

Abstract
Insects are the most diverse group of organisms on the planet. Variation in gene expression lies at the heart of this biodiversity and recent advances in sequencing technology have spawned a revolution in researchers' ability to survey tissue-specific transcriptional complexity across a wide range of insect taxa. Increasingly, studies are using a comparative approach (across species, sexes and life stages) that examines the transcriptional basis of phenotypic diversity within an evolutionary context. In the present review, we summarize much of this research, focusing in particular on three critical aspects of insect biology: morphological development and plasticity; physiological response to the environment; and sexual dimorphism. A common feature that is emerging from these investigations concerns the dynamic nature of transcriptome evolution as indicated by rapid changes in the overall pattern of gene expression, the differential expression of numerous genes with unknown function, and the incorporation of novel, lineage-specific genes into the transcriptional profile.
Keywords: RNA-Seq, non-model organism, differential expression, comparative transcriptomics, NGS, insects
Introduction
How phenotypic diversity is generated is a major question in modern biology. The basis of such diversity in insects is an interesting question not only evolutionarily, but also in an applied context, because insects are major agricultural pests and vectors of many human diseases. As the genomics revolution has progressed, it has become clear that simply detecting the genetic differences between two phenotypically divergent forms often tells us little about the underlying basis of the divergent trait. This is particularly true in cases where highly similar (or even identical) genomes give rise to different phenotypes, as is seen in the polyphenism of eusocial species and in the radical sexual dimorphism observed in many insect orders. Above the species level, two taxa may exhibit minimal differences at the nucleotide level yet show extreme phenotypic divergence. King & Wilson (1975) first pointed out that the basis of many phenotypic changes might not be harboured in simple point mutations in structural proteins, but rather centred in regulatory gene expression. Recently this phenomenon has been invoked in insect evolution (Catalan et al., 2012; Hardison & Taylor, 2012; Saminadin-Peter et al., 2012; Glaser-Schmitt et al., 2013), and a full understanding of the striking phenotypic evolution observed in insects will require both genomic and transcriptomic approaches.
In the very recent past comparative studies of gene expression were limited to a few organisms, but the advent of next-generation sequencing, combined with continuous progress in RNA sequencing (RNA-Seq) data quantity and quality (Picelli et al., 2013), have democratized the technology. It is now possible for researchers outside the model organism canon to use comparative transcriptomics, and studies of a variety of insect traits have recently benefited from this approach. Because neither a reference genome nor extensive knowledge of the biology of the targeted insect are needed to accomplish a detailed transcriptome analysis, numerous and phylogenetically diverse insect species have been examined in the past decade. By tracking the expression of genes across several species or under various conditions and developmental stages, entomologists have gained valuable insight into the evolution and molecular basis of insect development, physiology and behaviour.
Many of the earliest insect RNA-Seq studies involved simple annotation analysis to categorize the genes expressed in a single tissue in one species. As methods of assembly, annotation and expression quantitation have improved, researchers are increasingly using a comparative approach (among species, sexes, tissue types, life stages, etc.) to isolate the transcriptional basis of phenotypic diversity. In the present review, we examine the areas of insect biology employing this approach, with particular focus on studies highlighting the dynamic nature of transcriptome evolution through rapid changes in gene expression and incorporation of new genes into transcriptional profiles. We include all insect species examined to date, with the exception of the ‘supermodel’ species Drosophila melanogaster, whose transcriptomics are discussed in detail elsewhere (Graveley et al., 2011; and summary databases at http://flybase.org/static_pages/feature/previous/articles/2010_03/RNA-seq_data.html). Important technical aspects of the RNA-Seq method (Box 1) have been discussed in detail in several reviews (Wang et al., 2009; Martin and Wang 2011; Wolf, 2013; etc.), and there are some pitfalls to the approach that must be considered (Box 2). Overall, however, this approach has provided a broadening of opportunities for research at the level of gene expression in an increasingly diverse group of non-model insects.
