

Abstract
Thiazolidinediones (TZDs) are synthetic ligands of Peroxisome-Proliferator-Activated Receptor gamma (PPARγ). Troglitazone, rosiglitazone, and pioglitazone have been approved for treatment of diabetes mellitus type II. All three compounds, together with the first TZD ciglitazone, also showed an antitumor effect in preclinical studies and a beneficial effect in some clinical trials. This review summarizes hypotheses on the role of PPARγ in tumors, on cellular targets of TZDs, antitumor effects of monotherapy and of TZDs in combination with other compounds, with a focus on their role in the treatment of differentiated thyroid carcinoma. The results of chemopreventive effects of TZDs are also considered. Existing data suggest that the action of TZDs is highly complex and that actions do not correlate with cellular PPARγ expression status. Effects are cell-, species-, and compound-specific and concentration-dependent. Data from human trials suggest the efficacy of TZDs as monotherapy in prostate cancer and glioma and as chemopreventive agent in colon, lung, and breast cancer. TZDs in combination with other therapies might increase antitumor effects in thyroid cancer, soft tissue sarcoma, and melanoma.
1. Introduction
Glitazones, also called thiazolidinediones (TZDs), are five-membered carbon ring molecules containing two heteroatoms (nitrogen and sulfur). One carbonyl group in the thiazole at position 4 and another at position 2 make the heterocyclic compound a thiazolidine-2,4-dione [1]. TZDs are ligands of the Peroxisome Proliferator Activated Receptor gamma (PPARγ), a nuclear receptor inducing upregulation of specific genes that decrease insulin resistance, inflammation, VEGF-induced angiogenesis, proliferation, and leptin levels, inducing differentiation of adipocytes, and increasing adiponectin levels. This spectrum of actions led to the approval of TZDs for treatment of diabetes mellitus type II. TZDs differ according to the substitution at C5 (Figure 1).
Figure 1.

Chemical formulae of the most common TZDs with antitumor action.
Ciglitazone (CIGLI) is the prototype of all TZDs but has never been approved for medication of diabetes mellitus because its clinical activity was too weak. Troglitazone (TRO) was the first TZD approved for treatment of diabetes mellitus in 1997 [2]. The compound showed beneficial effects on glucose levels, insulin sensitivity, and free fatty acid concentration but was withdrawn from the market in 2000 due to severe hepatotoxicity. The second TZD, rosiglitazone (ROSI), has been banned in Europe and restricted in the USA because of increased cardiovascular morbidity. Also the use of pioglitazone (PIO) as the third TZD with antidiabetic action is restricted due to concerns about a potential facilitation of bladder cancer development. The fourth substance with an antidiabetic profile, rivoglitazone, is still under investigation [3]. Reasons for the troubled history of antidiabetic TZDs are manifold and appear to be due to the highly pleiotropic action of these PPARγ agonists and crosstalk of PPARγ with other signaling pathways.
In addition to diabetes mellitus treatment, ligands to PPARγ could also be exploited for treating other diseases, for instance, in cancer treatment. This idea originated from the finding that PPARγ is involved in cell proliferation and PPARγ expression levels change from normal to transformed tissues. Effects of PPARγ activation are ligand-specific. TZDs with potent PPARγ agonist activity can display, like rivoglitazone, strong antidiabetic activity, or, like efatutazone (EFA), predominantly antitumor effects. TZDs, such as netoglitazone, can also activate other PPARs and cause antitumor effects [4]. This review will focus on the effects of selective PPARγ TZDs in tumors.
2. Role of PPARγ Expression in Neoplasms
PPARγ expression compared to normal tissue tends to be increased in precursor lesions and differentiated tumors and decreased in the poorly differentiated cancers. This pattern has been reported for instance for gastric, ureteric, and breast cancer [5–7]. In ovarian cancer, however, PPARγ levels independent from tumor differentiation are increased [8]. Upregulation of PPARγ may be an early event in tumorigenesis and a marker for differentiated cancer lesions [9]. Methylation (silencing) of the PPARγ promotor, which is detected in 30% of colorectal tumors, however, correlated with poor prognosis [10]. Studies linking tumor prognosis and PPARγ expression were mainly based on immunohistochemical detection of the PPARγ antigen in paraffin-embedded tissue. Since antigenicity is low and may decrease during storage of the paraffin samples, the absence of PPARγ staining in archival tissues may be a false negative due to methodological problems [11].
Identification of the contribution of PPARγ to tumor development and progression is further complicated by crosstalk with other pathways. Akt phosphorylation in the endometrium, for instance, is directly regulated by PPARγ and indirectly through induction of PTEN by PPARγ, where PTEN decreases p-Akt via inhibition of PI3K [12].
3. Mechanism of Antitumor Action by TZDs
Although all TZDs are PPARγ ligands, the observed antitumor effects can only be explained in part by genomic PPARγ activation. Genomic activation is defined as the binding of a nuclear receptor to a response element, which activates the transcription of certain genes. The process is also termed transactivation. Another DNA-mediated effect is transrepression, which describes the binding of receptors to transcription factors (e.g., nuclear factor kappa B (NFκB) or activator protein 1 (AP-1)).
PPARγ ligands trigger a conformational change of the PPARγ receptor that attracts transcriptional coactivators of the steroid receptor coactivator family. Once activated by ligand binding, the PPARγ receptor forms heterodimers with the retinoid X-receptor and transcription is initiated. Transcriptional activation may result in decreased proliferation, migration and inflammation and increased differentiation and apoptosis (Figure 2). Inflammatory effects are usually mediated by transrepression [13].
Figure 2.

Effects of TZDs on apoptosis, migration, invasion, and proliferation of cancer cells and on inflammation. In some ellipses, only one representative is listed; Bax and p53 react similarly, as well as p27 and p21. MMPs represents MMP-2 and MMP-9 and Cyclin E represents cyclin D1, cyclin B1, CDK2, and CDK4. Abbreviations: EGF: epithelial growth factor receptor; PPRE: PPARγ response element, Surv: survivin, E-cad: E-cadherin, b-cat: β-catenin, Cytok: cytokines.
Figure 2 illustrates the variety of pathways influenced by genomic activation of PPARγ by TZDs, resulting in downregulation of migration, proliferation, inflammation, and invasion and in upregulation of apoptosis. Common mechanisms involve influence on EGF signaling, cyclins, Ki-67, c-myc, cyclin-dependent kinases, p53 and PTEN expression, adhesion proteins, metalloproteinases, and cytokines [14–19].
Hormone-dependent cancers react through different mechanisms to TZDs depending on the hormone receptor status. In androgen-dependent prostate carcinoma, for instance, CIGLI downregulated aromatase activity, while in androgen-independent tumors proliferation was reduced [20].
Different TZDs may act by different mechanisms; while CIGLI downregulated cyclin D1 and upregulated p21 by PPARγ independent pathways, ROSI used PPARγ signaling to induce these effects in androgen-independent prostate carcinoma cells [21].
The description of all mechanisms of TZDs is beyond the scope of this review but one important signaling pathway for tumor cells and for surrounding tissue (tumor microenvironment) each illustrates the variety of PPARγ effects. Tumor biology is not only determined by tumor cells but to a high extent by properties of stromal cells in the tumor microenvironment. Among the diverse cells in the tumor stroma (endothelial cells, cancer-associated fibroblasts, leukocytes, myofibroblasts, and mesenchymal stem cells), tumor-associated macrophages play the most decisive role in tumor progression [22].
For tumor cells, signaling by Epidermal Growth Factor receptor (EGF-receptor, Figure 2) is highly relevant. The signaling cascade of the EGF-receptor involves the ERK cascade, consisting of Ras-Raf-MEK1/MEK2-ERK1/ERK2 and is seen in several cancer types [23]. ERK may phosphorylate PPARγ and reduce its genomic activity. This effect occurs in cancer cell lines and a variety of normal cells alike [24]. TRO, for example, was reported to bind to the EGF receptor and trigger its internalization in EGF-receptor transfected endothelial cells [25]. This action is an example of nongenomic effects of TZDs since no ligand binding to response element occurred.
Normal macrophages can transform into tumor-associated macrophages under stimulation of PPARγ ligands [26]. ROSI decreased activation of macrophages and thereby reduced inflammation in nondiabetic patients with symptomatic carotid artery stenosis [27]. In murine macrophages, these effects are mediated by interaction of PPARγ with NfκB [28]. In these effects, transrepression appears to be the main mechanism.
Finally, MEK1 action by ROSI may lead to nuclear export and cytoplasmic retention of PPARγ and off-DNA interaction with proteins in MEK1-GFP and PPARγ (wild-type and mutant) cotransfected HEK-293 cells [29]. In this effect no genomic action of TZDs was involved.
4. Therapeutic Efficacy of TZDs in Specific Cancers
Decrease of cell proliferation, cytotoxicity, and proapoptotic effects induced by CIGLI, TRO, ROSI, and PIO has been reported in a variety of cell lines (sarcoma, melanoma, glioblastoma, breast carcinoma, colorectal cancer, gastric cancer, pancreatic cancer, prostate, bladder cancer, hepatic cancer, thyroid cancer, ovarian cancer, endometrial cancer, and lung cancer cells), which will not be listed in detail. Based on promising cellular action, animal experiments and clinical trials have been conducted in several common cancers.
EFA, which was developed as a chemostatic rather than an antidiabetic drug, has also been studied in some of these cancers. EFA is 500x more potent an activator of PPARγ than TRO and 50x stronger than ROSI. EFA was studied in a preclinical murine model for breast cancer based on BRCA1 (BReast CAncer 1) deficiency. In the MMTV-Cr BRCA1flox/flox p53+/− model, exon 11 of the BRCA1 gene is deleted by Mouse Mammary Tumor Virus (MMTV)-Cre transgene. The deletion is accompanied by loss of one germline copy of TP53. EFA reduced the incidence of noninvasive and well-differentiated tumors in this model [30].
Cell proliferation and xenograft size of pancreatic, anaplastic thyroid, and colorectal cancer were reduced by EFA administration [31].
Based on these promising preclinical effects, phase I trials were initiated either as monotherapy or in combination with other compounds. After monotherapy with EFA, stable disease was induced in 10/22 patients with advanced liposarcoma [14]. A phase 1 study evaluating the combination of bexarotene with EFA in solid tumors is currently recruiting patients (NCT01504490).
