mixture of beta distributions corresponding to each element of sq, plus an additional component S corresponding to subclonal states:
| (12) |
where wSq ∈ wq specify mixture weights for each state in sq, and wSc specifies the subclonal component weight. The subclonal component S is specified by composing a Beta distribution (modeling sampling noise) with an exponential distribution over subclonal cancer-cell fractions, having a single parameter λ:
Note the change of coordinates in the exponential component using δ; this allows modeling in consistent units of cancer-cell fractions, regardless of tumor purity and local copy-number (note this distribution is renormalized on the unit interval). The probability of a given integer copy-state sq may then be calculated as:
Similarly, the probability that a given mutation is subclonal is calculated as:
For the computations in this study we fixed λ = 25, wsq to 0.25, and wSc to 0.75, which produced a fit to the combined-sample mutation-fraction distribution (Fig. 4b). The results presented in Fig. 4 were robust to various settings.
Optimization of mixture components weights corresponding to integer somatic multiplicities may be accomplished in a manner similar to that described for the SNCA mixture model in eq. (6). A Dirichlet prior may be specified as a vector of pseudo-counts equivalent to prior observations of each multiplicity value. Weights are then calculated as the mode of the posterior Dirichlet calculated from the observed counts. These computations are used to calculate a mutation-score likely for each purity ploidy mode when ABSOLUTE is run with paired SCNA and somatic point mutation data.
Simulation of cancer-genome evolution to support genome-doubling inferences
A simple simulation was performed to obtain P-values for the probability that an observed configuration of homologous copy-numbers could be produced from a serial process of independent gains and losses. Genome-wide homologous copy-numbers are summarized at chromosome-arm resolution as integer gains/losses (total of 78 states). We then fix the total number of gains/losses N for the sample, and calculate rates for each arm, which are normalized to probabilities. Simulation of the sample is performed by independently sampling N gains and losses from these probabilities. This was repeated 1000 times for each sample, keeping track of the number of times M that the extent of even high homologous copy-number present in the observed sample was attained or exceeded. The P-value is then: P = M/1000, if M > 0, otherwise P < 0.001.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Cancer Institute as part of The Cancer Genome Atlas project: U24CA126546 (M.M.), U24CA143867 (M.M.), and U24CA143845 (G.G.). S.L.C. and E.H. were supported by training grant T32 HG002295 from the National Human Genome Research Institute (NHGRI). In addition, D.P. was supported by NIH - 5R01 GM083299-14, D.A.L. by DoD W81XWH-10-1-0222, T.Z. by NIH/NIGMS 5 T32 GM008313, R.B. by NIH K08CA122833 and U54CA143798. B.A.W. was supported by NRSA grant F32CA113126.
Footnotes
Author Contributions: M.M. posed the concept of using allelic copy number analysis to assess tumor genomic purity. G.G. and S.L.C. conceived and designed the analysis. S.L.C. designed and implemented the ABSOLUTE algorithm, nd performed data analysis. K.C. assisted with mutation calling and validation. E.H. performed analysis with ASCAT and assisted with multiplicity analysis. A.M. assisted with the determination of statistical power for the ovarian cancer data. T.Z. assisted with analysis of the SCNA length distribution vs. genome doubling. R.O. and W.W. processed the SNP array experiments. W.W. designed and executed the DNA mixing experiment. H.S. and P.W.L. conceived and executed the leukocyte methylation signature analysis. B.A.W. contributed insight into allelic analysis of SNP array data. D.A.L. managed the FACS analysis of specimens for ploidy determination. All authors contributed to the final manuscript. D.P. and R.B. provided critical review of the manuscript. S.L.C., M.M., G.G., and E.S.L. wrote the manuscript. R.B., M.M., and G.G. provided leadership for the project.