Box 1. Next-generation sequencing RNA sequencing primer
Platform
There are several platforms that are categorized as next-generation sequencing (NGS; aka massively parallel sequencing methods), but the Illumina platform is the most widely used and flexible for RNA sequencing (RNA-Seq) studies (Van Verk et al., 2013). Other methods such as 454, PacBio, IonTorrent and SOLID platforms are also available. In RNA-Seq studies, the RNA from a single cell, tissue or whole animal (as with small insects) is isolated and purified by taking advantage of the polyA tail on the mRNA, which is present in most translated transcripts. This step separates the RNAs that will eventually be translated into proteins from nontranslated cellular RNAs (e.g. ribosomal RNAs). The purified RNA is then made into cDNA, and this cDNA is amplified using standard PCR approaches. The PCR product is fragmented to a specific length that is dependent on the platform used (Illumina, 454, etc.) and the size selected cDNA is converted into a ‘library’ by the addition of platform-specific adaptors to the ends of the cDNA.
The Illumina method has several platforms with different capacities. For instance, the miSEQ platform generates paired end fragment size reads (currently 300 base pairs each read), and 25 000 000 reads per run, while the Illumina HiSEQ2500 can generate paired end reads of 125 bases each but generates 4 billion reads. Both the Illumina and 454 methods allow samples to be multiplexed, meaning that several tissues or tissues from several organisms can be run at the same time. In this approach, the target cDNA is amplified with primers that harbour specific DNA sequences called ‘barcodes’. The sequences from the various mRNA samples from different developmental stages, tissues or even individuals can then be sorted using informatics techniques. In this review we focus mostly on studies where NGS approaches have been used to address questions about insect biology, though some previous studies using expressed sequence tag analysis and microarrays are also discussed.
Assembly and Annotation
Assembly refers to the process of piecing the small NGS sequence reads back together to get full-length reads for the different RNAs present in the tissue. Assembling short reads (<200 base pairs) is in general more difficult than assembling longer reads unless a decent scaffold sequence is available. Thus, model organisms whose genomes and transcriptomes are already well characterized are much easier to work with, especially when short read lengths are used. Commonly used assembly programs include Trinity (Grabherr et al. 2011), Trans-ABySS (Birol et al., 2009), Velvet-Oases (Schulz et al., 2012), and SOAPdenovo (Xie et al., 2014). Another advance in the assembly area are the occurrence of ‘Assemblathons’ (https://genome10k.soe.ucsc.edu/assemblathon; Bradnam et al., 2013). These are semi-competitions where different informatics teams in the field use their software to assemble a target genome provided by the organizers of the assemblathon, with each participant having the same amount of time to do the assembly. These assemblathons should be consulted regularly for advances that occur as a result of these competitions (Bradnam et al., 2013).
Quantitation of transcript abundance
Knowing what transcripts are present in a tissue or cell type supplies only one dimension of data for an RNA-Seq study. Additional information can be obtained by looking at the relative abundance of different transcripts to generate an ‘expression profile’ for different genes or gene categories. Phylogenetic studies usually ignore transcript abundance and focus on the identification and comparison of orthologous sequences present in different taxa. Other types of studies explicitly compare expression profiles as well as enumerating which transcripts are present. Most traits related to insect biology are potentially affected by variation in expression levels, and studies of such traits require fairly accurate estimation of abundance of transcripts. An updated list of read mapping software is provided by http://wwwdev.ebi.ac.uk/fg/hts_mappers/.
Box 2. Pitfalls of RNA-Seq
Non-Model Problem: Applying RNA-Seq methods to non-model organisms poses specific problems with assembly that are not faced in model organisms. Because assembly must be conducted de novo rather than based on an existing reference, specialized methods are required (Grabherr et al., 2011; Martin & Wang, 2011). While these issues are less extreme with longer reads (because a sufficiently long read will cover the entire transcript, eliminating the need for assembly), it is currently impossible in most non-model studies to know whether the ‘right’ assembly has been found. The overall quality of an assembly is assessed by looking at the number and length of the transcripts identified, and any sequencing errors that appear in the final assembly can be corrected. Following assembly, an attempt is made to determine the identity and function of the final transcripts. This annotation relies on finding orthology between the assembled transcript sequence and sequences previously characterized in other organisms. Orthology determination may rely on sequence similarity, predicted structural similarity between proteins, or other traits, based on Bayesian statistics and phylogenetic tree approaches. Transcripts that cannot be annotated (i.e. those with no significant similarity to any previously identified sequence) are characterized as novel, and may be unique to the taxa being investigated.