The first trial of antitumor effects of the antidiabetic TZDs was conducted in three liposarcoma patients, where decrease of proliferation with TRO has been reported [32]. No beneficial effects, however, were obtained in a trial with ROSI in 9 liposarcoma patients [33]. Despite the negative outcome of this trial, another phase II trial on ROSI is ongoing (NCT00004180; http://www.cancer.gov/clinicaltrials/).
TZDs showed variable efficacy in studies of common cancers using xenograft and transgenic mouse models, in case studies and clinical trials (an overview is provided in Table 1).
Table 1.
Relationship between protective role of PPARγ expression and efficacy of TZDs in therapy.
| Cancer type | Role of PPARγ | TZD | Experimental model | Result | Reference |
|---|---|---|---|---|---|
| PIO | Xenograft (HT-29) in mice with APC mutation, sc | Increased tumor growth | [41] | ||
| Colon | ⇓/⇑ | Azoxymethane-induced murine tumors | Reduced tumor growth | [39] | |
| TRO | HT-29 xenografts, sc | Reduced tumor growth and metastasis | [40] | ||
| Metastatic colon cancer, 25 patients | All progressive disease | [43] | |||
|
| |||||
| Lung | ⇓/⇑ | ROSI | Chemically-induced mouse model | Decrease in adenoma formation | [46] |
|
| |||||
| ROSI | LMM3 injection into mice, sc | Decreased tumor growth | [48] | ||
| Breast | ⇓ | Chemically induced rat model | Decreased tumor growth and incidence | [49] | |
| TRO | Advanced chemotherapy breast refractory cancer, 22 patients | No CR or PR, 3 SD | [50] | ||
| ROSI | Early stage breast cancer, 38 patients | No decrease in proliferation | [51] | ||
|
| |||||
| PIO | PC3 xenografts, sc. | Decrease of bone-invasive potential | [53] | ||
| Prostate | ⇓ | TRO | Advanced prostate carcinoma, 41 patients | Stabilization of PSA levels | [54] |
| ROSI | Recurrent prostate carcinoma, 1 patient | Delayed increase of PSA levels | [55] | ||
|
| |||||
| Glioma | ⟺ | PIO | LN229 orthotopic xenografts | Reduced tumor volume, invasion | [58] |
| Chemorefractory glioma, 14 patients | Disease stabilization (29%) | [59] | |||
|
| |||||
| Melanoma | ⟺ | CIGLI | A375 xenografts, sc. | Growth inhibition, pro-apoptotic effects | [62] |
|
| |||||
| PIO | Transgenic mouse model (PPAR fusion protein/PTEN deletion) | Decreased tumor growth and metastasis | [106] | ||
| Thyroid | ⇓ | ROSI | Transgenic mouse model (Thyroid hormone receptor-β negative) | Delayed progression | [107] |
| Metastatic thyroid cancer, 1 patient | Decrease in metastasis size | [109] | |||
PPARγ expression on tumor progression: promotion: ⇑; protection: ⇓; no effect: ⟺; CR: complete response; PR: partial response; SD: stable disease; sc: subcutaneous implantation of tumor cells.
4.1. Colorectal Cancer
Studies on human tumor samples support the hypothesis that PPARγ expression has protective effects in colorectal cancer [34]; patients with PPARγ expression usually showed a better prognosis [11]. Accordingly, reduction of β-catenin and PPARγ was associated with high numbers of tumor-associated macrophages, increased metastasis, and poor survival [35]. On the other hand, loss of function point mutations of the PPARγ gene and polymorphisms in PPARγ genes were encountered in 8% of colorectal carcinoma patients, but some studies on PPARγ expression in colorectal samples did not find any relation of PPARγ immunoreactivity and tumor parameters [36, 37]. The role of PPARγ activation in the progression of malignant lesions is questioned by the fact that heterozygous and homozygous intestinal-specific PPARγ deficiency promoted tumor formation [38]. This suggests that murine models might not be representative for the study of TZDs in colorectal cancer.
Consistent with the unclear role of PPARγ in tumor samples, TZDs showed variable effects in vivo. PPARγ activation inhibited xenograft growth in mice and PPARγ agonists reduced the number of aberrant cryptal foci in chemically induced inflammatory bowel disease in mice [39, 40]. On the other hand, PIO induced increased polyp numbers in mice with APC mutation, prone to developing colon adenoma (APCmin), not in wild-type mice, suggesting that, under certain genetic conditions, TZDs could also promote colon cancer development [41]. The disparate results might be explained by in vitro studies in colon cancer cell lines showing that the level of PPARγ expression correlated to cells' sensitivity to proliferation inhibition [42].
A phase II trial with TRO did not increase progression-free survival in 25 colorectal cancer patients [43].
4.2. Lung Cancer
PPARγ expression in well-differentiated lung adenocarcinoma was higher than in poorly differentiated tumors, suggesting that it promotes tumor formation but is not a marker for aggressive growth [44]. In another study, expression was linked to poor prognosis, showing the opposite trend [45]. ROSI decreased progression of chemically induced murine cancer model [46].
4.3. Breast Cancer
In breast cancer PPARγ mRNA levels did not correlate with nodal involvement and tumor grade but significantly lower PPARγ levels were seen in large metastatic tumors, patients with local recurrence and poor survival [47]. Despite the fact that samples of aggressive tumors showed increased PPARγ expression, TZDs displayed moderate positive effects in breast cancer models. ROSI reduced tumor growth in a chemically induced rat and in a syngenic murine tumor model [48, 49]. Both in patients with advanced breast carcinoma and in patients with early mammary cancer treatment with TZDs did not cause therapeutic effects [50, 51].
4.4. Prostate Cancer
In the majority of prostate cancers (73%), immunoreactivity and expression of PPARγ correlated inversely with tumor size and PSA levels [52]. Data obtained in prostate cancer xenografts as well as results from a phase II trial and a case report showed efficacy of PIO and TRO [53–55].
4.5. Glioblastoma
No correlation of PPARγ expression has been established with glioma [56]. Diabetes mellitus patients under TZD medication, however, showed lower incidence of high-grade glioma than the control group (patients with hip fractures), while survival of patients with glioma was similar in both groups [57]. Efficacy of PIO has been shown in glioma xenografts and in a phase II trial [58, 59].
4.6. Melanoma
No correlation of PPARγ expression and melanoma prognosis was seen [60]. In a cohort study of diabetes mellitus patients under PIO medication, an increased hazard ratio for melanoma (1.3) was reported [61]. It is not clear whether these data represent an increased incidence of tumors because the maximum duration of follow-up was <6 years after the initiation of PIO. Studies on monotherapy with TZDs in melanoma are limited: only CIGLI was reported to inhibit growth of melanoma xenografts [62].
Higher mRNA or protein expression in well-differentiated tumors compared to poorly differentiated tumors and tumors with poor prognosis is interpreted as protective effect of PPARγ in tumor development. In prostate cancer patients, protective effects of PPARγ and therapeutic effect of TZDs were in line (Table 1). In glioma samples, PPARγ expression was not linked to good prognosis but TZDs showed therapeutic efficacy.
5. Role of TZDs in Chemoprevention
While therapeutic efficacy of monotherapy with TZDs was relatively low, data obtained from meta-analysis of diabetes studies as well as in vitro data suggested that TZDs could be efficient in chemoprevention (Table 2).
Table 2.
Summary of data on chemopreventive effects of TZDs in animal and human epidemiological studies.
| Cancer type | Role of PPARγ expression | TZD | Experimental model | Result | Reference |
|---|---|---|---|---|---|
| PIO | Chemically-induced rat cancer model | Reduction of tumor incidence | [121] | ||
| Colon | ⇓/⇑ | Transgenic murine cancer model (nonsense mutation in the adenomatous polyposis coli) | Increase of tumor incidence | [122] | |
| TRO | Chemically-induced rat cancer model | Reduction of tumor incidence | [123] | ||
| ROSI | Meta-analysis of diabetes trials | Reduced colon cancer incidence | [64] | ||
|
| |||||
| Lung | ⇓/⇑ | PIO | Chemically induced murine cancer model | Reduction of tumor incidence | [72] |
| PIO | Observational study | Reduced lung cancer incidence | [63] | ||
|
| |||||
| Breast | ⇓ | PIO | Meta-analysis of diabetes trials | Reduced breast cancer incidence | [64] |
|
| |||||
| Liver | ⇓ | PIO | Chemically induced rat cancer model | Reduced tumor incidence | [73] |
|
| |||||
| Endometrium | ⇓ | ROSI | Transgenic murine cancer model | Reduced tumor incidence | [12] |
|
| |||||
| Oral (squamous cancer) | ⇓ | PIO | Transgenic rat cancer model | Reduced tumor incidence | [77] |
| TRO | Chemically induced rat cancer model | Reduced tumor incidence | [78] | ||
PPARγ expression on tumor progression: promotion: ⇑; protection: ⇓.
5.1. Data from Diabetes Trials
Medication with TZDs for >1 year decreased the incidence of head and neck cancers by 40% and lung cancer by 33% in diabetes mellitus patients [63]. The reduction of lung cancer reached 75% in the African-American population. The reduction was specific for lung cancer, as prostate and colorectal cancer incidence was not changed. Of note, in this study, patients with preexisting malignancies were excluded. The largest meta-analysis on cancer incidence and cancer mortality included data of 46 trials. The number of malignancies was disclosed in 28/33 trials with ROSI and in 18/33 trials with PIO [64]. This meta-analysis reported less cancer cases (342 versus 457) in patients treated with TZDs compared to other medications. Overall, treatment with TZDs was associated with a significantly lower incidence of cancer cases (Mantel-Haenszel odds ratio (MH-OR) 0.85; P = 0.027). For ROSI this effect was significant for colorectal cancer (MH-OR 0.63; P = 0.03). PIO treatment significantly reduced the incidence of breast cancer (MH-OR 0.28; P = 0.004). An increase in the incidence of bladder cancer by PIO treatment was not seen (MH-OR 2.05; P = 0.12), but cancer mortality was increased upon TZD treatment. Since this mortality most probably is due to preexisting cancers, the question remains whether treatment with TZDs could promote the growth of already existing malignant lesions.
