

Abstract
Over 70% of patients with head and neck squamous cell carcinoma (HNSCC) present with locoregionally advanced stage III and IV disease. In spite of aggressive therapy, locoregional disease recurs in 60% and metastatic disease develops in 15% to 25% of patients causing a major decline in quality and length of life. Therefore, there is a need to identify and understand genes that are responsible for inducing an aggressive HNSCC phenotype. Evidence has shown that protein kinase C (PKC) ε is a transforming oncogene and may play a role in HNSCC progression. In this study, we determine the downstream signaling pathway mediated by PKCε to promote an aggressive HNSCC phenotype. RNA interference knockdown of PKCε in UMSCC11A and UMSCC36, two highly invasive and motile HNSCC cell lines with elevated endogenous PKCε levels, resulted in cells that were significantly less invasive and motile than the small interfering RNA–scrambled control transfectants; 51 ± 5% (P < 0.006) and 49 ± 3% (P < 0.010) inhibition in invasion and 69 ± 1% (P < 0.0005) and 66 ± 3% (P < 0.0001) inhibition in motility, respectively. PKCε-deficient UMSCC11A clones had reduced levels of active and serine-phosphorylated RhoA and RhoC. Moreover, constitutive active RhoA completely rescued the invasion and motility defect, whereas constitutive active RhoC completely rescued the invasion and partially rescued the motility defect of PKCε-deficient UMSCC11A clones. These results indicate that RhoA and RhoC are downstream of PKCε and critical for PKCε-mediated cell invasion and motility. Our study shows, for the first time, that PKCε is involved in a coordinated regulation of RhoA and RhoC activation, possibly through direct post-translational phosphorylation.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most frequent cancer worldwide, comprising ~50% of all malignancies in some developing nations. Surgery and radiotherapy are highly effective in the treatment of stage I and II disease; however, >70% of patients present with locoregionally advanced stage III and IV disease. In spite of initial aggressive therapy for HNSCC patients with advanced disease, local recurrence rates are upwards of 60% and metastatic disease develops in 15% to 25% of patients, causing a major decline in quality and length of life (1). Fewer than 30% of HNSCC patients are free of disease after 3 years and the 5-year survival rates have remained largely unchanged in the past three decades (2). Thus, critical issues in HNSCC are disease recurrence and metastasis, accounting for the high incidence of morbidity and mortality.
Protein kinase C (PKC) is a family of serine/threonine kinases known to play critical roles in the signal transduction pathways involved in proliferation, differentiation, apoptosis, and migration (3). Decades of work on PKCs have shown that PKC isoforms play heterologous, sometimes paradoxically antagonistic roles in cancer initiation and progression. Thus, it is necessary to understand the role of each individual PKC isoform in oncogenesis. Overexpression of PKCε in normal fibroblasts resulted in malignant transformation with changes in morphology, serum-and anchorage-dependent growth, cell cycle progression, and the ability to form tumors in experimental animals (4, 5). Epidermis-specific PKCε transgenic mice developed highly malignant and metastatic squamous cell carcinomas in response to 12-O-tetradecanoylphorbol-13-acetate stimulation (6). PKCε was shown to regulate hepatocyte growth factor/c-Met signaling, a pathway implicated in angiogenesis, tumorigenesis, and metastasis in HNSCC (7–9). Our laboratory reported that elevated PKCε is prognostic of lower overall and disease-free survival in patients with invasive breast cancer (10). Moreover, higher levels of PKCε were found to correlate with an increase in disease recurrence and a decrease in overall survival in HNSCC (11). In this study, we report that specific inhibition of PKCε is sufficient to dampen the invasive and motile phenotype of aggressive HNSCC. Our results show that targeted disruption of PKCε leads to inactivation of RhoA and RhoC, indicating that the PKCε-Rho GTPase signaling axis is critical for promoting an invasive and motile tumor cell phenotype in HNSCC.
Materials and Methods
Cell lines
UMSCC HNSCC cell lines were provided by Dr. Thomas Carey (University of Michigan, Ann Arbor, MI) and cultured in DMEM supplemented with 10% fetal bovine serum. Immortalized normal oral epithelial cells (E6/E7-NOE) were provided by Drs. William Foulkes and Ala-Eddin Al Moustafa (McGill University, Montreal, Quebec, Canada) and cultured in keratinocyte serum-free medium without supplement.
