

Abstract
Lipoxin A4 has been described as a major signal for the resolution of inflammation and is abnormally produced in the lungs of patients with cystic fibrosis (CF). In CF, the loss of chloride transport caused by the mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel gene results in dehydration, mucus plugging, and reduction of the airway surface liquid layer (ASL) height which favour chronic lung infection and neutrophil based inflammation leading to progressive lung destruction and early death of people with CF. This review highlights the unique ability of LXA4 to restore airway surface hydration, to stimulate airway epithelial repair, and to antagonise the proinflammatory program of the CF airway, circumventing some of the most difficult aspects of CF pathophysiology. The report points out novel aspects of the cellular mechanism involved in the physiological response to LXA4, including release of ATP from airway epithelial cell via pannexin channel and subsequent activation of and P2Y11 purinoreceptor. Therefore, inadequate endogenous LXA4 biosynthesis reported in CF exacerbates the ion transport abnormality and defective mucociliary clearance, in addition to impairing the resolution of inflammation, thus amplifying the vicious circle of airway dehydration, chronic infection, and inflammation.
1. Lipoxin A4
1.1. Lipoxin A4 and Eicosanoid Class Switching
Lipoxin A4 (LXA4) belongs to a class of newly identified specialised proresolution lipid mediators playing a central role in the resolution of inflammation which results from the sequential production of characteristic eicosanoids in a process termed “class switching” [1, 2]. Prostaglandins are biosynthesized early, initiating the acute inflammatory response. Then Leukotrienes typified by Leukotriene B4 (LTB4) play a role in the amplification and propagation of inflammation [1] acting in concert with the peptide Interleukin 8 (IL8) as a potent neutrophil chemoattractant [3, 4]. Both LTB4 and IL8 are negatively correlated with pulmonary function in CF. LXA4 is the first eicosanoid expressed in the active resolution phase of inflammation [5] followed by biosynthesis of the Resolvins and Protectins. LTB4 and LXA4 are closely related metabolites of arachidonic acid and can be synthesised from a common unstable intermediate [3].
1.2. Lipoxin A4 Synthesis
LXA4 is produced by multistep enzymatic process resulting from lipoxygenase (LO) activities in different cell types [6]. Neutrophils [7], eosinophils [8], alveolar macrophages [9], platelets [10], or airway epithelial cells [11] express different LO which act in sequence in LXA4 biosynthesis [3, 12].
Two main pathways will result in LXA4 synthesis. One involves lipoxygenation of arachidonic acid by 15-LO in macrophages and epithelial cells. The 5-LO expressed by neutrophils can then utilise the 15(S)-hydroxyeicosatetranoic acid (15S-HETE) released as a substrate to synthesize LXA4 [7] (Figure 1, blue arrows). Alternatively, platelet 12-LO [10] and macrophage or epithelial 15-LO [13, 14] are each able to transform Leukotriene A4, released by neutrophils, into LXA4 (Figure 1, brown arrows). The activity of 15-LO promotes LXA4 biosynthesis and blocks leukotriene biosynthesis, both as a result of 15-LO products competing for flux at the 5-LO level and by diversion of the intermediate Leukotriene A4 away from LTB4 towards LXA4 biosynthesis [1, 11, 15].
Figure 1.
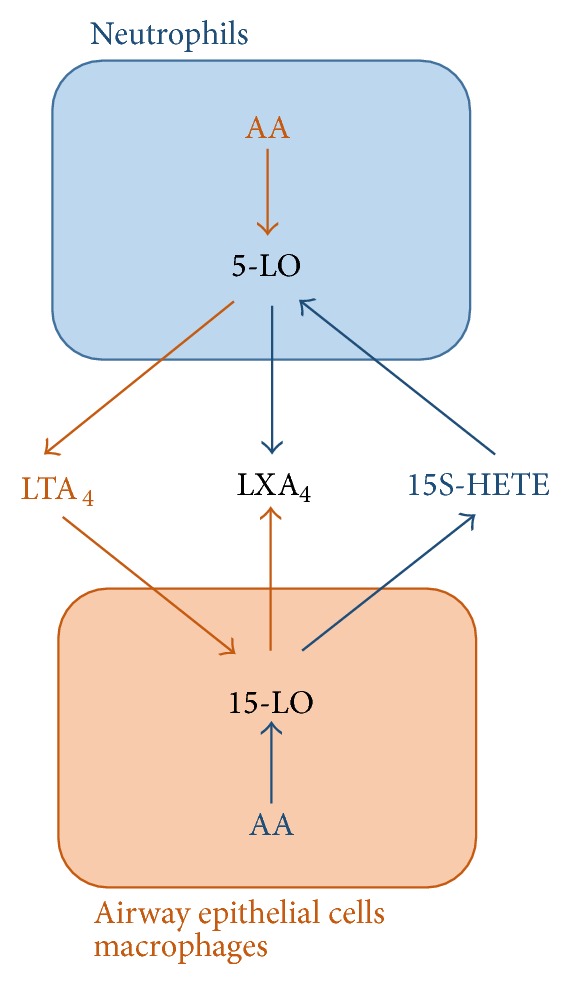
Lipoxin A4 biosynthesis by trans-cellular cooperation in the airways. The neutrophil donates LTA4 intermediate formed by the action of 5 lipoxygenase (5-LO) on arachidonic acid (AA) to the acceptor airway epithelial cell or alveolar macrophage whereby 15 lipoxygenase (15-LO) catalyses LXA4 formation (brown arrows). Airway epithelial cell or alveolar macrophage 15-LO activity catalyses the conversion of AA to 15S-HETE which is donated to the acceptor neutrophil and converted to LXA4 by 5-LO catalysis (blue arrows).
1.3. Lipoxin A4 Anti-Inflammatory Actions
The anti-inflammatory action of LXA4 is mainly mediated by the formyl-peptide receptor 2 (FPR2) which is one member of a subgroup of receptors linked to inhibitory G-proteins, also called ALX [16, 17]. FPR2 receptor activation by specific agonists results in transient Ca2+ flux, phosphorylation of extracellular signal regulated kinases (ERK), and chemotaxis [18]. The molecular and pharmacological characterization of FPR2 receptor have been previously reviewed [19, 20]. Briefly, the seventh transmembrane domain of the FPR2 receptor is essential for LXA4 recognition, whereas the additional regions of the receptor (e.g., extracellular loops) are required for high affinity binding of the peptide ligands [17, 19, 20]. LXA4 also interacts directly with the cysLTI receptor to transduce signals that prevent the proinflammatory response and contributes to the active resolution of inflammation [18, 21].
LXA4 inhibits neutrophil effector functions [5] and in particular inhibits LTB4 induced neutrophil transmigration [22–24]. LXA4 suppresses IL8 production by leukocytes and bronchial epithelial cells including airway epithelial cells from patients with cystic fibrosis [25–28]. Mice treated with analogues of LXA4 and subsequently challenged with P. aeruginosa contained the bacterial challenge more effectively [29]. LXA4 affects leukocytes in a cell type specific manner, inhibiting the activation of polymorphonuclears (PMNs) and eosinophils whilst activating monocytes and macrophages. PMN recruitment is a multistep process that involves chemotaxis, adhesion, and transmigration. In in vitro models LXA4, LXB4, and ATLS inhibit PMN chemotaxis in response to the chemoattractant LTB4 and inhibit eosinophil responses to platelet activating factor. Stimulation of macrophages with LXA4 significantly enhances phagocytosis of apoptotic PMN, suggesting that LXA4 can promote the clearance of apoptotic leukocytes by macrophages at an inflammatory site [30, 31].