5.2. In Vitro Differentiation Studies
Morphological differentiation (duct formation in collagen gels) increased in pancreatic carcinoma cells treated with TRO [65] and increases of villin and mucin mRNA were observed in colon cancer cell lines [66]. ROSI induced PTEN expression in Caco-2 cells and restored glandular morphogenesis [67]. It increased tyrosinase expression, an indication for differentiation, in a melanoma cell line [68]. ROSI also caused reversal of epithelial-mesenchymal transition in anaplastic thyroid cancer cell lines and increased expression of thyroglobulin, TSH receptor, sodium-iodide symporter, and thyroperoxidase mRNA [69]. CIGLI induced brain tumor stem cell differentiation [70]. In cultures of metaplastic urothelial cells, differentiation markers were increased after treatment with TRO [71].
5.3. TZD Effects in Animal Studies
PIO prevented lung tumor development in carcinogen-induced mouse models [72]. In a similar manner, PIO protected rats against chemically-induced (diethylnitrosamine and acetylaminofluorene) hepatocarcinogenesis [73]. PPARγ could play a tumor-promoting role in hepatoma, because expression is significantly reduced in hepatocellular carcinoma with poor prognosis [74]. A similar situation is seen in endometrium carcinoma, where benign lesions show strong PPARγ immunoreactivity but malignant lesions low to absent PPARγ expression [12]. Chemoprevention of endometrial cancer by ROSI was observed in PTEN heterozygous mice [75]. Increased PPARγ expression was predominantly seen in less invasive oral squamous cancer [76]. Chemically-induced oral squamous carcinoma in rats was reduced by 40% through administration of PIO [77] and tongue carcinoma formation was reduced by 40% by TRO [78].
On the other hand, tumor-promoting effects of PIO were observed in the APCmin murine colon cancer model [41]. Because tumor-promoting effects were not seen in all cancer models, a model-specific effect cannot be excluded. The complex and, in part, opposing effects of TZDs on cancer development and progression can be explained by their cell-specific and species-specific action (tumor cells versus tumor environment). Effects of TZDs on immune cells may be the reason for the tumor-promoting effect of PIO in the APCmin mouse model and the reduced tumor growth in immune-compromised mice and in the azoxymethane-induced tumor model [79]. While PPARγ activation may decrease proliferation of tumor cells, it may increase macrophage polarization towards the M2 phenotype (TAM) and induce anti-inflammatory effects, also mediated by PPARγ activation (see Section 3)
5.4. Human Data
One phase II trial on prevention of lung, head, and neck carcinoma in 21 patients with oral leukoplakia using PIO has been completed. Fifteen patients showed partial responses, 2 stable disease and 4 patients had progressive disease (NCT00099021; http://www.cancer.gov/clinicaltrials/). Based on these promising results, another trial on prevention of lung cancer is recruiting patients (NCT00780234; http://www.cancer.gov/clinicaltrials/).
In human trials, no general correlation of the protective effect of PPARγ expression against tumor progression and chemopreventive effects of TZDs was obvious. While a protective role of PPARγ expression was postulated in breast tumors and TZDs also acted preventive on the development of breast cancer in humans, the chemopreventive effect on colon cancer was not consistent with a protective role of PPARγ expression in tumor samples.
6. Combined Treatments of TZDs with Other Drug Compounds
6.1. In Vitro Studies
Several studies evaluated the effect of combined therapies with TZDs and other agents. A large variety of combinations of TZDs have been evaluated in vitro. The observed antitumor effects include cytotoxicity/decrease of cell viability, growth inhibition, and apoptosis (for overview see Table 3).
Table 3.
Results of therapies combining TZDs with other antitumor treatments.
| TZD | Additional compound | Model | Effect | Reference |
|---|---|---|---|---|
| Gamma-radiation | Lung carcinoma cell lines (A549, H460) | DNA damage, apoptosis | [124] | |
| RXR-α ligands (SR11237, 6-OH-11-O-hydroxyphenanthrene) | Breast carcinoma cell line (MDA-MB231), lung carcinoma cell line (Calu-3), glioblastoma cell line (U87MG), melanoma cell line (G361) | Growth inhibition; apoptosis | [125–127] | |
| CIGLI | TNF-α-related apoptosis inducing ligand | Ovarian cancer cell line (HEY) | Decrease of proliferation | [128] |
| Lovastatin | Pancreatic carcinoma cell lines (Panc02, MIA, PACa-2), breast carcinoma cell lines (EMT6, MDA-MB-316), colon cancer cell line (C26) | Decrease of cell viability; decrease of proliferation | [129] | |
| Phenylbutyrate | Lung carcinoma cell lines (A549, H157) | Growth inhibition | [130] | |
|
| ||||
| 9-cis retinoic acid | Gastric carcinoma cell line (SGC7901) | Apoptosis | [131] | |
| Cisplatin | Lung cancer cell lines (A549, H522); mesotheloma cell line (EHMES-10) | Growth inhibition | [87, 132] | |
| Paclitaxel | Lung carcinoma cell lines (A549, H522) | Growth inhibition | [132] | |
| RXR-α ligands (bexarotene, all-trans retinoic acid) | Breast cancer cell lines (MCF-7, T-47D, ZR-75-1) | Growth inhibition | [133] | |
| TRO | Cell signalling molecules (TRAIL, heregulin) | Ovarian cancer cell line (HEY); breast cancer cell lines (MCF-7, SKBR-3, MDA-MB-453) | Decrease of cell number; apoptosis | [128, 134] |
| Lovastatin | Glioblastoma cell line (DBTRG05MG), lung cancer cell line (CL1-0) | Cell cycle inhibitor expression | [135] | |
| Aspirin | Lung cancer cell lines (CL1-0, A549) | Decrease of proliferation | [136] | |
| Tamoxifen | Breast cancer cell line (MCF-7) | Growth inhibition | [137] | |
| X-rays | Cervix cancer cell lines (HeLa, Me180) | Decrease of cell viability | [138] | |
|
| ||||
| Platinium-based compounds (cisplatin, carboplatin) | Ovarian cancer cell lines (OVCA420, OVCA429, ES), lung cancer cell lines (A549, Calu-1, H23, H596, H1650) | Growth inhibition | [85] | |
| 5-Fluorouracil | Hepatoma cell lines (BEL7402, Huh-7); colon cancer cell line (HT-29) | Decrease of cell viability, apoptosis | [82, 139] | |
| RXR-α ligands (bexarotene, 9-cis retinoic acid) | Breast cancer cell lines (MCF-7TR1, SKBR-3, T47D), colon cancer cell line (Moser) | Increase of differentiation, growth inhibition; decrease of cell viability | [80, 140] | |
| ROSI | Cell signalling molecules (TNF-α, anti-Fas IgM, Seliciclib) | Breast cancer cell line (MDA-MB-231) | Growth inhibition | [141] |
| Gemcitabine | Pancreas cancer cell lines (PANC-1, Panc02) | Decrease of cell viability, growth inhibition | [142] | |
| Gefitinib | Lung cancer cell line (A549) | Growth inhibition | [83] | |
| Herceptin | Breast cancer cell line (MCF-7) | Growth inhibition | [84] | |
| Bortezomib | Melanoma cell lines (MV3, FemX-1, G361) | Growth inhibition | [143] | |
|
| ||||
| Paclitaxel | Lung cancer cell lines (A549, H522) | Growth inhibition | [132] | |
| RXR-α ligands (LG268) | Liposarcoma cells (primary) | Increase of differentiation | [81] | |
| PIO | Statins (Simvastin, lovastatin) | Glioblastoma cell lines (U87, U138, LN405, RGII); meningeoma cell lines (IOMM-Lee, KT21-MG1) | Decrease of cell viability | [144, 145] |
| Gemcitabine | Pancreas cancer cell line (PANC-1) | Decrease of cell viability | [142] | |
| 2-Deoxyglucose | Prostate cancer cell lines (PC-3, LNCaP) | Decrease in tumor spheroid formation | [146] | |
|
| ||||
| EFA | Paclitaxel | Anaplastic thyroid carcinoma cell lines (DRO, BHT-101, ARO) | Growth inhibition | [88] |
In combination treatment with RXR-α ligands, increased cellular differentiation was reported [80, 81]. Some combined therapies take advantage of the cross-talk of PPARγ with other signaling pathways. For instance, the upregulation of PTEN by ROSI rendered hepatoma cells more sensitive to the action of 5-fluorouracil [82]. Based on the idea of cross-talk between the ERK and PPARγ pathways, combinations of ERK inhibitors and PPARγ agonists could be useful in tumors with deleterious elevation of PPARγ. Experimental data corroborate such an idea: gefitinib and ROSI increased growth inhibition of lung cancer cells and increased PPARγ and PTEN expression [83]. Herceptin, an antibody against the EGF-receptor HER2, sensitized breast cancer cells for the differentiating action of TRO [84].
6.2. Animal Studies
The following examples show that improved antitumor responses were also obtained in vivo: growth of lung carcinoma xenografts and of chemically-induced breast tumors was inhibited by a combination of ROSI and platinum-based compounds [85, 86]. ROSI in combination with suberoylanilidehydroxamic acid (SAHA) decreased progression of preinvasive lung cancer in a murine model by 77% [46]. Similarly, a combination of TRO and platinum-based compounds increased survival of mesothelioma-xenografted mice [87]. The combination of EFA and paclitaxel reduced the size of anaplasic thyroid carcinoma xenografts [88]. Progression of ovarian carcinoma xenografts was slower when a combination of CIGLI and cisplatin was administered. Synergistic effects were reduction of angiogenesis and increased proapoptotic effects [89]. Aerosolized budesonide and oral PIO decreased lung cancer mass by 90% in a benz(a)pyrene-induced murine lung cancer model [90].
6.3. Human Data
Phase II trials of combination with the COX-2 inhibitor rofecoxib and PIO were able to induce complete response, partial responses, or stable disease in 5/5 angiosarcoma, 1/1 hemangioendothelioma, 4/19 metastatic melanoma, 10/40 soft tissue sarcoma, and 4/14 glioma patients [59, 91, 92]. Combination of PIO with other chemostatic drugs induced one complete response and prolonged disease-free survival in 2 of 19 patients with advanced melanoma enrolled in this phase II trial [91]. These data suggest potential efficacy of TZDs combined with other compounds in melanoma. For further evaluation of comedication with TZDs in patients, a prospective phase I/II trial of PIO combined with lenalidomide, dexamethasone, and treosulfan (NCT01614301) is currently recruiting patients (http://www.cancer.gov/clinicaltrials/).