Generation of stable small interfering RNA-PKCε UMSCC11A and UMSCC36 clones
Double-stranded oligonucleotides, 5′-GATCGATC-CAAGTCAGCAC-3′ of PKCε were synthesized (Invitrogen, Carlsbad, CA) and cloned into pSilencer2.1-U6 hygro expression vector (Ambion, Austin, TX) and named small interfering RNA (siRNA)-PKCε. A 19-bp scrambled sequence with no significant sequence homology to any known human gene sequences (silencer-negative control 1; Ambion) was cloned into pSilencer2.1-U6 hygro expression vector and named siRNA scrambled. Sequencing of siRNA-PKCε and siRNA-scrambled expression vectors was done by the University of Michigan DNA Sequencing Core and verified. UMSCC11A and UMSCC36 cells were transfected with siRNA scrambled or siRNA-PKCε using electroporation (Nucleofector device, Amaxa Biosystems, Gaithersburg, MD). Single clones were established by culturing transfected cells in the described medium supplemented with 100 μg/mL hygromycin (Invitrogen) for 21 days. Protein levels of PKCε were determined by Western blot analysis.
Generation of PKCε-deficient/G14V-RhoA and PKCε-deficient/G14V-RhoC UMSCC11A clones
NH2-terminal 3× hemagglutinin (HA)–tagged constitutive active (G14V mutant) RhoA and RhoC were obtained from Gutherie cDNA Resource center (Sayre, PA). 3× HA-tagged G14V-RhoA, 3×-HA-tagged G14V-RhoC, or empty vector (pcDNA3.1) was transfected into siRNA-PKCε UMSCC11A clones using electroporation. Polyclonal cell populations were established by culturing transfected cells in the described medium supplemented with 100 μg/mL hygromycin and 300 μg/mL G418 for 21 to 28 days. Protein levels of PKCε, HA-tagged RhoA, and HA-tagged RhoC were determined by Western blot analysis.
Western blot analysis
Whole-cell lysates (50 μg) were mixed with Laemelli buffer, heat denatured for 3 minutes, separated by 10% SDS-PAGE, and transferred to polyvinylidene difluoride (PVDF) membrane. Nonspecific binding was blocked by overnight incubation with 2% bovine serum albumin in TBS with 0.05% Tween 20. Immobilized proteins were probed using antibodies specific for PKCε (Upstate Biotechnology, Charlottesville, VA), RhoA (Cytoskeleton, Denver, CO), RhoC (Santa Cruz Biotechnology, Santa Cruz, CA), HA (Covance, Princeton, NJ), or actin (Santa Cruz Biotechnology) and visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ).
Cell invasion and random motility assays
Cell invasion was determined as described from the cell invasion assay kit (Chemicon International, Temecula, CA). Cells were harvested and resuspended in serum-free medium. An aliquot (1 × 105 cells) of the prepared cell suspension was added into the chamber and incubated for 24 hours at 37°C in a 10% CO2 tissue culture incubator. Noninvading cells were gently removed from the interior of the inserts with a cotton-tipped swab. Invasive cells were stained and quantified by colorimetric reading at 560 nm. Random cell motility was determined as described from the motility assay kit (Cellomics, Pittsburgh, PA). Cells were harvested, suspended in serum-free medium, and plated on top of a field of microscopic fluorescent beads. After a 16-hour incubation period, cells were fixed and areas of clearing in the fluorescent bead field corresponding to phagokinetic cell tracks were quantified using NIH ScionImager.
Rho GTPase activation assay
Cells were lysed in 300 μL of 50 mmol/L Tris (pH 7.4), 10 mmol/L MgCl2, 500 mmol/L NaCl, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate, and protease inhibitors. Lysates (1–2 mg) were cleared at 16,000 × g for 5 minutes, and the supernatants were rotated for 2 hours at 4°C with 60 μg glutathione S-transferase (GST)–Rho-binding domain (RBD; GST fusion protein containing the RBD of rhotekin) bound to glutathione-Sepharose beads. Samples were washed in 50 mmol/L Tris (pH 7.4), 10 mmol/L MgCl2, 150 mmol/L NaCl, 1% Triton X-100, and protease inhibitors. Western blot analyses were done on GST-RBD pull-downs with antibodies specific to RhoA, RhoC, or HA.