2. Cystic Fibrosis
2.1. Cystic Fibrosis Disease and the CF Gene
Cystic fibrosis (CF) is the most common lethal genetic disorder in Caucasians caused by a mutation in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR). The disease was first characterised in 1938 by Andersen who described the cystic fibrosis of the pancreas and correlated it with the lung and intestinal disease that occurs in CF [32]. In 1953, the observation of excessive salt loss in the sweat of CF patients was noted; however, it was not until 1983 that it was first shown that sufferers of CF displayed abnormal chloride transport. This discovery was not sufficient for the identification of the defective protein in CF patients. In 1985 polymorphic markers associated with the disease were identified and finally the CFTR gene was identified [33–35].
The CFTR protein is principally expressed in the apical membranes of epithelia where it acts as an anion channel providing a pathway for Cl− and bicarbonate (HCO3 −) movement, controlling the rate of fluid flow, and also regulating the function of other ion channels and transporters in epithelial cells [36, 37]. A number of different CFTR mutations have been identified that lead to differing outcomes in terms of protein synthesis, trafficking, regulation, and CFTR levels within the cell [38, 39]. CFTR is abundantly expressed in epithelial cell membranes and, as such, CF disease particularly affects epithelial sites: the submucosal glands, airway surface epithelium [40], pancreatic ductal epithelium, the epithelium of the crypts of Lieberkuhn throughout the gastrointestinal tract [41], the epithelium of sweat glands [42], the epithelium of the developing genital ducts, adult epididymis and vas deferens, and the cervical and the uterine epithelial surfaces [43, 44]. However, there are exceptions among epithelial tissues where CF related dysfunction is not prominent, such as kidney collecting ducts, the epithelium of Burners gland, and the submucosal glands of the duodenum [44]. The major clinical features of CF are chronic pulmonary disease, exocrine pancreatic insufficiency, and male infertility. CF lung disease reflects the failure of airway defence against chronic bacterial infection, leading to an aggravated immune response, bronchial epithelial remodelling, and ultimately lung destruction. The progressive lung destruction is the main cause of morbidity and mortality in CF [45, 46]. Whilst it was initially believed that the pulmonary complaints associated with CF were directly related to the CFTR dysfunction in epithelial cells, it is now recognised that other cell types including neutrophils [47, 48], macrophages [49, 50], and dendritic cells [51] are directly affected by the absence or dysfunctional CFTR.
2.2. Abnormal Production of Lipoxin A4 in Cystic Fibrosis
In addition to CFTR dysfunction, other abnormalities have been described in chronically inflamed and infected CF airways, including intrinsic proinflammatory properties, amplified inflammatory responses to infections, and reduced bacterial clearance. More specifically, the levels of LXA4 have been reported to be decreased in CF, like in other chronic airway inflammatory diseases such as asthma [29, 52–55]. A significant suppression in LXA4/neutrophil ratios in bronchoalveolar lavages (BAL) fluid of patients with CF compared with pulmonary inflammatory controls was reported [29, 56]. Furthermore, in paediatric CF BAL even in the absence of infection, the ratio of LXA4 to LTB4 is depressed and this correlates with a significant lower level of 15 LO-2 transcripts in CF BAL [2]. A decreased proportion of proresolving compounds (LXA4) compared to proinflammatory (LTB4) is associated with decreased lung function parameters [57]. In addition, in vitro studies support a role for CFTR in LXA4 production. The inhibition of CFTR reduces LXA4 synthesis by 50% during platelets/PMN coincubation by inhibiting the lipoxin synthase activity of platelets 12-LO. This correlated with the observation that platelets from patients with CF generated 40% less LXA4 compared to healthy subjects [58]. The decreased LXA4 production in CF provides a mechanistic explanation of the failure to actively resolve acute airway inflammation seen in these patients.
3. Regulation of Ion Transport and Airway Surface Liquid Layer in Cystic Fibrosis
3.1. Abnormal Ion and Fluid Transport in Cystic Fibrosis
The lung must continually defend itself against bacteria that deposit on the airway surfaces during normal tidal breathing. Mucus clearance is a primary form of pulmonary defence and the efficiency of mucociliary clearance in large part depends upon the volume of the airway surface liquid layer (ASL). The ASL allows for mucus containing foreign bodies to be transported away from the lung to the oropharynx where it is either expelled from the body or swallowed and destroyed by the gut. The ASL provides a low viscosity solution allowing free ciliary beat and mucus transport [59]. The normal hydration of the airway surface is maintained (in the highly water permeable airway epithelium) by active ion-transport controlling the quantity of salt (NaCl) delivered to airway surfaces, with water following passively by osmosis [60]. The NaCl concentration of the airway surface liquid is tightly regulated in normal airway epithelia by the epithelial sodium channel (ENaC) mediated Na+ absorption and Cl− secretion. Cl− is secreted by epithelial cells via the apical CFTR Cl− channel and calcium activated Cl− channels, with Cl− entering the cell through the Na+–K+–2Cl− cotransporter located in the basolateral membrane. Regulation of Cl− secretion determines the net transport of ions across the epithelium and hence the mass of salt on the epithelial surfaces. CFTR was also found to regulate ENaC suggesting that CFTR acted both as a Cl− channel and as a regulator of other ion transport processes. In CF, mutations of the CFTR gene result in defective Cl− secretion and Na+ hyperabsorption by airway epithelia [61, 62]. Studies in CF airway epithelium cultures, transgenic mice, and people with CF suggest that the initiating event in CF airway disease is a reduced ASL volume resulting from dehydration. This dehydration leads to reduced mucus clearance, adhesion of mucus to airway surfaces, and chronic bacterial infection of the lung (Figure 2). The chronic bacterial infection leads to an aggravated immune response, bronchial epithelial remodelling, and ultimately lung destruction [59, 63–70].
Figure 2.
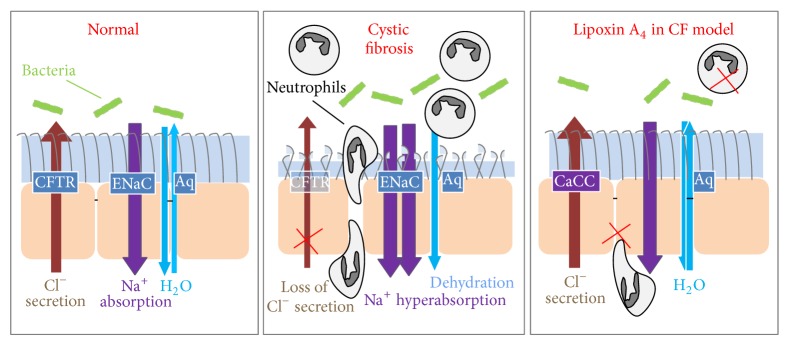
In normal airways the airway surface liquid layer (ASL) provides an adequate mucociliary clearance which is maintained by a combination of Cl− secretion through the cystic fibrosis transmembrane conductance regulator (CFTR), Na+ absorption via the epithelial sodium channel (ENaC), and water transport through a paracellular pathway and membrane bound aquaporins (Aq). In CF, a defective CFTR leads to loss of Cl− secretion and Na+ hyperabsorption. The concomitant dehydration of the airway lumen favours bacterial infection and inflammation (mainly neutrophilic). LXA4 mediates an increase in ASL height and restores it to normal levels in CF bronchial epithelium. LXA4 also increase tight junction formation, reestablishing the epithelial barrier function. Taken together this work provides evidence for LXA4 as potentially a new therapy for CF patients.