According to human trials, only soft tissue sarcoma and melanoma might be sensitive to combinations of TZDs and COX-2 inhibitors and TZDs in polytherapy, respectively.
7. Specific Role of TZDs in Differentiated Thyroid Carcinoma (DTC)
PPARγ has a specific role in thyroid cancer because follicular thyroid cancer is the only known neoplasm to be associated with a PPARγ fusion gene product [93]. PAX8/PPARγ is expressed in 30–35% of follicular thyroid carcinoma and 2–13% of follicular adenomas [94]. This chimeric protein is the result of a genetic translocation between chromosomes 2 and 3 and can activate the PPARγ response element and induce proliferation. The mutation acts both as a gain and loss of function mutant in thyroid cancer and determines thyroid tumor differentiation; in more aggressive tumors gain of function predominates [93].
Thyroid cancer incidence in the United States has increased in the last thirty years not only apparently because of enhanced detection but probably also as a true increase [95]. DTC is the most common type of thyroid carcinoma, mainly in the form of papillary thyroid carcinoma, accounting for 80–90% of all thyroid cancer cases. The second-most common form of DTC is follicular thyroid cancer with 10–15% incidence. The prognosis of DTC is generally good, with a 10-year survival rate of 85% [96]. A total of 10–20% of patients develops distant metastases [97]. In this group, the 10-year survival rate drops to 40%. Recurrence in DTC, however, occurs in up to a third of patients and only 30% of patients with distant metastases respond to radioiodine (RAI) therapy with complete remission [98, 99]. First-line treatment of DTC is by total or near total removal of the thyroid and if necessary lymph node dissection (Figure 3). This is generally followed by RAI treatment for thyroid remnant ablation and elimination of metastases. In case of insufficient efficacy of this treatment, doxorubicin is initiated [100]. Because doxorubicin treatment is not highly efficient, it is expected that, in the future, differentiating therapies will play a prominent role in cancer treatment. Redifferentiating compounds include retinoids, histone deacetylase inhibitors, DNA methyltransferase inhibitors, and TZDs. Somatostatin analogues such as 68Ga-DOTATOC are additional options for RAI-negative thyroid cancer [101].
Figure 3.
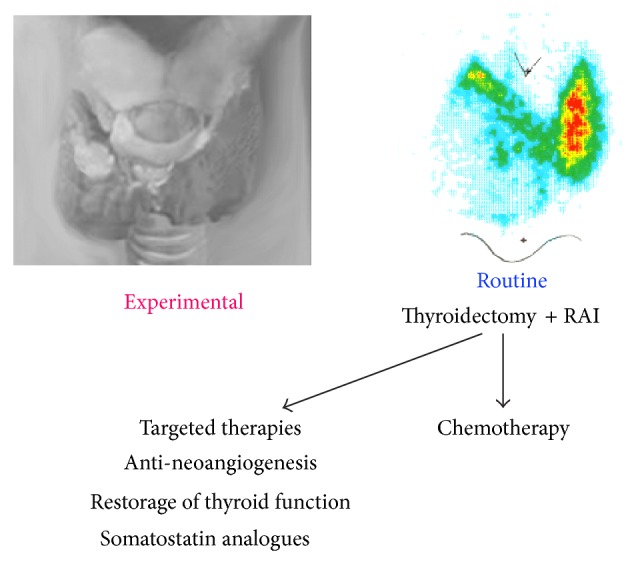
Overview of treatment options for DTC. Scheme of thyroid tumor (upper left) and scintigraphy with 123Iodide showing lack of uptake in the lower part of the right lobe (upper right).
PIO and CIGLI did not increase differentiation in a study on the human papillary carcinoma cell line NPA [102]. In another, TRO, ROSI, and PIO showed antiproliferative, proapoptotic, and differentiating effects on DTC cells [103]; TRO could increase expression of sodium-iodide symporter in DTC lines [104] and restore radioiodine-uptake in vitro [105].
7.1. Animal Studies
PIO was effective in reducing metastatic disease in a tumor model where the effect of PAX8/PPARγ fusion protein is mimicked [106]. ROSI was also able to reduce thyrocyte growth by 40% in a murine knock-in model of thyroid hormone receptor β [107].
7.2. Human Data
In a small cohort of 5 patients treated with PIO for 6 months, no increase in RAI-uptake was seen [108]. Two case reports described successful induction of RAI-uptake after treatment with ROSI in a patient with noniodide avid metastases of DTC [109, 110]. Decreased thyroglobulin levels and tumor size indicated partial success of this treatment. Evidence for increased RAI-uptake upon treatment with ROSI was obtained in one of five patients enrolled in a pilot study [111]. In another pilot study, ROSI treatment resulted in positive RAI scans in 4/10 patients and a clinical trial showed increased RAI-uptake in therapeutic 131I scans in 5/23 patients [112, 113]. Despite reinduction of RAI-uptake in 5/20 patients of another phase II trial, none had a complete or partial response to ROSI after 3 months [114] by RECIST criteria [115]. The status of a current trial (NCT00098852) with ROSI for reinduction of radioiodine-uptake is not yet known (http://www.clinicaltrial.gov/). Also the redifferentiating action of PIO is being reassessed in a trial focused on follicular variants of PTC (NCT01655719; http://www.clinicaltrial.gov/). Interpretation of the results is complicated by limited accuracy of the technique of 131I scans and unknown status of receptor expression of the treated tumors, too low levels of expression by the target cells, inhomogeneity of RAI-uptake into the tumor, and the generally poor correlation between RAI-uptake and clinical remission, all of which may be reasons for lack of efficacy. In addition, observation time of less than one year may not be enough to monitor effects in slow-growing DTC.
8. Conclusion
Current data do not suggest a correlation of clinical efficacy and high PPARγ expression according to mRNA and protein expression in tumor samples. This lack of relation could be due to methodical problems of PPARγ detection in archived tumor samples and in the complexity of TZD action. First, TZDs show a variety of genomic and nongenomic effects and several antitumor effects occur independent of PPARγ. This is particularly obvious in experiments where combination of PPARγ agonists and antagonists act synergistically on inhibition of proliferation [116]. Cell specific effects of TZDs are particularly important in cancer because their action on immune cells may antagonize their effects on tumor cells. This suggests that administration of TZDs after tumor initiation may be inefficient or even deleterious and could explain why cancer mortality was increased in the meta-analysis of cancer incidence in patients with TZD treatment. Species-specific action was reported between human and murine endothelial cells where increase of proliferation was seen in the mouse cells and an antiproliferative effect in human cells [117]. Furthermore, TZDs show compound-specificity. TRO and CIGLI acted as antiproliferatives on ovarian cancer cell lines, while ROSI and PIO did not. This could be due to additional targets and/or PPARγ independent effects; TRO for instance has stronger Akt/mTOR activity than the other TZDs. Finally, the effect of TZDs is concentration-dependent. Low concentrations of TZDs induced cell cycle arrest, while higher doses (>100 µM) caused apoptosis. Effects at higher concentrations can be explained by transactivation of PPARγ by cross-talk between signaling pathways where one receptor activates a receptor for a different ligand. Alternatively, TZDs may activate a specific subunit within a receptor oligomer [118]. As to the concentration, other coactivators may be involved in the effect and different downstream processes may be activated. PPARγ agonists can also change the cell's expression of PPARγ to different extents.
Against the background of limitations of traditional as well as new (transgenic) mouse models [119, 120] for human cancer, only efficacy in human trials is included in our final assessment. Use of TZDs in cancer might be therapeutic in prostate cancer and glioma, chemopreventive in colon, lung, and breast cancer, and increase therapeutic efficacy combined with other therapies in thyroid cancer, soft tissue sarcoma, and melanoma.
Conflict of Interests
The authors declare that there is no conflict of interests.