Statistical analysis
Data are presented as mean ± SE and analyzed using Student’s t test. P < 0.05 was considered statistically significant.
Results and Discussion
A recent report by our group showed that PKCε is a predictive biomarker of survival in invasive breast cancer patients and specific disruption of PKCε resulted in significant inhibition in tumorigenesis and metastasis in an orthotopic model of breast cancer (10). To date, there is only one publication of note focusing on the role of PKCs in HNSCC. In this study, PKCα, PKCβ, PKCε, PKCγ, and PKCζ protein levels were shown to be elevated in the primary tumor tissue of oral cavity patients; however, only PKCε was found to be a prognostic marker in this small cohort of 29 patients, even better than the traditional gold standard of tumor-node-metastasis staging (11). Elevated PKCε was reported to be significantly associated with an increase in disease recurrence (P < 0.04) and a decrease in overall survival (P < 0.02). These results provide evidence that PKCε promotes an aggressive cancer phenotype and that further studies on the role of PKCε in HNSCC are warranted.
In our initial experiment, we determined PKCε protein levels in a panel of HNSCC cell lines. PKCε levels were dramatically elevated in all of the HNSCC cell lines, with the exception of UMSCC38, compared with E6/E7 immortalized oral epithelial cells (NOE; Fig. 1A). We decided to focus our work on UMSCC11A and UMSCC36 to further examine the role of PKCε in promoting an invasive and motile phenotype in HNSCC. As shown in Fig. 1B, UMSCC11A and UMSCC36 cells were significantly more motile than NOE cells; 471 ± 15% and 602 ± 26%, respectively (n = 3; P < 0.001). Moreover, UMSCC11A and UMSCC36 cells were ~2-fold more invasive than NOE cells; 116 ± 6% and 110 ± 5%, respectively (n = 5; P < 0.001; Fig. 1C). These results clearly show that UMSCC11A and UMSCC36 cells, two HNSCC cell lines with elevated PKCε levels, are significantly more motile and invasive than oral epithelial cells.
Figure 1.
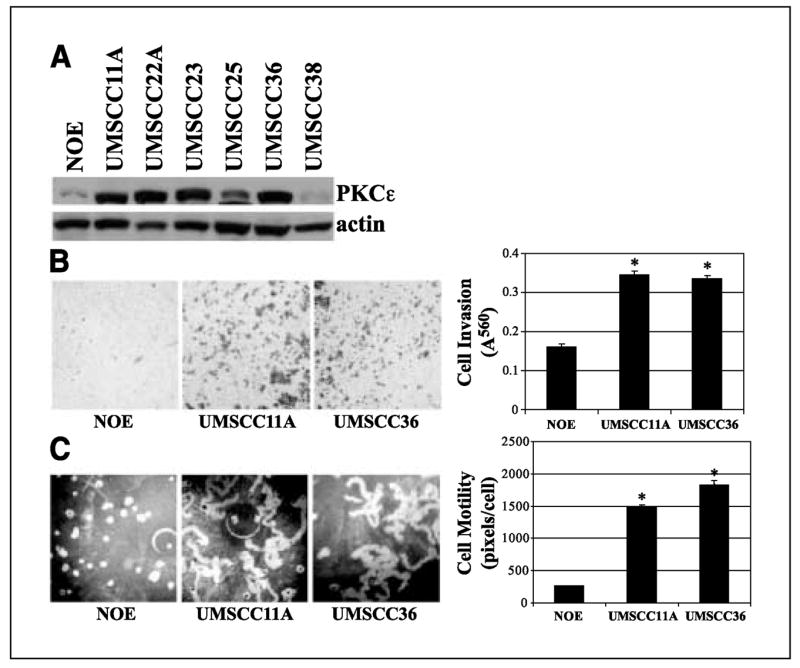
Elevated PKCε levels are associated with a highly invasive and motile phenotype in HNSCC. A, PKCε proteins levels of E6/E7 immortalized oral epithelial cells (NOE) and a panel of HNSCC cell lines (UMSCC series). B, UMSCC11A and UMSCC36 cells are more invasive than NOE cells. A reconstituted basement membrane assay was used to assess for cell invasion. The number of invaded cells was counted in five fields and the mean values were determined. *, P < 0.001. C, UMSCC11A and UMSCC36 cells are more motile than NOE cells. Areas of clearing in the fluorescent bead field corresponding to phagokinetic cell tracks were quantified using NIH ScionImager. *, P < 0.001.