3.2. Lipoxin A4 Restores Fluid Transport in Cystic Fibrosis
One of the greatest challenges of fundamental research into reversing the CF defect in the lung has been to design a strategy to overcome the absence of functional CFTR by stimulating chloride secretion via alternative pathways, thus restoring airway hydration and mucociliary clearance. This can be achieved via the stimulation of calcium activated Cl− by agents that raise the intracellular concentration of calcium. Yet, this strategy has been plagued by the side effects of the amplification of the calcium-dependent proinflammatory response resulting in undesirable activation of NFκB. In addition to its anti-inflammatory properties, LXA4 stimulates a rapid and transient intracellular Ca2+ increase in normal and CF bronchial epithelial cells expressing the FPR2 receptor [71, 72]. This intracellular calcium signal is mainly due to calcium mobilisation from intracellular calcium stores in non-CF airway epithelial cells and due to calcium entry and intracellular calcium release in CF airway epithelial cells. In both, non-CF and CF bronchial epithelia, LXA4 stimulates whole-cell Cl− currents which are inhibited by NPPB (calcium-activated Cl− channel inhibitor) and BAPTA-AM (chelator of intracellular Ca2+) but not by CFTRinh-172 (CFTR inhibitor) [71, 72]. Furthermore, in models of fully differentiated bronchial epithelia derived from primary culture of bronchial brushings from patients with CF and cultured under air-liquid interface, LXA4's effects on ion transport result in an increase of the airway surface liquid (ASL) layer height. LXA4 exerts this effect on the ASL dynamics via the FPR2 receptor. The sustained increase in ASL height induced by LXA4 in non-CF and CF bronchial epithelia results from inhibition of amiloride-sensitive Na+ absorption and stimulation of an intracellular calcium signal and Ca2+-activated Cl− secretion independent from CFTR [72, 73]. LXA4 thus restores Cl− secretion and normal ASL height both central to the pathophysiology of CF airway disease, highlighting a role for LXA4 in the restoration of normal innate immune defence (Figure 2).
4. Nucleotides and CF Airway Disease
4.1. Regulation of ASL and Mucociliary Clearance by Nucleotides
Mason et al. first proposed that extracellular ATP regulates ion transport rates when added to either the apical or basolateral surface of human airway epithelium and found that these effects appear to be mediated by cell surface receptors that respond to ATP by regulating ion transport rates through the release of Ca2+ from internal stores and extracellular Ca2+ influx [74]. As agonists were being screened to restore Cl− and fluid secretion in CF airway epithelium, nucleotide agonists emerged quickly as stimulants of Cl− and fluid secretion independent of CFTR. Knowles et al. showed that extracellular nucleotides stimulated Cl− secretion in CF patients. Purinergic agonists in addition to ATP such as UTP, UDP, and ADP also had the power to stimulate Cl− secretion in CF and non-CF airway epithelial models [75]. In addition, adenosine receptors can also stimulate Cl− secretion in airway epithelial cells by activating the cAMP/PKA signal transduction pathway and eventually CFTR [76, 77]. ATP signalling through purinergic P2Y receptors is effective in airway epithelia in inhibiting ENaC activity and initiating Ca2+-activated Cl− secretion [78]. All functionally defined P2Y receptors are able to couple through the IP3 pathway consisting of activation of PLC increase in inositol phosphates and mobilization of Ca2+ from intracellular stores. In addition and secondary to the activation of the PLC, multiple signal transduction pathways including PKC, phospholipase A2, Ca2+ sensitive ion channels, and formation of endothelium derived relaxing factors have been shown to be involved in the responses to activation of native P2Y-receptors. Another function of the P2Y receptors is the activation of ciliary beat frequency. In hydrated airways, the rate of mucociliary clearance is determined by ciliary beat frequency and nonsaturating concentrations of ATP generates alternating Ca2+ signals in ciliated cells which in turn increases ciliary beat frequency [79, 80].
Pharmacological data has shown that the P2Y11 receptor is preferentially activated by ATP and is uniquely coupled to both the phosphoinositide and the cAMP pathways [81]. Evidence is available that ATP and ADP, two physiologic nucleotides that can be released into the extracellular space, are able to raise cAMP levels in native cells via activation of P2Y11 receptors. Those results provide a mechanism in addition to activation of P2Y2 or adenosine receptors, by which exogenous or endogenously related nucleotides can increase cellular levels of this important cyclic nucleotide. Given the evidence that a number of types of cells both release ATP and possess P2Y11 receptor, then nucleotide mediated activation of P2Y11 receptors provides a means for autocrine regulation of epithelial and other cell types. Activation of the P2Y11 receptor in different cell types has a number of different outcomes. For example, the P2Y11 receptor mediates the inhibition of neutrophil apoptosis, impaired endothelial cell proliferation or regulation of secretory function of pancreatic ductal cells by ATP [82–84].
4.2. Nucleotides Release by Pannexin Channel
The complex cellular composition of the airways that is ciliated cells and mucin-secretory goblet cells suggests that several mechanisms and pathways are involved in the release of nucleotides into the airways. Two general mechanisms for the release of ATP from cells have been proposed as vesicular release and channel-mediated release. While vesicular release of ATP is well documented, ATP release can also occur in the absence of vesicules. For example, human erythrocyte which is devoid of cytoplasmic vesicle can release ATP in low oxygen content or in response to shear stress [85]. Pannexins belong to the family of connexin channels that have been proposed as diffusion pathways for ATP release under various experimental conditions. The Pannexins primarily form oligomeric structures embedded in a single plasma membrane that when open provide a conduction pathway between cytosol and extracellular space. They are mechanosensitive and are highly permeable to ATP [86]. Exposure of the alveolar A549 cells to thrombin resulted in a strong ATP release response that was inhibited by the nonselective blockers of pannexin channels suggesting that ATP release from thrombin-stimulated lung epithelial cells occurs through pannexin channels [87]. A study by Ransford et al. 2009 showed ATP release induced by hypotonic shock of human bronchial epithelial cells was inhibited after silencing pannexin-1 (Panx1) via shRNA [88]. The large pores of Panx1 (the most studied pannexin channel) are permeable to ions, second messengers, and neurotransmitters such as ATP, IP3, and amino acids. Panx1 is also implicated in secretion of arachidonic acid and its metabolites and it is now widely regarded that Panx1 membrane channels are also involved in the extracellular mode of wave propagation. Panx1 channels open in response to mechanical stress or other stimuli such as depolarization and release ATP to the extracellular medium. ATP binding to purinergic receptors triggers an increase of cytoplasmic Ca2+ via the IP3 pathway. The Ca2+ increase is not restricted to the same cell but also includes cells within diffusion distance for the released ATP also stimulating cells that are coupled to the stimulated cell by gap junction channels permitting the flux of IP3.
The increase in Ca2+ can activate Panx1 channels and subsequent release of ATP provides a new source for extracellular ATP to reach more distant cells [86]. The application of micromolar concentrations of Ca2+ to the cytoplasmic side of Panx1 channels in excised membrane patches activated the channels at negative membrane potentials where the channels are normally closed [86].
4.3. Lipoxin A4 Increases the Airway Surface Liquid Height via P2Y11 Activation
The mechanism by which LXA4 stimulates Ca2+-activated Cl− secretion and ASL height increase has been elucidated. Higgins et al. reported that LXA4 induces an apical ATP release from non-CF and CF airway epithelial cell lines and CF primary cultures. This ATP release induced by LXA4 is completely inhibited by antagonists of the FPR2 receptor and Panx1 channels suggesting a major role of Panx1 in this effect. Furthermore, LXA4 induces an increase in intracellular cAMP and calcium, which are abolished by the selective inhibition of the P2RY11 purinoreceptor. Panx1 and ATP hydrolysis inhibition and P2RY11 purinoreceptor knockdown all abolish the increase of ASL height induced by LXA4. Inhibition of the A2b adenosine receptor does not affect the ASL height increase induced by LXA4, whereas the PKA inhibitor partially inhibits this response. Taken together this report provides evidence for a novel role of LXA4 in stimulating apical ATP secretion via Panx1 channel and subsequent P2RY11 purinoreceptor activation in airway epithelial cells leading to an ASL height increase (Figure 3).
Figure 3.