Because well-annotated genomes and transcriptomes are available for so few non-model organisms, researchers will often find that 50 to 80% of the transcripts they identify are unannotated (Riesgo et al. 2012). Until additional resources are available, it is difficult to know what role, if any, these transcripts might play in the trait under study. While this problem can be avoided by using a candidate gene approach (which ensures, ipso facto, that no unknown transcripts will be found), candidate gene studies run the risk of finding a ‘shared’ genetic basis for traits in different taxa because they only look at a small fraction of the transcripts present. While unbiased whole transcriptome studies are challenging, their potential for uncovering new molecular pathways and processes make them the preferred approach.
Transcript Length and Source Problems: There are several problems that researchers need to consider in this context. Because many of the current methods are dependent on a manipulation of target mRNA (such as cDNA synthesis, amplification steps and ligation), accurate downstream quantitation of transcript abundance can be problematic. Another problem is that transcript length bias can occur because of the high degree of fragmentation and size selection of transcripts or cDNA fragments used in RNA-Seq methods. Yet another concern involves depth of coverage in an RNA-Seq study: rare or infrequently expressed transcripts are difficult to detect, and at low to moderate coverage levels it is impossible to know whether the absence of a transcript results from its absence in the genome, or from its rarity/low level of expression (Tarazona et al., 2011; Maza et al., 2013). The tissue chosen for comparison is also a critical aspect of RNA-Seq studies; for instance, if one is comparing expression of digestive transcripts between organisms, it is critical that only digestive tract tissue be sampled. Even with appropriate tissue sampling, highly composite parts of the anatomy such as the abdomen (which may contain multiple specialized structures) can result in many false-positives (Johnson et al., 2013), so great care must be taken in extracting and processing the tissue of interests. For this reason, RNA-Seq studies that seek to characterize transcriptome abundance should not use composite structures or whole insects as a tissue source (though for studies examining transcript presence rather than abundance, these tissue sources are acceptable).
Quantitating Abundance Problems: Another problem that arises when estimating abundance of transcripts involves isoforms (alternative versions of a specific transcript) and spliced transcripts. Identification of isoforms is sometimes difficult, and the validation of the existence of an alternative transcript produced by splicing is also a shortcoming in RNA-Seq studies. A promising alternative to cDNA RNA-Seq methods is to directly sequence RNA (DRS) for a sample (Ozsolak & Milos, 2011; Mannello et al., 2012). This approach requires single molecule sequencing approaches, which are currently on the horizon, or the development of direct RNA amplification without cDNA synthesis steps.
RNA-Seq applied to insect biology
RNA-Seq (also known as whole-transcriptome shotgun sequencing) technology now permeates numerous fields in evolutionary biology and has been used to address questions concerning differential gene expression, phylogenetic relationships, gene creation and gene family evolution, rates of protein evolution, genotype–phenotype association, chromosome organization and the regulation of development. To quantify the escalation of studies examining insect transcriptomes we conducted a literature search using the terms ‘expressed sequence tags’ (‘EST’), ‘microarray’, ‘RNA-Seq’ and ‘transcriptome’. We excluded marker-based analyses such as phylogenomic and population genomic studies from our survey and instead focused on research that examines changes in transcriptional profiles associated with phenotypic differences. This search revealed that most branches of insect diversity research now use transcriptome analysis (Fig. 1). As shown in Fig. 2A, RNA-Seq technology has quickly supplanted previous genomic approaches (e.g. EST sequencing and microarray cDNA hybridization) as the preferred method to examine gene expression patterns.
Figure 1.