An important question to address from a clinical and translational prospective is whether specific disruption of PKCε would be sufficient to dampen the invasive and motile phenotype of UMSCC11A and UMSCC36 cells. Stable PKCε-deficient UMSCC11A and UMSCC36 clones were generated using a H1 RNA polymerase III promoter-PKCε targeting siRNA expression vector. As shown in Fig. 2A, PKCε protein levels were significantly lower in the siRNA-PKCε UMSCC11A and UMSCC36 clones than in untransfected or siRNA-scrambled control cells. PKCε-deficient UMSCC11A and UMSCC36 clones were significantly less invasive and motile than the parental or siRNA-scrambled control cells (Fig. 2B and C). Cell invasion was decreased by 42% to 59% (n = 3; P < 0.006) and cell motility was suppressed by 68% to 70% (n = 3; P < 0.0005) for siRNA-PKCε UMSCC11A clones compared with siRNA-scrambled UMSCC11A cells. Moreover, cell invasion and cell motility for siRNA-PKCε UMSCC36 clones were inhibited by 34% to 57% (n = 3; P < 0.01) and 62% to 75% (n = 3; P < 0.0001) relative to siRNA-scrambled UMSCC36 cells, respectively.
Figure 2.

RNAi-mediated disruption of PKCε inhibits invasion and motility in UMSCC11A and UMSCC36. A, siRNA-PKCε UMSCC11A and UMSCC36 clones have reduced PKCε protein levels. B, PKCε-deficient UMSCC11A and UMSCC36 clones are significantly less invasive than siRNA-scrambled control cells or untransfected parental cells. *, P < 0.006 for siRNA-PKCε UMSCC11A and P < 0.01 for siRNA-PKCε UMSCC36 compared with siRNA-scrambled control cells. C, PKCε-deficient UMSCC11A and UMSCC36 clones are significantly less motile than siRNA-scrambled control cells or untransfected parental cells. *, P < 0.0005 for siRNA-PKCε UMSCC11A and P < 0.0001 for siRNA-PKCε UMSCC36 compared with siRNA-scrambled control cells. D, representative cell motility field for each cell line and PKCε-deficient clone.
The downstream signaling pathway used by PKCε to promote an invasive and motile phenotype is still not completely understood. Our laboratory reported that RNA interference (RNAi) knockdown of PKCε in MDA-MB-231 cells, a highly metastatic breast cancer cell line with elevated endogenous PKCε levels, resulted in a significant reduction in the levels of activate RhoC compared with siRNA-scrambled control cells (10). RhoC shares significant sequence homology, 82% nucleotide identity and 91% amino acid identity, with RhoA, so we determined if RhoA and RhoC were modulated through a PKCε-dependent mechanism in HNSCC. PKCε-deficient UMSCC11A clones had significantly lower amounts of active RhoA and RhoC levels compared with siRNA-scrambled control cells (Fig. 3A). In silico prediction of phosphorylation sites identified multiple serine and threonine residues as putative PKC phosphorylation sites on RhoA and RhoC, suggesting that phosphorylation of RhoA and RhoC through PKCε may be a possibility. So, we determined if the levels of serine- and threonine-phosphorylated RhoA and RhoC were modulated in our PKCε-deficient cells. As shown in Fig. 3B, PKCε-deficient UMSCC11A clones had reduced levels of serine-phosphorylated RhoA and RhoC compared with siRNA-scrambled control cells. Threonine-phosphorylated RhoA and RhoC were not detected for siRNA-scrambled control cells or PKCε-deficient clones (data not shown). These results suggest that PKCε-mediated regulation of RhoA and RhoC may be at the post-translational level, most likely through serine phosphorylation. Ongoing research in the laboratory is to use mass spectrometry to identify the serine residues on RhoA and RhoC that are phosphorylated by PKCε.
Figure 3.