Lipoxin A4 enhances epithelial barrier integrity by stimulating an increase in airway surface liquid (ASL) layer height, epithelial repair, and tight junction formation. Stimulation of the FPR2 receptor by LXA4 induces an apical ATP release through the pannexin (Panx1) channel activating a purinoreceptor pathway. Activation of P2Y11 receptors stimulates chloride secretion out of the cell by calcium activated chloride channels (CaCC) and inhibition of sodium absorption by amiloride sensitive epithelial sodium channels (ENaC) which result in a restored ASL height in CF bronchial epithelial cells. The calcium signal induced by P2Y11 activation also stimulates epithelial repair and tight junction formation. Taken together, the physiological effects induced by LXA4 have the potential to delay the invasion of bronchial epithelial cells by bacteria (green and orange structures).
5. Epithelial Repair in CF Airway
5.1. Altered Epithelial Repair in CF
In CF, recurrent infections and inflammatory insults result in damage to the airways and trigger the repair process [89]. Epithelial repair initially involves cell migration and cell proliferation to repopulate the injured area [90–92]. This process is then followed by differentiation of the epithelium [93]. Recent research suggests that epithelial repair as well as differentiation of the CF airway epithelium is downregulated or delayed [94–98]. More specifically, cell migration and proliferation both appear to be reduced during repair of CF bronchial epithelial cells compared to non-CF cells [99]. This delay in repair of the CF epithelium renders the lung more susceptible to ongoing bacterial infection and thus may trigger more epithelial damage [100].
5.2. Lipoxin A4 Regulates Airway Epithelial Integrity in CF Airway Epithelium
The lipid mediator LXA4 triggers epithelial cell migration and proliferation and thus plays a role in repair of epithelia including bronchial epithelium from patients with CF [22, 99, 101–104]. The effects of LXA4 in stimulating cell proliferation, cell migration, and wound repair are mediated by the apical ATP release and P2Y11 activation [105]. Stimulation of P2Y11 purinoreceptors induces calcium release and ERK phosphorylation, both of which play a key role in initiating cell proliferation and migration [106–113]. Furthermore, consistent with the role of potassium channels in two key processes of repair, migration, and proliferation in numerous cell types, the responses to LXA4 on the repair process are mediated by the downstream activation of KATP potassium channels [96, 97, 99, 114–117]. Additionally, LXA4 enhances airway epithelial tight junction formation which is a main factor of epithelial barrier integrity. LXA4 stimulates ZO-1, claudin-1, and occludin expression and trafficking at the apical membrane resulting in enhanced transepithelial electrical resistance in human airway epithelia [118] (Figure 2). Taken together, these effects of LXA4 on airway epithelial structure suggest the abnormal levels of LXA4 in CF airways may account for the reduced capacity for epithelial repair in CF.
6. Treatments of CF Airway Disease
6.1. Current Treatments and Opportunities
There is currently no treatment available that fully corrects the biochemical abnormality in CF and leads to a cessation of the typical pathobiology seen in the condition. Therapies to date have been centred on slowing the decline in pulmonary function over time to prolong survival. Medication is predominantly used to optimise nutrition (pancreatic enzymes, fat soluble vitamin supplementation), treat infection (oral, inhaled, and intravenous antibiotics), and facilitate effective mucociliary clearance (DNAse, hypertonic saline). Several anti-inflammatory approaches have been examined in CF; however, the ideal anti-inflammatory drug is not yet available [119]. A recent systematic review of the risks and benefits of inhaled corticosteroids in CF, examining evidence from 13 trials, concluded that there is insufficient evidence to establish whether they are beneficial in CF while it is established that ICS use can have adverse effects [120]. A systematic review of the efficacy of nonsteroidal anti-inflammatory drugs in CF concluded that treatment with high-dose ibuprofen was associated with a significantly lower annual rate of decline in lung function (especially in children); however, the adoption of ibuprofen into therapy has not been universally accepted [121]. Correcting the imbalance in fatty acid metabolism described in CF by supplementation of Docosahexaenoic Acid may be helpful, and efforts are ongoing to evaluate the potential therapeutic benefit [122].
Two promising avenues of therapy have recently emerged: small molecule correctors and gene therapy. The flagship small molecule corrector has been Ivacaftor (VX-770). This compound facilitates gating of defective CFTR where the cause of CFTR dysfunction is a gating mutation—predominantly G551D. This has been remarkably clinically successful but can be taken by only approximately 5% of patients worldwide [123, 124]. The manufacturers of Ivacaftor, Vertex Pharmaceuticals, are currently developing correctors for the commonest mutation Phe508del. Phase 2 trials of this compound have been shown to lead to positive changes in CFTR function, but not to the same degree as VX-770 [125]. Gene therapy was considered an obvious target for disease modifying treatment after the discovery of the CFTR gene; however initial attempts at this approach were unsuccessful, prompting a comprehensive review of the process of selection of endpoints, vectors, and delivery modes. A consortium in the UK has developed a comprehensive approach in this regard and will report shortly on multidose trials of gene therapy in individuals with CF [126]. A treatment approach capable of effectively preventing lung damage and decline in pulmonary function is currently absent despite the obvious hope relating to new developments.
For now, we continue to search for new and effective therapies to slow or prevent the decline in pulmonary function in CF.
6.2. Therapeutic Potential for LXA4 in the Treatment of CF Airway Disease
A variety of airway clearance therapies have been developed for patients with CF [127, 128]. Thus identification of agents, particularly endogenous biologicals that stimulate non-CFTR Cl− secretory pathways and promote ASL height recovery while providing anti-inflammatory effects are likely to be of therapeutic benefit in improving mucociliary clearance in patients with CF. The effect of LXA4 inhalation has been evaluated in a pilot study of eight asthmatic and healthy adult subjects. The challenge was tolerated, had no adverse effect on pulse or blood pressure, and demonstrated favourable effects on specific airway conductance [129].
In conclusion, the discovery of the multiple impacts of LXA4 in restoring bronchial epithelium ion transport, in enhancing ASL height, in restoring epithelial barrier function, and in reducing inflammation might provide significant advance in treatment of the CF airway disease (Figure 3).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Levy B. D., Clish C. B., Schmidt B., Gronert K., Serhan C. N. Lipid mediator class switching during acute inflammation: signals in resolution. Nature Immunology. 2001;2(7):612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 2.Ringholz F. C., Buchanan P. J., Clarke D. T., et al. Reduced 15-lipoxygenase 2 and lipoxin A4/leukotriene B4 ratio in children with cystic fibrosis. The European Respiratory Journal. 2014;44(2):394–404. doi: 10.1183/09031936.00106013. [DOI] [PubMed] [Google Scholar]