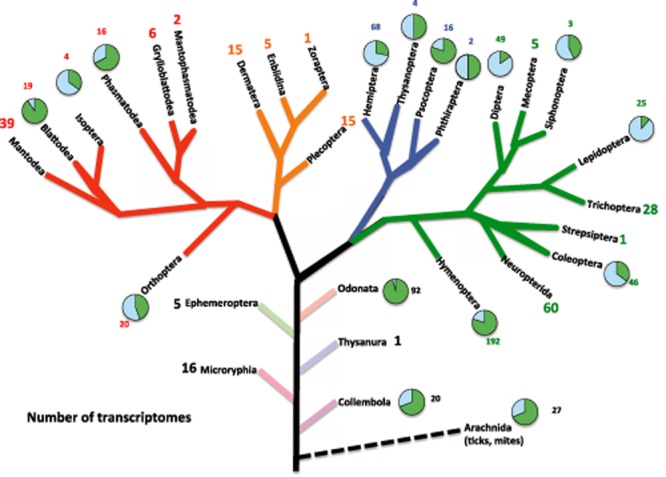
Phylogenetic tree showing the broad phylogenetic distribution of insect transcriptomes. Tree modified after Bugwood (http://wiki.bugwood.org/Main_Page). The number of transcriptomes for each insect order was compiled from the literature. Each order with transcriptomes generated independent of 1KITE (http://www.1kite.org) and surveyed as part of our literature search (key words: ‘transcriptome’, ‘RNA-Seq’, ‘microarray’, or ‘454’; November 2013) has a pie graph associated with it. The green (dark) section of the graph represents the number of species with transcriptomes generated for that order by 1KITE (http://www.1kite.org/downloads/1KITE_species.txt; November 2013). The blue (light) section represents the number of taxa with transcriptional studies done independently of the 1KITE initiative. The number in small font indicates the number of taxa slated for study by the 1KITE initiative. The number in large font represents the number of 1KITE species examined or slated for examination in a given order, where no studies independent of 1KITE have been done.
Figure 2.
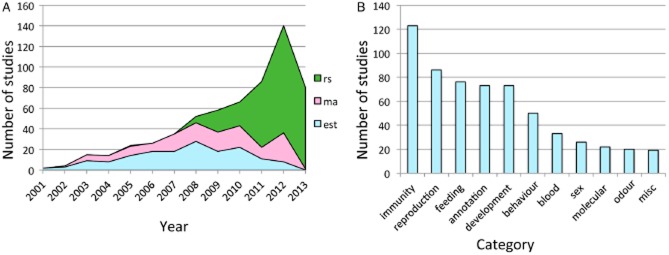
(A) Compilation of studies using transcriptomics in non-model (i.e. non Droosophila melanogaster) insects. The graph is a cumulative count of number of insect transcriptomes analyzed by year since 2001. Blue indicates clone and sequence expressed sequence tag studies, red indicates microarray studies and green inidicates the number of RNA-Seq studies done with next generation sequencing. (B) Bar graph showing the number of insect transcriptomes analyzed and the category of research the transcriptome was generated for.
The broad utility of RNA-Seq is illustrated by the diversity of insect groups and research questions addressed with this methodology (Fig. 2B). Much of the insect research using RNA-Seq reflects an applied focus on traits of importance to human health or agriculture (e.g. blood meal dynamics, vector-borne infection and resistance to pesticides), but other areas in which RNA-Seq is popular involve a more basic understanding of the evolutionary and behavioural biology of insects (e.g. caste differentiation, gregariousness, olfaction and diapause). We review recent work in these fields by focusing on three critical aspects of insects' evolutionary success: morphological development and plasticity, physiological response to the environment, and sexual dimorphism.