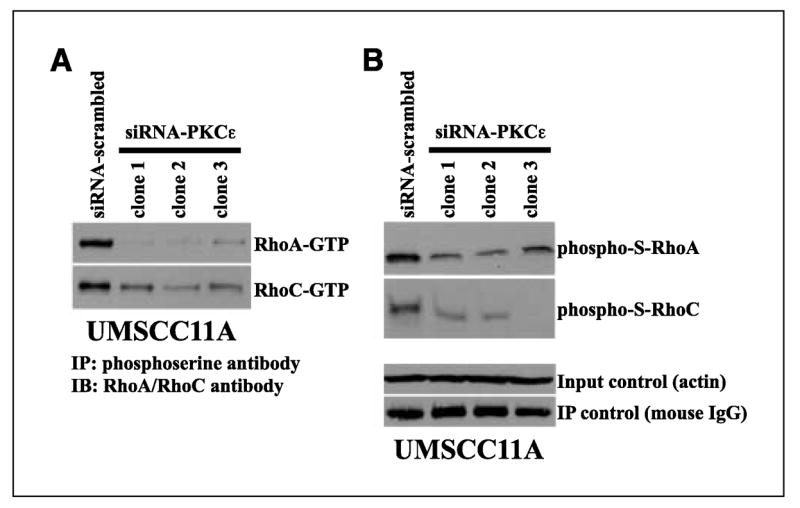
PKCε-deficient UMSCC11A cells have lower levels of active and serine-phosphorylated RhoA and RhoC. A, RhoA and RhoC activation levels. Pull-down assays using GST-RBD of rhotekin was used to determine the amount of active GTP-bound RhoA and RhoC. Western blot analyses were done on GST-RBD pull-downs with antibody specific to RhoA or RhoC. B, serine-phosphorylated RhoA (phospho-S-rhoA) and RhoC (phospho-S-rhoC) levels. Whole-cell lysates were immunoprecipitated with a phosphoserine antibody (Qiagen, Valencia, CA). Protein-antibody complexes were separated by SDS-PAGE and transferred to PVDF membrane, and Western blot (IB) analysis was done with a RhoA- or RhoC-specific antibody. Loading control (actin) is 12.5% of total protein used for immunoprecipitation (IP). Immunoprecipitation control detects the amount of mouse phosphoserine IgG antibody used for immunoprecipitation. Representative of several, independent experiments.
To provide direct evidence that RhoA and/or RhoC are required for PKCε-mediated cell invasion and motility, constitutive active RhoA or RhoC (G14V-RhoA or G14V-RhoC) was overexpressed in PKCε-deficient UMSCC11A clones to determine if restoring active RhoA or RhoC will be sufficient to rescue the PKCε knockdown loss-of-function phenotype. PKCε-deficient UMSCC11A clones that were generated previously were transfected with a 3× HA-tagged G14V-RhoA or G14V-RhoC neomycin-resistant expression vector and grown in selection antibiotics for 14 days, and stable polyclonal cell populations were isolated. The double-transfected PKCε-deficient/G14V-RhoA-overexpressing or PKCε-deficient/G14V-RhoC-overexpressing UMSCC11A cells had the proper genetic alterations and thus had elevated HA-tagged RhoA or RhoC protein levels in a PKCε-deficient background (Fig. 4A). Moreover, PKCε-deficient/G14V-RhoA or PKCε-deficient/G14V-RhoC cells had increased levels of active HA-tagged RhoA and RhoC, respectively. siRNA-scrambled control UMSCC11A cells are PKCε positive and used as the benchmark for our phenotype rescue experiments. Empty vector transfection of PKCε-deficient clones (siRNA-PKCε clones 1–3) had minimal effect on cell phenotype as these cells maintained their PKCε-deficient loss-of-function invasion and motility defect compared with siRNA-scrambled control (PKCεpositive) cells. Importantly, ectopic overexpression of constitutive active RhoA or RhoC in PKCε-deficient UMSCC11A clones resulted in a significant increase in cell invasion and motility compared with PKCε-deficient/empty vector control cells (P < 0.006 for invasion; P < 0.0004 for motility). As shown in Fig. 4C and D, overexpression of active RhoA was able to completely restore the invasion and motility defect of PKCε-deficient UMSCC11A cells to levels comparable with PKCε positive, siRNA-scrambled UMSCC11A cells; no significant difference in cell invasion (P > 0.09) and motility (P > 0.15) was determined between PKCε-deficient C1-3/G14V-RhoA UMSCC11A cells and siRNA-scrambled UMSCC11A cells. Constitutive active RhoC (PKCε-deficient/G14V-RhoC) was able to completely rescue the invasion defect but only was able to partially (~44%) rescue the motility defect of PKCε-deficient UMSCC11A cells. These results reveal that RhoA and RhoC activation are downstream of the PKCε signaling cascade and required for PKCε-mediated cell invasion and motility. Active RhoA or RhoC was able to completely rescue the invasion defect, suggesting that RhoA and RhoC may have overlapping roles in promoting cell invasion. Interestingly, active RhoA was more effective than active RhoC in rescuing the motility defect, suggesting that RhoA may play a more involved role than RhoC in driving the cell motility phenotype. In any event, our work indicates that inactivation of one Rho GTPase, either RhoA or RhoC, may not be adequate to reduce the incidence of tumor metastasis because RhoA and RhoC seem to have redundant functions in regulating cell invasion and motility. It is likely that a coordinated inactivation of RhoA and RhoC may be necessary to dampen the metastatic potential of aggressive HNSCC.