References
- 1.Malik S., Upadhyaya P. K., Miglani S. Thiazolidinediones: a plethro of biological load. International Journal of PharmTech Research. 2011;3(1):62–75. [Google Scholar]
- 2.Youssed J., Badr M. PPAR ligands. In: Youssed J., Badr M., editors. Peroxisome Proliferator-Activated Receptors: Discovery and Recent Advances. New York, NY, USA: Springer Science+Business Media; 2013. [Google Scholar]
- 3.Koffarnus R. L., Wargo K. A., Phillippe H. M. Rivoglitazone: a new thiazolidinedione for the treatment of type 2 diabetes mellitus. Annals of Pharmacotherapy. 2013;47(6):877–885. doi: 10.1345/aph.1R754. [DOI] [PubMed] [Google Scholar]
- 4.Imchen T., Manasse J., Min K.-W., Baek S. J. Characterization of PPAR dual ligand MCC-555 in AOM-induced colorectal tumorigenesis. Experimental and Toxicologic Pathology. 2013;65(6):919–924. doi: 10.1016/j.etp.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mylona E., Giannopoulou I., Diamantopoulou K., et al. Peroxisome proliferator-activated receptor gamma expression in urothelial carcinomas of the bladder: association with differentiation, proliferation and clinical outcome. European Journal of Surgical Oncology. 2009;35(2):197–201. doi: 10.1016/j.ejso.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Nomura S., Nakajima A., Ishimine S., Matsuhashi N., Kadowaki T., Kaminishi M. Differential expression of peroxisome proliferator-activated receptor in histologically different human gastric cancer tissues. Journal of Experimental and Clinical Cancer Research. 2006;25(3):443–448. [PubMed] [Google Scholar]
- 7.Papadaki I., Mylona E., Giannopoulou I., Markaki S., Keramopoulos A., Nakopoulou L. PPARγ expression in breast cancer: clinical value and correlation with ERβ . Histopathology. 2005;46(1):37–42. doi: 10.1111/j.1365-2559.2005.02056.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhang G. Y., Ahmed N., Riley C., et al. Enhanced expression of peroxisome proliferator-activated receptor gamma in epithelial ovarian carcinoma. British Journal of Cancer. 2005;92(1):113–119. doi: 10.1038/sj.bjc.6602244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao L., Liu F., Sun L., et al. Upregulation of PPARγ in tissue with gastric carcinoma. Hybridoma. 2010;29(4):341–343. doi: 10.1089/hyb.2010.0013. [DOI] [PubMed] [Google Scholar]
- 10.Pancione M., Sabatino L., Fucci A., et al. Epigenetic silencing of peroxisome proliferator- activated receptor γ is a biomarker for colorectal cancer progression and adverse patients’ outcome. PLoS ONE. 2010;5(12) doi: 10.1371/journal.pone.0014229.e14229 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Ogino S., Shima K., Baba Y., et al. Colorectal cancer expression of peroxisome proliferator-activated receptor gamma (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology. 2009;136(4):1242–1250. doi: 10.1053/j.gastro.2008.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nickkho-Amiry M., McVey R., Holland C. Peroxisome proliferator-activated receptors modulate proliferation and angiogenesis in human endometrial carcinoma. Molecular Cancer Research. 2012;10(3):441–453. doi: 10.1158/1541-7786.MCR-11-0233. [DOI] [PubMed] [Google Scholar]
- 13.Glass C. K., Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nature Reviews Immunology. 2010;10(5):365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- 14.Pishvaian M. J., Marshall J. L., Wagner A. J., et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer. 2012;118(21):5403–5413. doi: 10.1002/cncr.27526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun X., Ritzenthaler J. D., Zheng Y., Roman J., Han S. Rosiglitazone inhibits α4 nicotinic acetylcholine receptor expression in human lung carcinoma cells through peroxisome proliferator-activated receptor γ-independent signals. Molecular Cancer Therapeutics. 2009;8(1):110–118. doi: 10.1158/1535-7163.MCT-08-0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen F., Harrison L. E. Ciglitazone-induced cellular anti-proliferation increases p27 kip1 protein levels through both increased transcriptional activity and inhibition of proteasome degradation. Cellular Signalling. 2005;17(7):809–816. doi: 10.1016/j.cellsig.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Panigrahy D., Singer S., Shen L. Q., et al. PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. The Journal of Clinical Investigation. 2002;110(7):923–932. doi: 10.1172/JCI200215634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen B., Chu E. S. H., Zhao G., et al. PPARgamma inhibits hepatocellular carcinoma metastases in vitro and in mice. British Journal of Cancer. 2012;106(9):1486–1494. doi: 10.1038/bjc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galli A., Ceni E., Crabb D. W., et al. Antidiabetic thiazolidinediones inhibit invasiveness of pancreatic cancer cells via PPARγ independent mechanisms. Gut. 2004;53(11):1688–1697. doi: 10.1136/gut.2003.031997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moss P. E., Lyles B. E., Stewart L. V. The PPARγ ligand ciglitazone regulates androgen receptor activation differently in androgen-dependent versus androgen-independent human prostate cancer cells. Experimental Cell Research. 2010;316(20):3478–3488. doi: 10.1016/j.yexcr.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyles B. E., Akinyeke T. O., Moss P. E., Stewart L. V. Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARγ-dependent and PPARγ-independent pathways. Cell Cycle. 2009;8(2):268–277. doi: 10.4161/cc.8.2.7584. [DOI] [PubMed] [Google Scholar]
- 22.Mantovani A., Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Current Opinion in Immunology. 2010;22(2):231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Seger R., Krebs E. G. The MAPK signaling cascade. FASEB Journal. 1995;9(9):726–735. [PubMed] [Google Scholar]
- 24.Burgermeister E., Seger R. PPARγ and MEK interactions in cancer. PPAR Research. 2008;2008:16. doi: 10.1155/2008/309469.309469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X., Yang X., Xu Y., Jiang X., Nan F., Tang H. Troglitazone inhibits cell proliferation by attenuation of epidermal growth factor receptor signaling independent of peroxisome proliferator-activated receptor γ . Cell Research. 2009;19(6):720–732. doi: 10.1038/cr.2009.53. [DOI] [PubMed] [Google Scholar]
- 26.Dall’Asta M., Derlindati E., Ardigò D., Zavaroni I., Brighenti F., Del Rio D. Macrophage polarization: the answer to the diet/inflammation conundrum? Nutrition, Metabolism and Cardiovascular Diseases. 2012;22(5):387–392. doi: 10.1016/j.numecd.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 27.Meisner F., Walcher D., Gizard F., et al. Effect of rosiglitazone treatment on plaque inflammation and collagen content in nondiabetic patients: data from a randomized placebo-controlled trial. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(4):845–850. doi: 10.1161/01.ATV.0000203511.66681.7f. [DOI] [PubMed] [Google Scholar]
- 28.Ao C., Huo Y., Qi L., Xiong Z., Xue L., Qi Y. Pioglitazone suppresses the lipopolysaccharide-induced production of inflammatory factors in mouse macrophages by inactivating NF-κB. Cell Biology International. 2010;34(7):723–730. doi: 10.1042/CBI20090005. [DOI] [PubMed] [Google Scholar]
- 29.Burgermeister E., Chuderland D., Hanoch T., Meyer M., Liscovitch M., Seger R. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor γ . Molecular and Cellular Biology. 2007;27(3):803–817. doi: 10.1128/MCB.00601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakles R. E., Kallakury B. V. S., Furth P. A. The PPARγ agonist efatutazone increases the spectrum of well-differentiated mammary cancer subtypes initiated by loss of full-length BRCA1 in association with TP53 haploinsufficiency. The American Journal of Pathology. 2013;182(6):1976–1985. doi: 10.1016/j.ajpath.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimazaki N., Togashi N., Hanai M., et al. Anti-tumour activity of CS-7017, a selective peroxisome proliferator-activated receptor gamma agonist of thiazolidinedione class, in human tumour xenografts and a syngeneic tumour implant model. European Journal of Cancer. 2008;44(12):1734–1743. doi: 10.1016/j.ejca.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 32.Demetri G. D., Fletcher C. D., Mueller E., et al. Induction of solid tumor differentiation by the peroxisome proliferator- activated receptor-γ ligand troglitazone in patients with liposarcoma. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(7):3951–3956. doi: 10.1073/pnas.96.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debrock G., Vanhentenrijk V., Sciot R., Debiec-Rychter M., Oyen R., van Oosterom A. A phase II trial with rosiglitazone in liposarcoma patients. British Journal of Cancer. 2003;89(8):1409–1412. doi: 10.1038/sj.bjc.6601306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai Y., Wang W. H. Peroxisome proliferator-activated receptor gamma and colorectal cancer. World Journal of Gastrointestinal Oncology. 2010;2(3):159–164. doi: 10.4251/wjgo.v2.i3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pancione M., Forte N., Sabatino L., et al. Reduced β-catenin and peroxisome proliferator-activated receptor-γ expression levels are associated with colorectal cancer metastatic progression: correlation with tumor-associated macrophages, cyclooxygenase 2, and patient outcome. Human Pathology. 2009;40(5):714–725. doi: 10.1016/j.humpath.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 36.Theocharis S., Giaginis C., Parasi A., et al. Expression of peroxisome proliferator-activated receptor-γ in colon cancer: correlation with histopathological parameters, cell cycle-related molecules, and patients’ survival. Digestive Diseases and Sciences. 2007;52(9):2305–2311. doi: 10.1007/s10620-007-9794-4. [DOI] [PubMed] [Google Scholar]
- 37.Gustafsson A., Hansson E., Kressner U., et al. EP1-4 subtype, COX and PPARγ receptor expression in colorectal cancer in prediction of disease-specific mortality. International Journal of Cancer. 2007;121(2):232–240. doi: 10.1002/ijc.22582. [DOI] [PubMed] [Google Scholar]