- 3.Haeggström J. Z., Funk C. D. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chemical Reviews. 2011;111(10):5866–5896. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 4.Mukaida N., Okamoto S., Ishikawa Y., Matsushima K. Molecular mechanism of interleukin-8 gene expression. Journal of Leukocyte Biology. 1994;56(5):554–558. [PubMed] [Google Scholar]
- 5.Serhan C. N., Yacoubian S., Yang R. Anti-inflammatory and proresolving lipid mediators. Annual Review of Pathology. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fierro I. M., Serhan C. N. Mechanisms in anti-inflammation and resolution: the role of lipoxins and aspirin-triggered lipoxins. Brazilian Journal of Medical and Biological Research. 2001;34(5):555–566. doi: 10.1590/s0100-879x2001000500002. [DOI] [PubMed] [Google Scholar]
- 7.Chavis C., Vachier I., Chanez P., Bousquet J., Godard P. 5(S), 15(S)-dihydroxyeicosatetraenoic acid and lipoxin generation in human polymorphonuclear cells: dual specificity of 5-lipoxygenase towards endogenous and exogenous precursors. Journal of Experimental Medicine. 1996;183(4):1633–1643. doi: 10.1084/jem.183.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serhan C. N., Hirsch U., Palmblad J., Samuelsson B. Formation of lipoxin A by granulocytes from eosinophilic donors. FEBS Letters. 1987;217(2):242–246. doi: 10.1016/0014-5793(87)80671-3. [DOI] [PubMed] [Google Scholar]
- 9.Levy B. D., Bertram S., Tai H. H., et al. Agonist-induced lipoxin A4 generation: detection by a novel lipoxin A4-ELISA. Lipids. 1993;28(12):1047–1053. doi: 10.1007/BF02537069. [DOI] [PubMed] [Google Scholar]
- 10.Serhan C. N., Sheppard K.-A. Lipoxin formation during human neutrophil-platelet interactions: evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. Journal of Clinical Investigation. 1990;85(3):772–780. doi: 10.1172/JCI114503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clària J., Lee M. H., Serhan C. N. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Molecular Medicine. 1996;2(5):583–596. [PMC free article] [PubMed] [Google Scholar]
- 12.Special issue: the lipoxins and the aspirin-triggered lipoxins. Prostaglandins, Leukotrienes & Essential Fatty Acids. 2005;73(3-4):139–321. doi: 10.1016/j.plefa.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Levy B. D., Romano M., Chapman H. A., Reilly J. J., Drazen J., Serhan C. N. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. Journal of Clinical Investigation. 1993;92(3):1572–1579. doi: 10.1172/JCI116738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vachier I., Chanez P., Bonnans C., Godard P., Bousquet J., Chavis C. Endogenous anti-inflammatory mediators from arachidonate in human neutrophils. Biochemical and Biophysical Research Communications. 2002;290(1):219–224. doi: 10.1006/bbrc.2001.6155. [DOI] [PubMed] [Google Scholar]
- 15.Chiang N., Arita M., Serhan C. N. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins, Leukotrienes & Essential Fatty Acids. 2005;73(3-4):163–177. doi: 10.1016/j.plefa.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Chiang N., Hurwitz S., Ridker P. M., Serhan C. N. Aspirin has a gender-dependent impact on antiinflammatory 15-epi-lipoxin A4 formation: a randomized human trial. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(2):e14–e17. doi: 10.1161/01.ATV.0000196729.98651.bf. [DOI] [PubMed] [Google Scholar]
- 17.Ying G., Iribarren P., Zhou Y., et al. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. Journal of Immunology. 2004;172(11):7078–7085. doi: 10.4049/jimmunol.172.11.7078. [DOI] [PubMed] [Google Scholar]
- 18.Cattaneo F., Parisi M., Ammendola R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. International Journal of Molecular Sciences. 2013;14(4):7193–7230. doi: 10.3390/ijms14047193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye R. D., Boulay F., Ji M. W., et al. International union of basic and clinical pharmacology. Lxxiii. Nomenclature for the formyl peptide receptor (fpr) family. Pharmacological Reviews. 2009;61(2):119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiang N., Serhan C. N., Dahlén S.-E., et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacological Reviews. 2006;58(3):463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 21.Serhan C. N. Lipoxins and aspirin-triggered 15-epi-lipoxin biosynthesis: an update and role in anti-inflammation and pro-resolution. Prostaglandins and Other Lipid Mediators. 2002;68-69:433–455. doi: 10.1016/S0090-6980(02)00047-3. [DOI] [PubMed] [Google Scholar]
- 22.Bonnans C., Fukunaga K., Levy M. A., Levy B. D. Lipoxin A4 regulates bronchial epithelial cell responses to acid injury. The American Journal of Pathology. 2006;168(4):1064–1072. doi: 10.2353/ajpath.2006.051056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colgan S. P., Serhan C. N., Parkos C. A., Delp-Archer C., Madara J. L. Lipoxin A4 modulates transmigration of human neutrophils across intestinal epithelial monolayers. Journal of Clinical Investigation. 1993;92(1):75–82. doi: 10.1172/JCI116601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takano T., Clish C. B., Gronert K., Petasis N., Serhan C. N. Neutrophil-mediated changes in vascular permeability are inhibited by topical application of aspirin-triggered 15-epi-lipoxin A4 and novel lipoxin B4 stable analogues. The Journal of Clinical Investigation. 1998;101(4):819–826. doi: 10.1172/JCI1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.József L., Zouki C., Petasis N. A., Serhan C. N., Filep J. G. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-κB and AP-1 activation, and IL-8 gene expression in human leukocytes. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(20):13266–13271. doi: 10.1073/pnas.202296999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonnans C., Levy B. D. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. The American Journal of Respiratory Cell and Molecular Biology. 2007;36(2):201–205. doi: 10.1165/rcmb.2006-0269TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gewirtz A. T., McCormick B., Neish A. S., et al. Pathogen-induced chemokine secretion for model intestinal epithelium is inhibited by lipoxin A4 analogs. Journal of Clinical Investigation. 1998;101(9):1860–1869. doi: 10.1172/JCI1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verriere V., Grumbach Y., Chiron R., Urbach V. LXA4 effect on intracellular Ca2+, Cl− secretion and IL-8 production in normal and CF airway epithelium. Revue des Maladies Respiratoires. 2006;23(5):p. 574. [Google Scholar]
- 29.Karp C. L., Flick L. M., Park K. W., et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nature Immunology. 2004;5(4):388–392. doi: 10.1038/ni1056. [DOI] [PubMed] [Google Scholar]
- 30.Godson C., Mitchell S., Harvey K., Petasis N. A., Hogg N., Brady H. R. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. Journal of Immunology. 2000;164(4):1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 31.McMahon B., Mitchell S., Brady H. R., Godson C. Lipoxins: revelations on resolution. Trends in Pharmacological Sciences. 2001;22(8):391–395. doi: 10.1016/S0165-6147(00)01771-5. [DOI] [PubMed] [Google Scholar]
- 32.Andersen D. H. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Archives of Pediatrics & Adolescent Medicine. 1938;56(2):344–399. doi: 10.1001/archpedi.1938.01980140114013. [DOI] [Google Scholar]
- 33.Tsui L.-C., Buchwald M., Barker D., et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science. 1985;230(4729):1054–1057. doi: 10.1126/science.2997931. [DOI] [PubMed] [Google Scholar]
- 34.Wainwright B. J., Scambler P. J., Schmidtke J., et al. Localization of cystic fibrosis locus to human chromosome 7cen-q22. Nature. 1985;318(6044):384–385. doi: 10.1038/318384a0. [DOI] [PubMed] [Google Scholar]
- 35.White R., Woodward S., Leppert M., et al. A closely linked genetic marker for cystic fibrosis. Nature. 1985;318(6044):382–384. doi: 10.1038/318382a0. [DOI] [PubMed] [Google Scholar]