Figure 4.

Constitutive active RhoA or RhoC rescues the invasion and motility defect of PKCε-deficient UMSCC11A cells. A, PKCε-deficient/G14V-RhoA or PKCε-deficient/G14V-RhoC cells have elevated levels of HA-tagged RhoA or RhoC under a PKCε-deficient background. PKCε-deficient UMSCC11A clones were transfected with empty, 3× HA-tagged G14V-RhoA, or 3XHA-tagged G14V-RhoC neomycin-resistant expression vector, grown in selection antibiotics for 14 days, and stable polyclonal cell populations were isolated. B, PKCε-deficient/G14V-RhoA or PKCε-deficient/G14V-RhoC has elevated RhoA or RhoC activation levels, respectively. Western blot analyses were done on GST-RBD pull-downs with antibody specific to HA. C, overexpression of G14V-RhoA or G14V-RhoC completely rescues the cell invasion defect of siRNA-PKCε UMSCC11A cells. Cell invasion of PKCε-deficient/G14V-RhoA or PKCε-deficient/G14V-RhoC cells is not statistically different than siRNA-scrambled (PKCε positive) UMSCC11A cells. siRNA-PKCε/empty vector cells are significantly less invasive than siRNA-scrambled (PKCε positive) UMSCC11A cells. *, P < 0.002. D, overexpression of G14V-RhoA completely rescues and G14V-RhoC partially rescues the cell motility defect of siRNA-PKCε UMSCC11A cells. Cell motility of PKCε-deficient/G14V-RhoA cells is not statistically different than PKCε-scrambled (PKCε positive) UMSCC11A cells. PKCε-deficient/G14V-RhoC cells are more motile than PKCε-deficient/empty vector cells. #, P < 0.0004. siRNA-PKCε/empty vector cells are significantly less motile than siRNA-scrambled (PKCε positive) UMSCC11A cells. *, P < 0.005.
The Rho GTPases family consists of small, 20- to 30-kDa GTP-binding proteins that are highly conserved throughout evolution in a variety of organisms. All aspects of cellular motility and invasion, including cellular polarity, cytoskeletal organization, and transduc-tion of signals from the outside environment, are controlled through interplay between the Rho GTPases (12–14). Rho GTPases have been implicated in the progression of cancer in various organs, including breast, lung, and colon (15–17). However, there is very limited literature on the role of Rho GTPases in HNSCC development and progression. RhoA, Rac2, and Cdc42 were found to be elevated in premalignant dysplastic and HNSCC cell lines compared with normal keratinocytes supporting the importance of Rho GTPases in head and neck cancer development (18). Furthermore, based on their immunohistochemistry analyses, RhoA and Rac2 were suggested to be promising biomarkers of malignancy and/or aggressiveness in HNSCC (18). In recent work, our group showed that elevated RhoC is associated with lymph node metastasis and advanced stage tumors in a cohort of previously untreated HNSCC patients (19). Taken together, these studies reveal that dysregulation of Rho GTPases, particularly RhoA, RhoC, and Rac2, results in an aggressive HNSCC phenotype.