- 38.McAlpine C. A., Barak Y., Matise I., Cormier R. T. Intestinal-specific PPARγ deficiency enhances tumorigenesis in ApcMin/+ mice. International Journal of Cancer. 2006;119(10):2339–2346. doi: 10.1002/ijc.22115. [DOI] [PubMed] [Google Scholar]
- 39.Osawa E., Nakajima A., Wada K., et al. Peroxisome proliferator-activated receptor γ ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology. 2003;124(2):361–367. doi: 10.1053/gast.2003.50067. [DOI] [PubMed] [Google Scholar]
- 40.Yoshizumi T., Ohta T., Ninomiya I., et al. Thiazolidinedione, a peroxisome proliferator-activated receptor-gamma ligand, inhibits growth and metastasis of HT-29 human colon cancer cells through differentiation-promoting effects. International Journal of Oncology. 2004;25(3):631–639. [PubMed] [Google Scholar]
- 41.Choi I. K., Kim Y. H., Kim J. S., Seo J. H. PPAR-γ ligand promotes the growth of APC-mutated HT-29 human colon cancer cells in vitro and in vivo. Investigational New Drugs. 2008;26(3):283–288. doi: 10.1007/s10637-007-9108-x. [DOI] [PubMed] [Google Scholar]
- 42.Tsukahara T., Haniu H. Peroxisome proliferator-activated receptor gamma overexpression suppresses proliferation of human colon cancer cells. Biochemical and Biophysical Research Communications. 2012;424(3):524–529. doi: 10.1016/j.bbrc.2012.06.149. [DOI] [PubMed] [Google Scholar]
- 43.Kulke M. H., Demetri G. D., Sharpless N. E., et al. A phase II study of troglitazone, an activator of the PPARγ receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer Journal. 2002;8(5):395–399. doi: 10.1097/00130404-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 44.Theocharis S., Kanelli H., Politi E., et al. Expression of peroxisome proliferator activated receptor-gamma in non-small cell lung carcinoma: correlation with histological type and grade. Lung Cancer. 2002;36(3):249–255. doi: 10.1016/S0169-5002(02)00013-2. [DOI] [PubMed] [Google Scholar]
- 45.Sasaki H., Tanahashi M., Yukiue H., et al. Decreased perioxisome proliferator-activated receptor gamma gene expression was correlated with poor prognosis in patients with lung cancer. Lung Cancer. 2002;36(1):71–76. doi: 10.1016/S0169-5002(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 46.Lyon C. M., Klinge D. M., Do K. C., et al. Rosiglitazone prevents the progression of preinvasive lung cancer in a murine model. Carcinogenesis. 2009;30(12):2095–2099. doi: 10.1093/carcin/bgp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang W. G., Douglas-Jones A., Mansel R. E. Expression of peroxisome-proliferator activated receptor-gamma (PPARγ) and the PPARγ co-activator, PGC-1, in human breast cancer correlates with clinical outcomes. International Journal of Cancer. 2003;106(5):752–757. doi: 10.1002/ijc.11302. [DOI] [PubMed] [Google Scholar]
- 48.Magenta G., Borenstein X., Rolando R., Jasnis M. A. Rosiglitazone inhibits metastasis development of a murine mammary tumor cell line LMM3. BMC Cancer. 2008;8, article 47 doi: 10.1186/1471-2407-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bojková B., Garajová M., Kajo K., et al. Pioglitazone in chemically induced mammary carcinogenesis in rats. European Journal of Cancer Prevention. 2010;19(5):379–384. doi: 10.1097/CEJ.0b013e32833ca233. [DOI] [PubMed] [Google Scholar]
- 50.Burstein H. J., Demetri G. D., Mueller E., Sarraf P., Spiegelman B. M., Winer E. P. Use of the peroxisome proliferator-activated receptor (PPAR) γ ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Research and Treatment. 2003;79(3):391–397. doi: 10.1023/A:1024038127156. [DOI] [PubMed] [Google Scholar]
- 51.Yee L. D., Williams N., Wen P., et al. Pilot study of rosiglitazone therapy in women with breast cancer: effects of short-term therapy on tumor tissue and serum markers. Clinical Cancer Research. 2007;13(1):246–252. doi: 10.1158/1078-0432.CCR-06-1947. [DOI] [PubMed] [Google Scholar]
- 52.Nakamura Y., Suzuki T., Sugawara A., Arai Y., Sasano H. Peroxisome proliferator-activated receptor gamma in human prostate carcinoma. Pathology International. 2009;59(5):288–293. doi: 10.1111/j.1440-1827.2009.02367.x. [DOI] [PubMed] [Google Scholar]
- 53.Annicotte J.-S., Iankova I., Miard S., et al. Peroxisome proliferator-activated receptor γ regulates E-cadherin expression and inhibits growth and invasion of prostate cancer. Molecular and Cellular Biology. 2006;26(20):7561–7574. doi: 10.1128/MCB.00605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mueller E., Smith M., Sarraf P., et al. Effects of ligand activation of peroxisome proliferator-activated receptor γ in human prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(20):10990–10995. doi: 10.1073/pnas.180329197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hisatake J. I., Ikezoe T., Carey M., Holden S., Tomoyasu S., Koeffler H. P. Down-regulation of prostate-specific antigen expression by ligands for peroxisome proliferator-activated receptor γ in human prostate cancer. Cancer Research. 2000;60(19):5494–5498. [PubMed] [Google Scholar]
- 56.Kato M., Nagaya T., Fujieda M., Saito K., Yoshida J., Seo H. Expression of PPARγ and its ligand-dependent growth inhibition in human brain tumor cell lines. Japanese Journal of Cancer Research. 2002;93(6):660–666. doi: 10.1111/j.1349-7006.2002.tb01304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grommes C., Conway D. S., Alshekhlee A., Barnholtz-Sloan J. S. Inverse association of PPARγ agonists use and high grade glioma development. Journal of Neuro-Oncology. 2010;100(2):233–239. doi: 10.1007/s11060-010-0185-x. [DOI] [PubMed] [Google Scholar]
- 58.Grommes C., Karlo J. C., Caprariello A., Blankenship D., Dechant A., Landreth G. E. The PPARγ agonist pioglitazone crosses the blood-brain barrier and reduces tumor growth in a human xenograft model. Cancer Chemotherapy and Pharmacology. 2013;71(4):929–936. doi: 10.1007/s00280-013-2084-2. [DOI] [PubMed] [Google Scholar]
- 59.Hau P., Kunz-Schughart L., Bogdahn U., et al. Low-dose chemotherapy in combination with COX-2 inhibitors and PPAR-gamma agonists in recurrent high-grade gliomas—a phase II study. Oncology. 2007;73(1-2):21–25. doi: 10.1159/000120028. [DOI] [PubMed] [Google Scholar]
- 60.Meyer S., Vogt T., Landthaler M., et al. Research article: Cyclooxygenase 2 (COX2) and peroxisome proliferator-activated receptor gamma (PPARG) are stage-dependent prognostic markers of malignant melanoma. PPAR Research. 2010;2010:10. doi: 10.1155/2010/848645.848645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferrara A., Lewis J. D., Quesenberry C. P., Jr., et al. Cohort study of pioglitazone and cancer incidence in patients with diabetes. Diabetes Care. 2011;34(4):923–929. doi: 10.2337/dc10-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Botton T., Puissant A., Bahadoran P., et al. In vitro and in vivo anti-melanoma effects of ciglitazone. Journal of Investigative Dermatology. 2009;129(5):1208–1218. doi: 10.1038/jid.2008.346. [DOI] [PubMed] [Google Scholar]
- 63.Govindarajan R., Ratnasinghe L., Simmons D. L., et al. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. Journal of Clinical Oncology. 2007;25(12):1476–1481. doi: 10.1200/JCO.2006.07.2777. [DOI] [PubMed] [Google Scholar]
- 64.Monami M., Dicembrini I., Mannucci E. Thiazolidinediones and cancer: results of a meta-analysis of randomized clinical trials. Acta Diabetologica. 2014;51(1):91–101. doi: 10.1007/s00592-013-0504-8. [DOI] [PubMed] [Google Scholar]
- 65.Kawa S., Nikaido T., Unno H., Usuda N., Nakayama K., Kiyosawa K. Growth inhibition and differentiation of pancreatic cancer cell lines by PPARγ ligand troglitazone. Pancreas. 2002;24(1):1–7. doi: 10.1097/00006676-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 66.Kato M., Kusumi T., Tsuchida S., Tanaka M., Sasaki M., Kudo H. Induction of differentiation and peroxisome proliferator-activated receptor γ expression in colon cancer cell lines by troglitazone. Journal of Cancer Research and Clinical Oncology. 2004;130(2):73–79. doi: 10.1007/s00432-003-0510-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jagan I., Fatehullah A., Deevi R. K., Bingham V., Campbell F. C. Rescue of glandular dysmorphogenesis in PTEN-deficient colorectal cancer epithelium by PPARγ-targeted therapy. Oncogene. 2013;32(10):1305–1315. doi: 10.1038/onc.2012.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y., Meng Y., Li H., et al. Growth inhibition and differentiation induced by peroxisome proliferator activated receptor gamma ligand rosiglitazone in human melanoma cell line A375. Medical Oncology. 2006;23(3):393–402. doi: 10.1385/MO:23:3:393. [DOI] [PubMed] [Google Scholar]
- 69.Aiello A., Pandini G., Frasca F., et al. Peroxisomal proliferator-activated receptor-γ agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology. 2006;147(9):4463–4475. doi: 10.1210/en.2005-1610. [DOI] [PubMed] [Google Scholar]
- 70.Pestereva E., Kanakasabai S., Bright J. J. PPARγ agonists regulate the expression of stemness and differentiation genes in brain tumour stem cells. British Journal of Cancer. 2012;106(10):1702–1712. doi: 10.1038/bjc.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Varley C. L., Stahlschmidt J., Smith B., Stower M., Southgate J. Activation of peroxisome proliferator-activated receptor-gamma reverses squamous metaplasia and induces transitional differentiation in normal human urothelial cells. The American Journal of Pathology. 2004;164(5):1789–1798. doi: 10.1016/S0002-9440(10)63737-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li M.-Y., Kong A. W. Y., Yuan H., et al. Pioglitazone prevents smoking carcinogen-induced lung tumor development in mice. Current Cancer Drug Targets. 2012;12(6):597–606. doi: 10.2174/156800912801784848. [DOI] [PubMed] [Google Scholar]
- 73.Borbath I., Leclercq I., Moulin P., Sempoux C., Horsmans Y. The PPARgamma agonist pioglitazone inhibits early neoplastic occurrence in the rat liver. European Journal of Cancer. 2007;43(11):1755–1763. doi: 10.1016/j.ejca.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 74.Yu J., Qiao L., Zimmermann L., et al. Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo. Hepatology. 2006;43(1):134–143. doi: 10.1002/hep.20994. [DOI] [PubMed] [Google Scholar]