- 36.Riordan J. R., Rommens J. M., Kerem B.-S., et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 37.Bush A., Alton E., Davies J. C., Griesenbach U., Jaffe A., editors. Cystic Fibrosis in the 21st Century: Progress in Respiratory Research. Basel, Switzerland: Karger; 2006. [Google Scholar]
- 38.Wilschanski M., Zielenski J., Markiewicz D., et al. Correlation of sweat chloride concentration with classes of the cystic fibrosis transmembrane conductance regulator gene mutations. The Journal of Pediatrics. 1995;127(5):705–710. doi: 10.1016/S0022-3476(95)70157-5. [DOI] [PubMed] [Google Scholar]
- 39.Welsh M. J., Smith A. E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73(7):1251–1254. doi: 10.1016/0092-8674(93)90353-R. [DOI] [PubMed] [Google Scholar]
- 40.Engelhardt J. F., Yankaskas J. R., Ernst S. A., et al. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nature genetics. 1992;2(3):240–248. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- 41.Trezise A. E. O., Buchwald M. In vivo cell-specific expression of the cystic fibrosis transmembrane conductance regulator. Nature. 1991;353(6343):434–437. doi: 10.1038/353434a0. [DOI] [PubMed] [Google Scholar]
- 42.Kartner N., Augustinas O., Jensen T. J., Naismith A. L., Riordan J. R. Mislocalization of ΔF508 CFTR in cystic fibrosis sweat gland. Nature Genetics. 1992;1(5):321–327. doi: 10.1038/ng0892-321. [DOI] [PubMed] [Google Scholar]
- 43.Trezise A. E., Chambers J. A., Wardle C. J., Gould S., Harris A. Expression of the cystic fibrosis gene in human foetal tissues. Human Molecular Genetics. 1993;2(3):213–218. doi: 10.1093/hmg/2.3.213. [DOI] [PubMed] [Google Scholar]
- 44.Manson A. L., Trezise A. E. O., MacVinish L. J., et al. Complementation of null CF mice with a human CFTR YAC transgene. The EMBO Journal. 1997;16(14):4238–4249. doi: 10.1093/emboj/16.14.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boat T. F., Cheng P. W. Epithelial cell dysfunction in cystic fibrosis: implications for airways disease. Acta Paediatrica Scandinavica, Supplement. 1989;363:25–29. doi: 10.1111/apa.1989.78.s363.25. [DOI] [PubMed] [Google Scholar]
- 46.Mickle J. E., Macek M., Jr., Fulmer-Smentek S. B., et al. A mutation in the cystic fibrosis transmembrane conductance regulator gene associated with elevated sweat chloride concentrations in the absence of cystic fibrosis. Human Molecular Genetics. 1998;7(4):729–735. doi: 10.1093/hmg/7.4.729. [DOI] [PubMed] [Google Scholar]
- 47.Painter R. G., Valentine V. G., Lanson N. A., Jr., et al. CFTR expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry. 2006;45(34):10260–10269. doi: 10.1021/bi060490t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Painter R. G., Bonvillain R. W., Valentine V. G., et al. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. Journal of Leukocyte Biology. 2008;83(6):1345–1353. doi: 10.1189/jlb.0907658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di A., Brown M. E., Deriy L. V., et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nature Cell Biology. 2006;8(9):933–944. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- 50.Bonfield T. L., Hodges C. A., Cotton C. U., Drumm M. L. Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. Journal of Leukocyte Biology. 2012;92(5):1111–1122. doi: 10.1189/jlb.0412188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Heeckeren A. M., Schluchter M. D. Murine models of chronic Pseudomonas aeruginosa lung infection. Laboratory Animals. 2002;36(3):291–312. doi: 10.1258/002367702320162405. [DOI] [PubMed] [Google Scholar]
- 52.Bonnans C., Vachier I., Chavis C., Godard P., Bousquet J., Chanez P. Lipoxins are potential endogenous antiinflammatory mediators in asthma. American Journal of Respiratory and Critical Care Medicine. 2002;165(11):1531–1535. doi: 10.1164/rccm.200201-053OC. [DOI] [PubMed] [Google Scholar]
- 53.Planagumà A., Kazani S., Marigowda G., et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. The American Journal of Respiratory and Critical Care Medicine. 2008;178(6):574–582. doi: 10.1164/rccm.200801-061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balode L., Strazda G., Jurka N., et al. Lipoxygenase-derived arachidonic acid metabolites in chronic obstructive pulmonary disease. Medicina. 2012;48(6):292–298. [PubMed] [Google Scholar]
- 55.Chiron R., Grumbach Y. Y., Quynh N. V. T., Verriere V., Urbach V. Lipoxin A4 and interleukin-8 levels in cystic fibrosis sputum after antibiotherapy. Journal of Cystic Fibrosis. 2008;7(6):463–468. doi: 10.1016/j.jcf.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 56.Starosta V., Ratjen F., Rietschel E., Paul K., Griese M. Anti-inflammatory cytokines in cystic fibrosis lung disease. European Respiratory Journal. 2006;28(3):581–587. doi: 10.1183/09031936.06.00071405. [DOI] [PubMed] [Google Scholar]
- 57.Carlo T., Kalwa H., Levy B. D. 15-Epi-lipoxin A4 inhibits human neutrophil superoxide anion generation by regulating polyisoprenyl diphosphate phosphatase 1. The FASEB Journal. 2013;27(7):2733–2741. doi: 10.1096/fj.12-223982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mattoscio D., Evangelista V., De Cristofaro R., et al. Cystic fibrosis transmembrane conductance regulator (CFTR) expression in human platelets: impact on mediators and mechanisms of the inflammatory response. The FASEB Journal. 2010;24(10):3970–3980. doi: 10.1096/fj.10-159921. [DOI] [PubMed] [Google Scholar]
- 59.Boucher R. C. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annual Review of Medicine. 2007;58:157–170. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- 60.Matsui H., Davis C. W., Tarran R., Boucher R. C. Osmotic water permeabilities of cultured, well-differentiated normal and cystic fibrosis airway epithelia. Journal of Clinical Investigation. 2000;105(10):1419–1427. doi: 10.1172/JCI4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kunzelmann K., Kiser G. L., Schreibe R., Riordan J. R. Inhibition of epithelial Na+ currents by intracellular domains of the cystic fibrosis transmembrane conductance regulator. FEBS Letters. 1997;400(3):341–344. doi: 10.1016/S0014-5793(96)01414-7. [DOI] [PubMed] [Google Scholar]
- 62.Stutts M. J., Canessa C. M., Olsen J. C., et al. CFTR as a cAMP-Dependent regulator of sodium channels. Science. 1995;269(5225):847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- 63.Davis P. B., Drumm M., Konstan M. W. Cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 1996;154(5):1229–1256. doi: 10.1164/ajrccm.154.5.8912731. [DOI] [PubMed] [Google Scholar]
- 64.Worlitzsch D., Tarran R., Ulrich M., et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. The Journal of Clinical Investigation. 2002;109(3):317–325. doi: 10.1172/JCI200213870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boucher R. C., Stutts M. J., Knowles M. R., Cantley L., Gatzy J. T. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. Journal of Clinical Investigation. 1986;78(5):1245–1252. doi: 10.1172/JCI112708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boucher R. C. Regulation of airway surface liquid volume by human airway epithelia. Pflügers Archiv. 2003;445(4):495–498. doi: 10.1007/s00424-002-0955-1. [DOI] [PubMed] [Google Scholar]
- 67.Antigny F., Norez C., Becq F., Vandebrouck C. CFTR and Ca2+ signaling in cystic fibrosis. Frontiers in Pharmacology. 2011;2(67) doi: 10.3389/fphar.2011.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pillarisetti N., Linnane B., Ranganathan S. Early bronchiectasis in cystic fibrosis detected by surveillance CT. Respirology. 2010;15(6):1009–1011. doi: 10.1111/j.1440-1843.2010.01765.x. [DOI] [PubMed] [Google Scholar]
- 69.Pillarisetti N., Williamson E., Linnane B., et al. Infection, inflammation,and lung function decline in infants with cystic fibrosis. The American Journal of Respiratory and Critical Care Medicine. 2011;184(1):75–81. doi: 10.1164/rccm.201011-1892OC. [DOI] [PubMed] [Google Scholar]