It is unclear at this time how PKCε modulates the activation of RhoA and RhoC in HNSCC. Several plausible explanations can be drawn from our results. Apparently, phosphorylation of RhoA and RhoC may enhance their interaction with their downstream Rho effectors through an increase in binding affinity or a decrease in binding dissociation leading to an extended Rho activation signal. Additionally, the binding affinities of the negative GDP/GTP cycle regulators, RhoGAPs and RhoGDIs, may be decreased and/or the binding affinities of the positive GDP/GTP cycle regulators, RhoGEFs, may be enhanced to phosphorylated RhoA and RhoC resulting in higher levels of RhoA and RhoC that is GTP bound. Another possibility is that PKCε-mediated phosphorylation of RhoA and RhoC may enhance their protein stability through inhibition of protein degradation mechanisms resulting in an increase in the amount of total protein available for activation. This hypothesis is supported by a report showing that cyclic GMP-dependent kinase-mediated phosphorylation of RhoA protected RhoA, particularly the GTP-bound active form, from ubiquitin/proteasome-mediated degradation (20). A logical assumption is that total and active levels of Rho GTPases are concordant; however, there is no clear consensus in the literature to support this notion. Rac3 activation was found to be elevated, whereas Rac3 protein levels were unchanged in human breast cancer cell lines and tumor tissues (21). Additionally, our laboratory showed that RhoC activation levels are independent of total RhoC protein levels in HNSCC (19). Alternatively, we propose that the reduced levels of active RhoA and RhoC observed for PKCε-deficient cells may be due, at least in part, to the inability of the regulatory proteins in the GDP/GDP cycle to be stimulated and/or inactivated through PKCε-mediated phosphorylation. This possibility is supported by numerous studies showing that PKCs are able to regulate the activities of p115RhoGEF and RhoGDI and modulate the localization of p190 RhoGAP through direct phosphorylation (22–24). Additional work will be necessary to thoroughly examine these possibilities to better understand the mechanism of RhoA and RhoC regulation by PKCε.
In summary, specific disruption of PKCε was found to inhibit cell invasion and motility in aggressive HNSCC. This study shows, for the first time, that PKCε is involved in a coordinated regulation of RhoA and RhoC activation; moreover, the PKCε-RhoA/RhoC signaling axis may be indispensable for driving PKCε-mediated cell invasion and motility.
Acknowledgments
Grant support: Head and Neck Cancer Specialized Programs of Research Excellence grant P50CA97248 (Q. Pan, T.N. Teknos, and S.D. Merajver), Burroughs-Wellcome Fund (S.D. Merajver), Breast Cancer Research Foundation (S.D. Merajver), and NIH grant CA77612 (S.D. Merajver).
References
- 1.Genden EM, Ferlito A, Bradley PJ, Rinaldo A, Scully C. Neck disease and distant metastases. Oral Oncol. 2003;39:207–12. doi: 10.1016/s1368-8375(02)00049-0. [DOI] [PubMed] [Google Scholar]
- 2.Dimery IW, Hong WK. Overview of combined modality therapies for head and neck cancer. J Natl Cancer Inst. 1993;85:95–111. doi: 10.1093/jnci/85.2.95. [DOI] [PubMed] [Google Scholar]
- 3.Carter CA. Protein kinase C as a drug target: implications for drug or diet prevention and treatment of cancer. Curr Drug Targets. 2000;1:163–83. doi: 10.2174/1389450003349317. [DOI] [PubMed] [Google Scholar]
- 4.Cacace AM, Guadagno SN, Krauss RS, Fabbro D, Weinstein IB. The ε isoform of protein kinase C is an oncogene when overexpressed in rat fibroblasts. Oncogene. 1993;8:2095–104. [PubMed] [Google Scholar]
- 5.Mischak H, Goodnight JA, Kolch W, et al. Overexpression of protein kinase C-δ and -ε in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J Biol Chem. 1993;268:6090–6. [PubMed] [Google Scholar]
- 6.Jansen AP, Verwiebe EG, Dreckschmidt NE, et al. Protein kinase C-ε transgenic mice: a unique model for metastatic squamous cell carcinoma. Cancer Res. 2001;61:808–12. [PubMed] [Google Scholar]