References
- 1.Pinkel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 2.Mei R, et al. Genome-wide detection of allelic imbalance using human SNPs and high-density DNA arrays. Genome Res. 2000;10:1126–1137. doi: 10.1101/gr.10.8.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindblad-Toh K, et al. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat Biotechnol. 2000;18:1001–1005. doi: 10.1038/79269. [DOI] [PubMed] [Google Scholar]
- 4.Zhao X, et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64:3060–3071. doi: 10.1158/0008-5472.can-03-3308. [DOI] [PubMed] [Google Scholar]
- 5.Bignell GR, et al. High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 2004;14:287–295. doi: 10.1101/gr.2012304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell PJ, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang DY, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kallioniemi A, et al. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- 9.Boveri T. Journal of cell science. 2008 doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- 10.Mitelman F. Mutation research. 2000;462:247–253. doi: 10.1016/s1383-5742(00)00006-5. [DOI] [PubMed] [Google Scholar]
- 11.Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nat Genet. 2003;34:369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- 12.Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol. 2004;5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- 13.Storchova Z, Kuffer C. The consequences of tetraploidy and aneuploidy. J Cell Sci. 2008;121:3859–3866. doi: 10.1242/jcs.039537. [DOI] [PubMed] [Google Scholar]
- 14.Navin N, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011 doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hicks J, et al. High-resolution ROMA CGH and FISH analysis of aneuploid and diploid breast tumors. Cold Spring Harb Symp Quant Biol. 2005;70:51–63. doi: 10.1101/sqb.2005.70.055. [DOI] [PubMed] [Google Scholar]
- 17.Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 18.Lyng H, et al. GeneCount: genome-wide calculation of absolute tumor DNA copy numbers from array comparative genomic hybridization data. Genome Biol. 2008;9:R86. doi: 10.1186/gb-2008-9-5-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hudson TJ, et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weir BA, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stransky N, et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science. 2011 doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bass AJ, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nature genetics. 2011;43:964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujiwara T, et al. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 26.Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attiyeh EF, et al. Genomic copy number determination in cancer cells from single nucleotide polymorphism microarrays based on quantitative genotyping corrected for aneuploidy. Genome Res. 2009;19:276–283. doi: 10.1101/gr.075671.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Popova T, et al. Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol. 2009;10:R128. doi: 10.1186/gb-2009-10-11-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenman CD, et al. PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics. 2010;11:164–175. doi: 10.1093/biostatistics/kxp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Loo P, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yau C, et al. A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome Biol. 2010;11:R92. doi: 10.1186/gb-2010-11-9-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li A, et al. GPHMM: an integrated hidden Markov model for identification of copy number alteration and loss of heterozygosity in complex tumor samples using whole genome SNP arrays. Nucleic acids research. 2011;39:4928–4941. doi: 10.1093/nar/gkr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell D, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roschke AV, et al. Karyotypic complexity of the NCI-60 drug-screening panel. Cancer Res. 2003;63:8634–8647. [PubMed] [Google Scholar]
- 35.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gnirke A, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol. 2008;26:5284–5293. doi: 10.1200/JCO.2008.18.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galipeau PC, et al. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc Natl Acad Sci U S A. 1996;93:7081–7084. doi: 10.1073/pnas.93.14.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrett MT, et al. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Genet. 1999;22:106–109. doi: 10.1038/8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mermel CH, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fudenberg G, Getz G, Meyerson M, Mirny LA. High order chromatin architecture shapes the landscape of chromosomal alterations in cancer. Nature biotechnology. 2011;29:1109–1113. doi: 10.1038/nbt.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell PJ, et al. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci U S A. 2008;105:13081–13086. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yachida S, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012 doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dutrillaux B, Gerbault-Seureau M, Remvikos Y, Zafrani B, Prieur M. Breast cancer genetic evolution: I. Data from cytogenetics and DNA content. Breast Cancer Res Treat. 1991;19:245–255. doi: 10.1007/BF01961161. [DOI] [PubMed] [Google Scholar]
- 47.Ganem NJ, Storchova Z, Pellman D. Curr Opin Genet Dev. 2007;17:157–162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 48.Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell. 2010;141:81–93. doi: 10.1016/j.cell.2010.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bazeley PS, et al. A model for random genetic damage directing selection of diploid or aneuploid tumours. Cell Prolif. 2011;44:212–223. doi: 10.1111/j.1365-2184.2011.00746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu W, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–565. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walter MJ, et al. Clonal architecture of secondary acute myeloid leukemia. The New England journal of medicine. 2012;366:1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter S, Meyerson Matthew, Getz Gad. Accurate estimation of homologue-specific DNA concentration-ratios in cancer samples allows long-range haplotyping. 2011 Available from Nature Precedings http://hdl.handle.net/10101/npre.2011.6494.1.
- 54.Altshuler DM, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Browning BL, Yu Z. Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. American journal of human genetics. 2009;85:847–861. doi: 10.1016/j.ajhg.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huber W, von Heydebreck A, Sultmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18 Suppl 1:S96–104. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- 57.Dempster AP, Laird NM, Rubin DB. Maximum Likelihood from Incomplete Data via the EM Algorithm. Journal of the Royal Statistical Society Series B (Methodological) 1977;39:1–38. [Google Scholar]
- 58.Noushmehr H, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Korn JM, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.