Development and plasticity
Morphological development
Even insects with apparently simple hemimetabolous development undergo striking changes as they mature from egg to adult, and recent studies have compared de novo assembled transcriptomes across different developmental stages to identify the genes and signalling pathways involved (Chen et al., 2010; Ewen-Campen et al., 2011; Zeng et al., 2013). Most studies to date have involved the Holometabola. Zheng et al. (2012) compared gene expression between larval and pulpal stages of the tephritid fruit fly Bactocerca dorsalis and identified a large number of differentially expressed genes, most involved in metabolic pathways (e.g. oxidative phosphorylation and glycolysis). Li et al. (2013) compared the transcriptomes of four developmental stages (embryos, larvae, pupae and adults) of the lepidopteran maize pest Athetis lepigone and found that most differentially expressed genes were downregulated in larvae and upregulated in pupae. These expression differences were consistent with characteristic features of each life stage; for example, cuticular protein genes were upregulated in larvae and downregulated in pupae. In the only study we are aware of involving hemimetabolous insect development, Bao et al. (2013) conducted sex- and stage-specific transcriptome-wide analysis of immune function genes in the brown planthopper, Nilaparvata lugens, and found that protein families that sense foreign microbial infection (e.g. Peptidoglycan recognition proteins), mediate innate immunity (e.g. clip-domain serine proteases), and immune response effector genes, had relatively high expression levels in nymphs and extremely low levels in eggs. In contrast, some genes contributing to antibacterial defence (e.g. i-type lysozymes) and regulation of immune-response effectors (e.g. Toll genes) had their highest expression level in eggs, suggesting that they may have roles in embryogenesis and development as well as in immune function.
Phenotypic plasticity
All insects must complete development to adulthood, but in some species the precise outcome of this developmen tal trajectory is flexible. Such phenotypic plasticity – the ability of a single genotype to give rise to a range of phenotypes – is an obvious candidate for transcriptomic approaches, because genomics alone cannot explain how various phenotypes are generated. Eusocial insects in the orders Hymenoptera, Isoptera and Hemiptera offer an extreme version of plasticity, known as polyphenism, in which a single genotype is responsible for morphologically and/or behaviourally distinct castes of workers and reproductives. Only slightly less striking is the ability of some Orthopterans to switch between two phases, solitary and gregarious, which differ in morphology, behaviour and physiology. In many cases, the proximal causes of phenotype determination are well established [e.g. diet-induced caste determination in honey bees (Shuel & Dixon, 1960) and pheromone-induced caste determination in termites (Wilson, 1971)], but only recently have the necessary tools emerged to allow for exploration of the gene expression patterns that underlie these phenotypes.
Polyphenism
Studies of eusociality in Hymenoptera abound (Amdam et al., 2004; Patel et al., 2007; Toth & Robinson, 2007; Boomsma, 2009; Linksvayer & Wade, 2009; Johnson & Linksvayer, 2010; Toth et al., 2010; Daugherty et al., 2011; Johnson & Tsutsui, 2011; Le Conte et al., 2011; Ometto et al., 2011; Woodard et al., 2011; Greenberg et al., 2012; Huang et al., 2012; Liang et al., 2012; Baek et al., 2013; Ferreira et al., 2013). In the honey bee, Apis mellifera, research on the basis of polyphenism is well advanced, and has been reviewed elsewhere (Evans & Wheeler, 2001; Smith et al., 2008; Simpson et al., 2011). We touch here only on more recent studies using transcriptomic approaches, which have confirmed existing knowledge of the genes involved in eusociality and also identified novel genes and mechanisms. For instance, Simola et al. (2013) conducted genome comparisons of eusocial and solitary species to examine the importance of regulatory vs. compositional changes in the evolution of eusociality. They found nearly 2000 genes (largely involved in neuronal and hormonal functions) with similar regulatory changes in all eusocial vs. solitary insects, and identified specific transcription factors (empty spiracles and cAMP response element-binding protein) that appear to be involved in the evolution of eusociality. In another study, Ferreira et al. (2013) investigated whether expression changes in a conserved toolkit of genes (e.g. vitellogenin, insulin, major royal jelly protein, juvenile hormone (JH), methyltransferases, hexamerin, Malvolio, Amfor and P450) were responsible for caste evolution across social taxa, as suggested by some investigators (Toth & Robinson, 2007; Johnson & Linksvayer, 2010). Using a candidate gene approach, previous studies have concluded that these toolkit genes and processes share conserved roles in caste regulation in many eusocial taxa (Amdam et al., 2004; Toth et al., 2010; Daugherty et al., 2011; Woodard et al., 2011; Huang et al., 2012).