- 7.Dong G, Lee TL, Yeh NT, et al. Metastatic squamous cell carcinoma cells that overexpress c-Met exhibit enhanced angiogenesis factor expression, scattering, and metastasis in response to hepatocyte growth factor. Oncogene. 2004;23:6199–208. doi: 10.1038/sj.onc.1207851. [DOI] [PubMed] [Google Scholar]
- 8.Worden B, Yang XP, Lee TL, et al. Hepatocyte growth factor/scatter factor differentially regulates expression of proangiogenic factors through Egr-1 in head and neck squamous cell carcinoma. Cancer Res. 2005;65:7071–80. doi: 10.1158/0008-5472.CAN-04-0989. [DOI] [PubMed] [Google Scholar]
- 9.Kermorgant S, Zicha D, Parker PJ. PKC controls HGF-dependent c-Met traffic, signalling, and cell migration. EMBO J. 2004;23:3721–34. doi: 10.1038/sj.emboj.7600396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan Q, Bao LW, Kleer CG, et al. Protein kinase Cε is a predictive biomarker of aggressive breast cancer and a validated target for RNA interference anti-cancer therapy. Cancer Res. 2005;65:8366–71. doi: 10.1158/0008-5472.CAN-05-0553. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Gimeno C, Diaz-Meco MT, Dominguez I, Moscat J. Alterations in levels of different protein kinase C isotypes and their influence on behavior of squamous cell carcinoma of the oral cavity: εPKC, a novel prognostic factor for relapse and survival. Head Neck. 1995;17:516–25. doi: 10.1002/hed.2880170609. [DOI] [PubMed] [Google Scholar]
- 12.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 13.Nobes CD, Hall A. Rho, rac, cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 14.Sahai E, Marshall CJ. Rho-GTPases and cancer. Nat Rev Cancer. 2002;2:133–42. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 15.Kleer CG, van Golen KL, Zhang Y, Wu ZF, Rubin MA, Merajver SD. Characterization of RhoC expression in benign and malignant breast disease: a potential new marker for small breast carcinomas with metastatic ability. Am J Pathol. 2002;160:579–84. doi: 10.1016/S0002-9440(10)64877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fritz G, Just I, Kaina B. Rho GTPases are over-expressed in human tumors. Int J Cancer. 1999;81:682–7. doi: 10.1002/(sici)1097-0215(19990531)81:5<682::aid-ijc2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 17.Fritz G, Brachetti C, Bahlmann F, Schmidt M, Kaina B. Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br J Cancer. 2002;87:635–44. doi: 10.1038/sj.bjc.6600510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abraham MT, Kuriakose MA, Sacks PG, et al. Motility-related proteins as markers for head and neck squamous cell carcinoma. Laryngoscope. 2001;111:1285–9. doi: 10.1097/00005537-200107000-00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleer CG, Teknos TN, Islam M, et al. RhoC-GTPase expression as a potential marker of lymph node metastasis in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2006;12:4485–90. doi: 10.1158/1078-0432.CCR-06-0376. [DOI] [PubMed] [Google Scholar]
- 20.Rolli-Derkinderen M, Sauzeau V, Boyer L, et al. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ Res. 2005;96:1152–60. doi: 10.1161/01.RES.0000170084.88780.ea. [DOI] [PubMed] [Google Scholar]
- 21.Mira JP, Benard V, Groffen J, Sanders LC, Knaus UG. Endogenous, hyperactive Rac3 controls the proliferation of breast cancer cells by a p21-activated kinase-dependent pathway. Proc Natl Acad Sci U S A. 2000;97:185–9. doi: 10.1073/pnas.97.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehta D, Rahman A, Malik AB. Protein kinase C-α signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem. 2001;25:22614–20. doi: 10.1074/jbc.M101927200. [DOI] [PubMed] [Google Scholar]
- 23.Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. Protein kinase Cα-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem. 2003;278:28793–8. doi: 10.1074/jbc.M303900200. [DOI] [PubMed] [Google Scholar]
- 24.Brouns MR, Matheson SF, Hu KQ, et al. The adhesion signaling molecule p190 RhoGAP is required for morphogenetic processes in neural development. Development. 2000;127:4891–903. doi: 10.1242/dev.127.22.4891. [DOI] [PubMed] [Google Scholar]