- 75.Wu W., Celestino J., Milam M. R., et al. Primary chemoprevention of endometrial hyperplasia with the peroxisome proliferator-activated receptor gamma agonist rosiglitazone in the PTEN heterozygote murine model. International Journal of Gynecological Cancer. 2008;18(2):329–338. doi: 10.1111/j.1525-1438.2007.01002.x. [DOI] [PubMed] [Google Scholar]
- 76.Theocharis S., Klijanienko J., Giaginis C., et al. Peroxisome proliferator-activated receptor-γ in mobile tongue squamous cell carcinoma: associations with clinicopathological parameters and patients survival. Journal of Cancer Research and Clinical Oncology. 2011;137(2):251–259. doi: 10.1007/s00432-010-0882-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suzuki R., Kohno H., Suzui M., et al. An animal model for the rapid induction of tongue neoplasms in human c-Ha-ras proto-oncogene transgenic rats by 4-nitroquinoline 1-oxide: its potential use for preclinical chemoprevention studies. Carcinogenesis. 2006;27(3):619–630. doi: 10.1093/carcin/bgi241. [DOI] [PubMed] [Google Scholar]
- 78.Yoshida K., Hirose Y., Tanaka T., et al. Inhibitory effects of troglitazone, a peroxisome proliferator-activated receptor γ ligand, in rat tongue carcinogenesis initiated with 4-nitroquinoline 1-oxide. Cancer Science. 2003;94(4):365–371. doi: 10.1111/j.1349-7006.2003.tb01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li H., Sorenson A. L., Poczobutt J., et al. Activation of PPARγ in myeloid cells promotes lung cancer progression and metastasis. PLoS ONE. 2011;6(12) doi: 10.1371/journal.pone.0028133.e28133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bonofiglio D., Cione E., Qi H., et al. Combined low doses of PPARγ and RXR ligands trigger an intrinsic apoptotic pathway in human breast cancer cells. The American Journal of Pathology. 2009;175(3):1270–1280. doi: 10.2353/ajpath.2009.081078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tontonoz P., Singer S., Forman B. M., et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor γ and the retinoid X receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(1):237–241. doi: 10.1073/pnas.94.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cao L. Q., Wang X. L., Wang Q., et al. Rosiglitazone sensitizes hepatocellular carcinoma cell lines to 5-fluorouracil antitumor activity through activation of the PPARγ signaling pathway. Acta Pharmacologica Sinica. 2009;30(9):1316–1322. doi: 10.1038/aps.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee S. Y., Hur G. Y., Jung K. H., et al. PPAR-γ agonist increase gefitinib’s antitumor activity through PTEN expression. Lung Cancer. 2006;51(3):297–301. doi: 10.1016/j.lungcan.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 84.Yang Z., Bagheri-Yarmand R., Balasenthil S., et al. HER2 regulation of peroxisome proliferator-activated receptor γ (PPARγ) expression and sensitivity of breast cancer cells to PPARγ ligand therapy. Clinical Cancer Research. 2003;9(8):3198–3203. [PubMed] [Google Scholar]
- 85.Girnun G. D., Naseri E., Vafai S. B., et al. Synergy between PPARγ ligands and platinum-based drugs in cancer. Cancer Cell. 2007;11(5):395–406. doi: 10.1016/j.ccr.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tikoo K., Kumar P., Gupta J. Rosiglitazone synergizes anticancer activity of cisplatin and reduces its nephrotoxicity in 7, 12-dimethyl benz{a}anthracene (DMBA) induced breast cancer rats. BMC Cancer. 2009;9, article 107 doi: 10.1186/1471-2407-9-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hamaguchi N., Hamada H., Miyoshi S., et al. In vitro and in vivo therapeutic efficacy of the PPAR-γ agonist troglitazone in combination with cisplatin against malignant pleural mesothelioma cell growth. Cancer Science. 2010;101(9):1955–1964. doi: 10.1111/j.1349-7006.2010.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Copland J. A., Marlow L. A., Kurakata S., et al. Novel high-affinity PPARγ agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21WAF1/CIP1. Oncogene. 2006;25(16):2304–2317. doi: 10.1038/sj.onc.1209267. [DOI] [PubMed] [Google Scholar]
- 89.Yokoyama Y., Xin B., Shigeto T., Mizunuma H. Combination of ciglitazone, a peroxisome proliferator-activated receptor gamma ligand, and cisplatin enhances the inhibition of growth of human ovarian cancers. Journal of Cancer Research and Clinical Oncology. 2011;137(8):1219–1228. doi: 10.1007/s00432-011-0993-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fu H., Zhang J., Pan J., et al. Chemoprevention of lung carcinogenesis by the combination of aerosolized budesonide and oral pioglitazone in A/J mice. Molecular Carcinogenesis. 2011;50(12):913–921. doi: 10.1002/mc.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reichle A., Bross K., Vogt T., et al. Pioglitazone and rofecoxib combined with angiostatically scheduled trofosfamide in the treatment of far-advanced melanoma and soft tissue sarcoma. Cancer. 2004;101(10):2247–2256. doi: 10.1002/cncr.20574. [DOI] [PubMed] [Google Scholar]
- 92.Vogt T., Hafner C., Bross K., et al. Antiangiogenetic therapy with pioglitazone, rofecoxib, and metronomic trofosfamide in patients with advanced malignant vascular tumors. Cancer. 2003;98(10):2251–2256. doi: 10.1002/cncr.11775. [DOI] [PubMed] [Google Scholar]
- 93.Ondrey F. Peroxisome proliferator-activated receptor γ pathway targeting in carcinogenesis: implications For Chemoprevention. Clinical Cancer Research. 2009;15(1):2–8. doi: 10.1158/1078-0432.CCR-08-0326. [DOI] [PubMed] [Google Scholar]
- 94.Omur O., Baran Y. An update on molecular biology of thyroid cancers. Critical Reviews in Oncology/Hematology. 2014;90(3):233–252. doi: 10.1016/j.critrevonc.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 95.Li N., Du X. L., Reitzel L. R., Xu L., Sturgis E. M. Impact of enhanced detection on the increase in thyroid cancer incidence in the United States: review of incidence trends by socioeconomic status within the surveillance, epidemiology, and end results registry, 1980–2008. Thyroid. 2013;23(1):103–110. doi: 10.1089/thy.2012.0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eustatia-Rutten C. F. A., Corssmit E. P. M., Biermasz N. R., Pereira A. M., Romijn J. A., Smit J. W. Survival and death causes in differentiated thyroid carcinoma. Journal of Clinical Endocrinology and Metabolism. 2006;91(1):313–319. doi: 10.1210/jc.2005-1322. [DOI] [PubMed] [Google Scholar]
- 97.Durante C., Haddy N., Baudin E., et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: Benefits and limits of radioiodine therapy. Journal of Clinical Endocrinology and Metabolism. 2006;91(8):2892–2899. doi: 10.1210/jc.2005-2838. [DOI] [PubMed] [Google Scholar]
- 98.Dohán O., De La Vieja A., Paroder V., et al. The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocrine Reviews. 2003;24(1):48–77. doi: 10.1210/er.2001-0029. [DOI] [PubMed] [Google Scholar]
- 99.Schlumberger M. J. Diagnostic follow-up of well-differentiated thyroid carcinoma: historical perspective and current status. Journal of Endocrinological Investigation. 1999;22(11):3–7. [PubMed] [Google Scholar]
- 100.Carling T., Udelsman R., editors. Thyroid Tumors. 9th. Philadelphia, Pa, USA: Lippincott Williams & Wilkins; 2011. [Google Scholar]
- 101.Versari A., Sollini M., Frasoldati A., et al. Differentiated thyroid cancer: a new perspective with radiolabeled somatostatin analogues for imaging and treatment of patients. Thyroid. 2014;24(4):715–726. doi: 10.1089/thy.2013.0225. [DOI] [PubMed] [Google Scholar]
- 102.Martelli M. L., Iuliano R., Le Pera I., et al. Inhibitory effects of peroxisome proliferator-activated receptor γ on thyroid carcinoma cell growth. Journal of Clinical Endocrinology and Metabolism. 2002;87(10):4728–4735. doi: 10.1210/jc.2001-012054. [DOI] [PubMed] [Google Scholar]
- 103.Fröhlich E., Brossart P., Wahl R. Induction of iodide uptake in transformed thyrocytes: a compound screening in cell lines. European Journal of Nuclear Medicine and Molecular Imaging. 2009;36(5):780–790. doi: 10.1007/s00259-008-1024-6. [DOI] [PubMed] [Google Scholar]
- 104.Park J.-W., Zarnegar R., Kanauchi H., et al. Troglitazone, the peroxisome proliferator-activated receptor-γ agonist, induces antiproliferation and redifferentiation in human thyroid cancer cell lines. Thyroid. 2005;15(3):222–231. doi: 10.1089/thy.2005.15.222. [DOI] [PubMed] [Google Scholar]
- 105.Fröhlich E., Machicao F., Wahl R. Action of thiazolidinediones on differentiation, proliferation and apoptosis of normal and transformed thyrocytes in culture. Endocrine-Related Cancer. 2005;12(2):291–303. doi: 10.1677/erc.1.00973. [DOI] [PubMed] [Google Scholar]
- 106.Dobson M. E., Diallo-Krou E., Grachtchouk V., et al. Pioglitazone induces a proadipogenic antitumor response in mice with PAX8-PPARγ fusion protein thyroid carcinoma. Endocrinology. 2011;152(11):4455–4465. doi: 10.1210/en.2011-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kato Y., Ying H., Zhao L., et al. PPARγ insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-κB signaling pathway. Oncogene. 2006;25(19):2736–2747. doi: 10.1038/sj.onc.1209299. [DOI] [PubMed] [Google Scholar]
- 108.Rosenbaum-Krumme S. J., Bockisch A., Nagarajah J. Pioglitazone therapy in progressive differentiated thyroid carcinoma. NuklearMedizin. 2012;51(4):111–115. doi: 10.3413/Nukmed-0474-12-01. [DOI] [PubMed] [Google Scholar]
- 109.Elola M., Yoldi A., Emparanza J. I., Matteucci T., Bilbao I., Goena M. Redifferentiation therapy with rosiglitazone in a case of differentiated thyroid cancer with pulmonary metastases and absence of radioiodine uptake. Revista Espanola de Medicina Nuclear. 2011;30(4):241–243. doi: 10.1016/j.remn.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 110.Elias A. N., Lizotte P. Enhanced radioiodine uptake in a patient with poorly differentiated papillary thyroid cancer after treatment with rosiglitazone. Clinical Nuclear Medicine. 2006;31(9):517–519. doi: 10.1097/01.rlu.0000233148.45744.44. [DOI] [PubMed] [Google Scholar]
- 111.Philips J.-C., Petite C., Willi J.-P., Buchegger F., Meier C. A. Effect of peroxisome proliferator-activated receptor γ agonist, rosiglitazone, on dedifferentiated thyroid cancers. Nuclear Medicine Communications. 2004;25(12):1183–1186. doi: 10.1097/00006231-200412000-00005. [DOI] [PubMed] [Google Scholar]