Unbiased surveys of entire transcriptomes of social insects are still rare, however, and experiments using a candidate gene approach are inherently unlikely to find novel explanations for the investigated phenotypes. Thus, some workers have suggested that many of the molecular processes responsible for polyphenism in different social lineages will differ substantially, because extensive molecular and developmental re-wiring may be required in the evolution of caste commitment and eusociality (Boomsma, 2009; Linksvayer & Wade, 2009), and that each lineage may have a unique set of genes responsible for polyphenism. In the case of the primitively eusocial wasp Polistes canadensis, Ferreira et al. (2013) identified 2442 genes with caste-biased expression levels. Seventy-five percent of these were novel genes, and there was very little overlap in the identity and expression pattern of caste-biased genes from P. canadensis and those in other social hymenopterans. Only 6.5% of honey bee caste-biased genes were also caste-biased in P. canadensis, and there was no correlation with caste-biased genes of the eusocial fire ant, Solenopsis invicta.
Isoptera (termites) have also been the focus of research on phenotypic plasticity with respect to social behaviour (Korb et al., 2009; Husseneder et al., 2012; Tarver et al., 2012; Sen et al., 2013). Termites exhibit JH-dependent caste differentiation, and Tarver et al. (2012) examined the role of a termite-specific cytochrome P450 gene (Cyp15F1) in this process. Genes in the cytochrome P450 family 15 are known to play a role in JH biosynthesis and degradation, and elevated JH titres in Reticulitermes flavipes workers induce differentiation into the soldier caste via the intermediate presoldier stage. Tarver et al. (2012) tested whether expression levels of Cyp15F1 change in response to primer pheromones derived from the heads of soldiers. They found that expression levels of Cyp15F1 were elevated by one soldier pheromone (CAD), and that RNA interference (RNAi) against Cyp15F1 reduced presoldier formation by 78% compared with the no-RNAi control. The authors conclude that primer pheromones, acting as socio-environmental cues, directly influence Cyp15F1 expression and help regulate caste differentiation by mediating JH signalling.
Phase switching
Phase change in locusts can be induced rapidly and is fully reversible. The solitary phase, in which locusts actively avoid encountering each other and are cryptically coloured, may persist for many generations. When population densities increase to the point where avoidance is impossible, a shift to the gregarious phase, in which locusts are brightly coloured and can accumulate into devastating swarms, is triggered. While the complete shift from one phase to another – which involves changes in behaviour, colour, morphology, life history and physiology (Uvarov, 1966) – takes several generations, behavioural changes are evident within hours of the phase change-inducing stimulus (Roessingh et al., 1998). Although the number and function of the genes involved in this transition are not yet thoroughly known, two recent studies detected similar numbers of transcripts with phase-related expression differences: Badisco et al. (2011) conducted a microarray-based comparison of genes expressed in the central nervous system and found 214 genes (out of 20 755) differentially expressed in solitary and gregarious phases, while Chen et al. (2010), using a de novo approach, found 242 (out of 11 490) genes with expression differences. Wang et al. (2014) examined methylomes (i.e. DNA methylation patterns), brain transcriptomes and alternative pre-messenger RNA splice transcripts of solitary and gregarious locusts. Many of the genes that displayed differences are involved in the regulation of cytoskeletal microtubules (essential neuronal structures involved in neuronal polarization, development and plasticity). The authors conclude that the abundant differences between solitary and gregarious locusts in expression levels, methylation states and splice forms contribute to the neuronal plasticity that underlies the behavioural changes required for phase switching.