- 70.Ratjen F. What's new in CF airway inflammation: an update. Paediatric Respiratory Reviews. 2006;7(supplement 1):S70–S72. doi: 10.1016/j.prrv.2006.04.170. [DOI] [PubMed] [Google Scholar]
- 71.Bonnans C., Chanez P., Meziane H., Godard P., Bousquet J., Vachier I. Glucocorticoid receptor-binding characteristics in severe asthma. European Respiratory Journal. 2003;21(6):985–988. doi: 10.1183/09031936.03.00059802. [DOI] [PubMed] [Google Scholar]
- 72.Verrière V., Higgins G., Al-Alawi M., et al. Lipoxin a4 stimulates calcium-activated chloride currents and increases airway surface liquid height in normal and cystic fibrosis airway epithelia. PLoS ONE. 2012;7(5) doi: 10.1371/journal.pone.0037746.e37746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al-Alawi M., Buchanan P., Verriere V., et al. Physiological levels of lipoxin a4 inhibit enac and restore airway surface liquid height in cystic fibrosis bronchial epithelium. Physiological Reports. 2014;2(8) doi: 10.14814/phy2.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mason S. J., Paradiso A. M., Boucher R. C. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. British Journal of Pharmacology. 1991;103(3):1649–1656. doi: 10.1111/j.1476-5381.1991.tb09842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Knowles M. R., Clarke L. L., Boucher R. C. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. The New England Journal of Medicine. 1991;325(8):533–538. doi: 10.1056/NEJM199108223250802. [DOI] [PubMed] [Google Scholar]
- 76.Lazarowski E. R., Tarran R., Grubb B. R., van Heusden C. A., Okada S., Boucher R. C. Nucleotide release provides a mechanism for airway surface liquid homeostasis. The Journal of Biological Chemistry. 2004;279(35):36855–36864. doi: 10.1074/jbc.M405367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rollins B. M., Burn M., Coakley R. D., et al. A2B adenosine receptors regulate the mucus clearance component of the lung's innate defense system. The American Journal of Respiratory Cell and Molecular Biology. 2008;39(2):190–197. doi: 10.1165/rcmb.2007-0450OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mall M., Wissner A., Gonska T., et al. Inhibition of amiloride-sensitive epithelial Na+ absorption by extracellular nucleotides in human normal and cystic fibrosis airways. The American Journal of Respiratory Cell and Molecular Biology. 2000;23(6):755–761. doi: 10.1165/ajrcmb.23.6.4207. [DOI] [PubMed] [Google Scholar]
- 79.Lazarowski E. R., Boucher R. C. Purinergic receptors in airway epithelia. Current Opinion in Pharmacology. 2009;9(3):262–267. doi: 10.1016/j.coph.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lorenzo I. M., Liedtke W., Sanderson M. J., Valverde M. A. TRPV4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(34):12611–12616. doi: 10.1073/pnas.0803970105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Communi D., Pirotton S., Parmentier M., Boeynaems J.-M. Cloning and functional expression of a human uridine nucleotide receptor. Journal of Biological Chemistry. 1995;270(52):30849–30852. doi: 10.1074/jbc.270.52.30849. [DOI] [PubMed] [Google Scholar]
- 82.Vaughan K. R., Stokes L., Prince L. R., et al. Inhibition of neutrophil apoptosis by ATP is mediated by the P2Y11 receptor. The Journal of Immunology. 2007;179(12):8544–8553. doi: 10.4049/jimmunol.179.12.8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xiao Z., Yang M., Lv Q., et al. P2Y11 impairs cell proliferation by induction of cell cycle arrest and sensitizes endothelial cells to cisplatin-induced cell death. Journal of Cellular Biochemistry. 2011;112(9):2257–2265. doi: 10.1002/jcb.23144. [DOI] [PubMed] [Google Scholar]
- 84.Nguyen T. D., Meichle S., Kim U. S., Wong T., Moody M. W. P2Y11, a purinergic receptor acting via cAMP, mediates secretion by pancreatic duct epithelial cells. American Journal of Physiology: Gastrointestinal and Liver Physiology. 2001;280(5):G795–G804. doi: 10.1152/ajpgi.2001.280.5.G795. [DOI] [PubMed] [Google Scholar]
- 85.Sprague R. S., Ellsworth M. L., Stephenson A. H., Lonigro A. J. ATP: the red blood cell link to NO and local control of the pulmonary circulation. The American Journal of Physiology—Heart and Circulatory Physiology. 1996;271(6, part 2):H2717–H2722. doi: 10.1152/ajpheart.1996.271.6.H2717. [DOI] [PubMed] [Google Scholar]
- 86.Dahl G., Locovei S. Pannexin: to gap or not to gap, is that a question? IUBMB Life. 2006;58(7):409–419. doi: 10.1080/15216540600794526. [DOI] [PubMed] [Google Scholar]
- 87.Seminario-Vidal L., Kreda S., Jones L., et al. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of Rho- and Ca2+-dependent signaling pathways. Journal of Biological Chemistry. 2009;284(31):20638–20648. doi: 10.1074/jbc.M109.004762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ransford G. A., Fregien N., Qiu F., Dahl G., Conner G. E., Salathe M. Pannexin 1 contributes to ATP release in airway epithelia. The American Journal of Respiratory Cell and Molecular Biology. 2009;41(5):525–534. doi: 10.1165/rcmb.2008-0367OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Regamey N., Jeffery P. K., Alton E. W., Bush A., Davies J. C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax. 2011;66(7):624–629. doi: 10.1136/thx.2009.134106. [DOI] [PubMed] [Google Scholar]
- 90.Stripp B. R., Reynolds S. D. Maintenance and repair of the bronchiolar epithelium. Proceedings of the American Thoracic Society. 2008;5(3):328–333. doi: 10.1513/pats.200711-167DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zahm J. M., Chevillard M., Puchelle E. Wound repair of human surface respiratory epithelium. The American Journal of Respiratory Cell and Molecular Biology. 1991;5(3):242–248. doi: 10.1165/ajrcmb/5.3.242. [DOI] [PubMed] [Google Scholar]
- 92.Zahm J. M., Kaplan H., Hérard A. L., et al. Cell migration and proliferation during the in vitro wound repair of the respiratory epithelium. Cell Motility and the Cytoskeleton. 1997;37(1):33–43. doi: 10.1002/(SICI)1097-0169(1997)37:1<33::AID-CM4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 93.Erjefalt J. S., Erjefalt I., Sundler F., Persson C. G. A. In vivo restitution of airway epithelium. Cell and Tissue Research. 1995;281(2):305–316. doi: 10.1007/s004410050427. [DOI] [PubMed] [Google Scholar]
- 94.Trinh N. T. N., Bardou O., Privé A., et al. Improvement of defective cystic fibrosis airway epithelial wound repair after CFTR rescue. European Respiratory Journal. 2012;40(6):1390–1400. doi: 10.1183/09031936.00221711. [DOI] [PubMed] [Google Scholar]
- 95.Maillé E., Trinh N. T. N., Privé A., et al. Regulation of normal and cystic fibrosis airway epithelial repair processes by TNF-κ after injury. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2011;301(6):L945–L955. doi: 10.1152/ajplung.00149.2011. [DOI] [PubMed] [Google Scholar]
- 96.Trinh N. T. N., Privé A., Maillé E., Noël J., Brochiero E. EGF and K+ channel activity control normal and cystic fibrosis bronchial epithelia repair. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2008;295(5):L866–L880. doi: 10.1152/ajplung.90224.2008. [DOI] [PubMed] [Google Scholar]
- 97.Trinh N. T. N., Privé A., Kheir L., et al. Involvement of KATP and KvLQT1 K+ channels in EGF-stimulated alveolar epithelial cell repair processes. The American Journal of Physiology—Lung Cellular and Molecular Physiology. 2007;293(4):L870–L882. doi: 10.1152/ajplung.00362.2006. [DOI] [PubMed] [Google Scholar]
- 98.Hajj R., Lesimple P., Nawrocki-Raby B., Birembaut P., Puchelle E., Coraux C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. Journal of Pathology. 2007;211(3):340–350. doi: 10.1002/path.2118. [DOI] [PubMed] [Google Scholar]