- 112.Kebebew E., Peng M., Reiff E., et al. A phase II trial of rosiglitazone in patients with thyroglobulin-positive and radioiodine-negative differentiated thyroid cancer. Surgery. 2006;140(6):960–967. doi: 10.1016/j.surg.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 113.Tepmongkol S., Keelawat S., Honsawek S., Ruangvejvorachai P. Rosiglitazone effect on radioiodine uptake in thyroid carcinoma patients with high thyroglobulin but negative total body scan: a correlation with the expression of peroxisome proliferator-activated receptor-gamma. Thyroid. 2008;18(7):697–704. doi: 10.1089/thy.2008.0056. [DOI] [PubMed] [Google Scholar]
- 114.Kebebew E., Lindsay S., Clark O. H., Woeber K. A., Hawkins R., Greenspan F. S. Results of rosiglitazone therapy in patients with thyroglobulin-positive and radioiodine-negative advanced differentiated thyroid cancer. Thyroid. 2009;19(9):953–956. doi: 10.1089/thy.2008.0371. [DOI] [PubMed] [Google Scholar]
- 115.Eisenhauer E. A., Therasse P., Bogaerts J., et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) European Journal of Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 116.Burton J. D., Goldenberg D. M., Blumenthal R. D. Potential of peroxisome proliferator-activated receptor gamma antagonist compounds as therapeutic agents for a wide range of cancer types. PPAR Research. 2008;2008:7. doi: 10.1155/2008/494161.494161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kakiuchi-Kiyota S., Vetro J. A., Suzuki S., et al. Effects of the PPARγ agonist troglitazone on endothelial cells in vivo and in vitro: Differences between human and mouse. Toxicology and Applied Pharmacology. 2009;237(1):83–90. doi: 10.1016/j.taap.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 118.Takano S., Kubota T., Nishibori H., et al. Pioglitazone, a ligand for peroxisome proliferator-activated receptor-γ acts as an inhibitor of colon cancer liver metastasis. Anticancer Research. 2008;28(6):3593–3599. [PubMed] [Google Scholar]
- 119.Diede S. J., Yao Z., Chip Keyes C., et al. Fundamental differences in promoter CpG island DNA hypermethylation between human cancer and genetically engineered mouse models of cancer. Epigenetics. 2013;8(12):1254–1260. doi: 10.4161/epi.26486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.De Jong M., Maina T. Of mice and humans: are they the same?—Implications in cancer translational research. Journal of Nuclear Medicine. 2010;51(4):501–504. doi: 10.2967/jnumed.109.065706. [DOI] [PubMed] [Google Scholar]
- 121.Vaish V., Tanwar L., Sanyal S. N. The role of NF-κB and PPARγ in experimentally induced colorectal cancer and chemoprevention by cyclooxygenase-2 inhibitors. Tumour Biology. 2010;31(5):427–436. doi: 10.1007/s13277-010-0051-7. [DOI] [PubMed] [Google Scholar]
- 122.Pino M. V., Kelley M. F., Jayyosi Z. Promotion of colon tumors in C57BL/6J-APCmin/+ mice by thiazolidinedione PPARγ agonists and a structurally unrelated PPARγ agonist. Toxicologic Pathology. 2004;32(1):58–63. doi: 10.1080/01926230490261320. [DOI] [PubMed] [Google Scholar]
- 123.Kohno H., Yoshitani S., Takashima S., et al. Troglitazone, a ligand for peroxisome proliferator-activated receptor γ, inhibits chemically-induced aberrant crypt foci in rats. Japanese Journal of Cancer Research. 2001;92(4):396–403. doi: 10.1111/j.1349-7006.2001.tb01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Han E. J., Im C. N., Park S. H., Moon E. Y., Hong S. H. Combined treatment with peroxisome proliferator-activated receptor (PPAR) gamma ligands and gamma radiation induces apoptosis by PPARγ-independent up-regulation of reactive oxygen species-induced deoxyribonucleic acid damage signals in non-small cell lung cancer cells. International Journal of Radiation Oncology Biology Physics. 2013;85(5):e239–e248. doi: 10.1016/j.ijrobp.2012.11.040. [DOI] [PubMed] [Google Scholar]
- 125.James S. Y., Lin F., Kolluri S. K., Dawson M. I., Zhang X.-K. Regulation of retinoic acid receptor β expression by peroxisome proliferator-activated receptor γ ligands in cancer cells. Cancer Research. 2003;63(13):3531–3538. [PubMed] [Google Scholar]
- 126.Papi A., Rochhi P., Ferreri A. M., Guerra F., Orlandi M. Enhanced effects of PPARγ ligands and RXR selective retinoids in combination to inhibit migration and invasiveness in cancer cells. Oncology Reports. 2009;21(4):1083–1089. doi: 10.3892/or_00000327. [DOI] [PubMed] [Google Scholar]
- 127.Papi A., Tatenhorst L., Terwel D., et al. PPARγ and RXRγ ligands act synergistically as potent antineoplastic agents in vitro and in vivo glioma models. Journal of Neurochemistry. 2009;109(6):1779–1790. doi: 10.1111/j.1471-4159.2009.06111.x. [DOI] [PubMed] [Google Scholar]
- 128.Bräutigam K., Biernath-Wüpping J., Bauerschlag D. O., et al. Combined treatment with TRAIL and PPARγ ligands overcomes chemoresistance of ovarian cancer cell lines. Journal of Cancer Research and Clinical Oncology. 2011;137(5):875–886. doi: 10.1007/s00432-010-0952-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mrówka P., Glodkowska E., Nowis D., et al. Ciglitazone, an agonist of peroxisome proliferator-activated receptor γ, exerts potentiated cytostatic/cytotoxic effects against tumor cells when combined with lovastatin. International Journal of Oncology. 2008;32(1):249–255. [PubMed] [Google Scholar]
- 130.Chang T. H., Szabo E. Enhanced growth inhibition by combination differentiation therapy with ligands of peroxisome proliferator-activated receptor-γ and inhibitors of histone deacetylase in adenocarcinoma of the lung. Clinical Cancer Research. 2002;8(4):1206–1212. [PubMed] [Google Scholar]
- 131.Liu Y., Zhu Z.-A., Zhang S.-N., et al. Combinational effect of PPARγ agonist and RXR agonist on the growth of SGC7901 gastric carcinoma cells in vitro. Tumor Biology. 2013;34(4):2409–2418. doi: 10.1007/s13277-013-0791-2. [DOI] [PubMed] [Google Scholar]
- 132.Reddy R. C., Srirangam A., Reddy K., et al. Chemotherapeutic drugs induce PPAR-γ expression and show sequence-specific synergy with PPAR-γ ligands in inhibition of non-small cell lung cancer. Neoplasia. 2008;10(6):597–603. doi: 10.1593/neo.08134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Elstner E., Williamson E. A., Zang C., et al. Novel therapeutic approach: ligands for PPARγ and retinoid receptors induce apoptosis in bcl-2-positive human breast cancer cells. Breast Cancer Research and Treatment. 2002;74(2):155–165. doi: 10.1023/A:1016114026769. [DOI] [PubMed] [Google Scholar]
- 134.Park B.-H., Lee S.-B., Stolz D. B., Lee Y. J., Lee B.-C. Synergistic interactions between heregulin and peroxisome proliferator-activated receptor-γ (PPARγ) agonist in breast cancer cells. Journal of Biological Chemistry. 2011;286(22):20087–20099. doi: 10.1074/jbc.M110.191718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yao C.-J., Lai G.-M., Chan C.-F., Cheng A.-L., Yang Y.-Y., Chuang S.-E. Dramatic synergistic anticancer effect of clinically achievable doses of lovastatin and troglitazone. International Journal of Cancer. 2006;118(3):773–779. doi: 10.1002/ijc.21361. [DOI] [PubMed] [Google Scholar]
- 136.Yan K.-H., Yao C.-J., Chang H.-Y., Lai G.-M., Cheng A.-L., Chuang S.-E. The synergistic anticancer effect of troglitazone combined with aspirin causes cell cycle arrest and apoptosis in human lung cancer cells. Molecular Carcinogenesis. 2010;49(3):235–246. doi: 10.1002/mc.20593. [DOI] [PubMed] [Google Scholar]
- 137.Yu H.-N., Noh E.-M., Lee Y.-R., et al. Troglitazone enhances tamoxifen-induced growth inhibitory activity of MCF-7 cells. Biochemical and Biophysical Research Communications. 2008;377(1):242–247. doi: 10.1016/j.bbrc.2008.09.111. [DOI] [PubMed] [Google Scholar]
- 138.An Z., Liu X., Song H., et al. Effect of troglitazone on radiation sensitivity in cervix cancer cells. Radiation Oncology Journal. 2012;30(2):78–87. doi: 10.3857/roj.2012.30.2.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang Y.-Q., Tang X.-Q., Sun L., et al. Rosiglitazone enhances fluorouracil-induced apoptosis of HT-29 cells by activating peroxisome proliferator-activated receptor γ . World Journal of Gastroenterology. 2007;13(10):1534–1540. doi: 10.3748/wjg.v13.i10.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cesario R. M., Stone J., Yen W.-C., Bissonnette R. P., Lamph W. W. Differentiation and growth inhibition mediated via the RXR:PPARγ heterodimer in colon cancer. Cancer Letters. 2006;240(2):225–233. doi: 10.1016/j.canlet.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 141.Mody M., Dharker N., Bloomston M., et al. Rosiglitazone sensitizes MDA-MB-231 breast cancer cells to anti-tumour effects of tumour necrosis factor-α, CH11 and CYC202. Endocrine-Related Cancer. 2007;14(2):305–315. doi: 10.1677/ERC-06-0003. [DOI] [PubMed] [Google Scholar]
- 142.Koga H., Selvendiran K., Sivakumar R., et al. PPARγ potentiates anticancer effects of gemcitabine on human pancreatic cancer cells. International Journal of Oncology. 2012;40(3):679–685. doi: 10.3892/ijo.2011.1237. [DOI] [PubMed] [Google Scholar]
- 143.Freudlsperger C., Thies A., Pfuller U., Schumacher U. The proteasome inhibitor bortezomib augments anti-proliferative effects of mistletoe lectin-I and the PPAR-γ agonist rosiglitazone in human melanoma cells. Anticancer Research. 2007;27(1):207–213. [PubMed] [Google Scholar]
- 144.Gehring S., Tapia-Pérez J. H., Kirches E., et al. Cytotoxic effects of statins and thiazolidinediones on meningioma cells. Journal of Neuro-Oncology. 2011;102(3):383–393. doi: 10.1007/s11060-010-0351-1. [DOI] [PubMed] [Google Scholar]
- 145.Tapia-Pérez J. H., Kirches E., Mawrin C., Firsching R., Schneider T. Cytotoxic effect of different statins and thiazolidinediones on malignant glioma cells. Cancer Chemotherapy and Pharmacology. 2011;67(5):1193–1201. doi: 10.1007/s00280-010-1535-2. [DOI] [PubMed] [Google Scholar]
- 146.Gottfried E., Rogenhofer S., Waibel H., et al. Pioglitazone modulates tumor cell metabolism and proliferation in multicellular tumor spheroids. Cancer Chemotherapy and Pharmacology. 2011;67(1):117–126. doi: 10.1007/s00280-010-1294-0. [DOI] [PubMed] [Google Scholar]