- 99.Buchanan P. J., McNally P., Harvey B. J., Urbach V. Lipoxin A4-mediated KATP potassium channel activation results in cystic fibrosis airway epithelial repair. The American Journal of Physiology—Lung Cellular and Molecular Physiology. 2013;305(2):L193–L201. doi: 10.1152/ajplung.00058.2013. [DOI] [PubMed] [Google Scholar]
- 100.Roger P., Puchelle E., Bajolet-Laudinat O., et al. Fibronectin and α5β1 integrin mediate binding of Pseudomonas aeruginosa to repairing airway epithelium. European Respiratory Journal. 1999;13(6):1301–1309. doi: 10.1034/j.1399-3003.1999.13f14.x. [DOI] [PubMed] [Google Scholar]
- 101.Gronert K. Lipoxins in the eye and their role in wound healing. Prostaglandins, Leukotrienes & Essential Fatty Acids. 2005;73(3-4):221–229. doi: 10.1016/j.plefa.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 102.Kenchegowda S., Bazan N. G., Bazan H. E. P. EGF stimulates lipoxin A4 synthesis and modulates repair in corneal epithelial cells through ERK and p38 activation. Investigative Ophthalmology and Visual Science. 2011;52(5):2240–2249. doi: 10.1167/iovs.10-6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang S. B., Hu K. M., Seamon K. J., Mani V., Chen Y., Gronert K. Estrogen negatively regulates epithelial wound healing and protective lipid mediator circuits in the cornea. FASEB Journal. 2012;26(4):1506–1516. doi: 10.1096/fj.11-198036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gronert K., Maheshwari N., Khan N., Hassan I. R., Dunn M., Schwartzman M. L. A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. Journal of Biological Chemistry. 2005;280(15):15267–15278. doi: 10.1074/jbc.M410638200. [DOI] [PubMed] [Google Scholar]
- 105.Higgins G., Buchanan P., Perriere M., et al. Activation of P2RY11 and ATP release by lipoxin A4 restores the airway surface liquid layer and epithelial repair in cystic fibrosis. American Journal of Respiratory Cell and Molecular Biology. 2014;51(2):178–190. doi: 10.1165/rcmb.2012-0424OC. [DOI] [PubMed] [Google Scholar]
- 106.Yang L., Cranson D., Trinkaus-Randall V. Cellular injury induces activation of MAPK via P2Y receptors. Journal of Cellular Biochemistry. 2004;91(5):938–950. doi: 10.1002/jcb.10774. [DOI] [PubMed] [Google Scholar]
- 107.Klepeis V. E., Weinger I., Kaczmarek E., Trinkaus-Randall V. P2Y receptors play a critical role in epithelial cell communication and migration. Journal of Cellular Biochemistry. 2004;93(6):1115–1133. doi: 10.1002/jcb.20258. [DOI] [PubMed] [Google Scholar]
- 108.Yin J., Yu F.-S. X. Erk1/2 mediate wounding- and g-protein-coupled receptor ligands-induced egfr activation via regulating adam17 and hb-egf shedding. Investigative Ophthalmology & Visual Science. 2009;50(1):132–139. doi: 10.1167/iovs.08-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sherwood C. L., Lantz R. C., Burgess J. L., Boitano S. Arsenic alters ATP-dependent Ca2+ signaling in human airway epithelial cell wound response. Toxicological Sciences. 2011;121(1):191–206. doi: 10.1093/toxsci/kfr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ko T., An H. J., Ji Y. G., Kim O. J., Lee D. H. P2Y receptors regulate proliferation of human pancreatic duct epithelial cells. Pancreas. 2012;41(5):797–803. doi: 10.1097/MPA.0b013e31823ba3b3. [DOI] [PubMed] [Google Scholar]
- 111.Boucher I., Rich C., Lee A., Marcincin M., Trinkaus-Randall V. The P2Y2 receptor mediates the epithelial injury response and cell migration. American Journal of Physiology—Cell Physiology. 2010;299(2):C411–C421. doi: 10.1152/ajpcell.00100.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weinger I., Klepeis V. E., Trinkaus-Randall V. Tri-nucleotide receptors play a critical role in epithelial cell wound repair. Purinergic Signalling. 2005;1(3):281–292. doi: 10.1007/s11302-005-8132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Degagné É., Degrandmaison J., Grbic D. M., Vinette V., Arguin G., Gendron F.-P. P2Y2 receptor promotes intestinal microtubule stabilization and mucosal re-epithelization in experimental colitis. Journal of Cellular Physiology. 2013;228(1):99–109. doi: 10.1002/jcp.24109. [DOI] [PubMed] [Google Scholar]
- 114.Rao J. N., Platoshyn O., Li L., et al. Activation of K+ channels and increased migration of differentiated intestinal epithelial cells after wounding. The American Journal of Physiology—Cell Physiology. 2002;282(4):C885–C898. doi: 10.1152/ajpcell.00361.2001. [DOI] [PubMed] [Google Scholar]
- 115.Pardo L. A. Voltage-gated potassium channels in cell proliferation. Physiology. 2004;19(5):285–292. doi: 10.1152/physiol.00011.2004. [DOI] [PubMed] [Google Scholar]
- 116.Kessler W., Budde T., Gekle M., Fabian A., Schwab A. Activation of cell migration with fibroblast growth factor-2 requires calcium-sensitive potassium channels. Pflugers Archiv European Journal of Physiology. 2008;456(5):813–823. doi: 10.1007/s00424-008-0452-2. [DOI] [PubMed] [Google Scholar]
- 117.Lotz M. M., Wang H., Song J. C., Pories S. E., Matthews J. B. K+ channel inhibition accelerates intestinal epithelial cell wound healing. Wound Repair and Regeneration. 2004;12(5):565–574. doi: 10.1111/j.1067-1927.2004.012509.x. [DOI] [PubMed] [Google Scholar]
- 118.Grumbach Y., Quynh N. V. T., Chiron R., Urbach V. LXA4 stimulates ZO-1 expression and transepithelial electrical resistance in human airway epithelial (16HBE14o-) cells. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2009;296(1):L101–L108. doi: 10.1152/ajplung.00018.2008. [DOI] [PubMed] [Google Scholar]
- 119.Balfour-Lynn I. M. Anti-inflammatory approaches to cystic fibrosis airways disease. Current Opinion in Pulmonary Medicine. 2007;13(6):522–528. doi: 10.1097/MCP.0b013e3282ef9806. [DOI] [PubMed] [Google Scholar]
- 120.Balfour-Lynn I. M., Welch K. Inhaled corticosteroids for cystic fibrosis. Cochrane Database of Systematic Reviews. 2012;11 doi: 10.1002/14651858.CD001915.pub3. [DOI] [PubMed] [Google Scholar]
- 121.Lands L. C., Stanojevic S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. The Cochrane Database of Systematic Reviews. 2013;6 doi: 10.1002/14651858.CD001505.pub3.CD001505 [DOI] [PubMed] [Google Scholar]
- 122.van Biervliet S., van Biervliet J. P., Robberecht E., Christophe A. Docosahexaenoic acid trials in cystic fibrosis: a review of the rationale behind the clinical trials. Journal of Cystic Fibrosis. 2005;4(1):27–34. doi: 10.1016/j.jcf.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 123.Accurso F. J., Rowe S. M., Clancy J. P., et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. The New England Journal of Medicine. 2010;363(21):1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ramsey B. W., Davies J., McElvaney N. G., et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England Journal of Medicine. 2011;365(18):1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Clancy J. P., Dupont L., Konstan M. W., et al. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax. 2013;68(9):818–825. doi: 10.1136/thoraxjnl-2012-202230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Armstrong D. K., Cunningham S., Davies J. C., Alton E. W. F. W. Gene therapy in cystic fibrosis. Archives of Disease in Childhood. 2014;99(5):465–468. doi: 10.1136/archdischild-2012-302158. [DOI] [PubMed] [Google Scholar]
- 127.Pisi G., Chetta A. Airway clearance therapy in cystic fibrosis patients. Acta Biomedica de l'Ateneo Parmense. 2009;80(2):102–106. [PubMed] [Google Scholar]
- 128.Tarran R., Grubb B. R., Parsons D., et al. The CF salt controversy: in vivo observations and therapeutic approaches. Molecular Cell. 2001;8(1):149–158. doi: 10.1016/S1097-2765(01)00286-6. [DOI] [PubMed] [Google Scholar]
- 129.Christie P. E., Spur B. W., Lee T. H. The effects of lipoxin a4 on airway responses in asthmatic subjects. The American Review of Respiratory Disease. 1992;145(6):1281–1284. doi: 10.1164/ajrccm/145.6.1281. [DOI] [PubMed] [Google Scholar]