Consistent with the need to respond to the diverse sensory inputs inherent in swarming and migration, genes involved in environmental interaction are upregulated in gregarious locusts (Chen et al., 2010; Badisco et al., 2011; Guo et al., 2011). Such genes include those associated with the regulation and development of neuronal structures; regulation of neurotransmitter activities; and detection of chemosensory cues. High expression levels in these categories might help gregarious locusts deal with sensory complexity, because enhanced nervous system plasticity should facilitate rapid adaptation to new environments and stimuli. Acute stress genes [HSPs and pathogen resistance proteins (Badisco et al., 2011)] are also upregulated in gregarious locusts, possibly mitigating the increase in disease risk associated with swarm life. In contrast, ‘anti-aging’ genes, including those associated with oxidative stress resistance, detoxification and anabolic renewal (Badisco et al., 2011) and biosynthetic pathways (Chen et al., 2010) are downregulated in gregarious locusts, which may represent a trade-off to free metabolic resources for other functions at the cost of a reduction in lifespan (Uvarov, 1966).
Physiological response to the environment
Response to environmental stress and infection
Insects can respond to environmental stress in many ways. Stressors may be predictable (e.g. seasonal changes in climate, bacteria associated with specific environments) or unpredictable (e.g. unseasonable temperatures, viral infection), and some may be constant (e.g. freezing temperatures in polar environments); thus, the particular environment of each species will determine the genes that are differentially expressed in response to stress. Several studies have explored changes in gene regulation in response to cold stress (Timmermans et al., 2009; Teets et al., 2012; Dunning et al., 2013; Teets & Denlinger, 2013). Teets et al. (2012) examined gene expression changes in response to extreme cold in the Antarctic midge, Belgica antarctica, which is the world's southernmost insect and the only insect known to be endemic to Antarctica. They found upregulation of cellular recycling pathways and coordinated upregulation of numerous genes involved in autophagy (Teets & Denlinger, 2013), suggesting that autophagy and related processes are important aspects of stress tolerance in the Antarctic environment. The authors compared these results with data from the Arctic collembolan, Megaphorura arctica, and found very little overlap in expression profiles, indicating that these polar species have developed different gene expression mechanisms to withstand similar desiccating conditions. High temperatures can be as dangerous as low ones, and recent transcriptome studies have shown that, in spite of their name, antifreeze proteins play a role in tolerance of both high and low temperatures (Ma et al., 2012; Qiu et al., 2013).
In predictable environments, dormancy for part of the year might be a reasonable response to environmental stress. Diapause and quiescence are both forms of dormancy that allow insects to survive stressful conditions, but they are distinct in that diapause involves a pre-programmed developmental arrest that is anticipatory in nature, while quiescence is characterized by an immediate developmental arrest in response to unfavourable conditions. These processes have been examined by comparing the transcriptomes of diapausing and quiescent organisms (Poelchau et al., 2013b), and of developmental stages in different phases of diapause (Poelchau et al., 2013a). For instance, Gong et al. (2013) compared the transcriptomes of the diapausing and non-diapausing orange wheat blossom midge, Sitodiplosis mosellana, an economically important wheat pest. In diapausing individuals, Gong et al. found upregulation of heat shock proteins, carbohydrate metabolism genes, and genes involved in the suppression of JH signalling and response.
The expression of immune response genes (i.e. those transcribed in response to infection by viruses, bacteria or parasites) has been examined in several species (Table S1). In the harlequin ladybird beetle, Harmomia axyridis, a widespread invasive species that has acquired strong resistance to bacterial and fungal infection, Vilcinskas et al. (2013) compared transcriptome profiles before and after infection. Their findings indicate that immunity evolved through the rapid expansion of genes coding for antimicrobial peptides and proteins. Similarly rapid evolution of immune response genes was also found in studies of Nasonia vitripennis. Sackton et al. (2013) found that many novel (i.e. found only N. vitripennis) immune genes were induced by infection, producing a species-specific immune system for this wasp.
Blood meal dynamics
Because blood-feeding mosquitos serve as vectors for malaria, West Nile Virus, and other diseases, the transcriptional regulation of blood feeding is of particular interest to human health. Two recent studies examined the role of olfaction in blood meal timing in Anopheles gambiae: Rinker et al. (2013) found that moderate changes in odorant receptor (Or) transcript abundance can modulate oviposition behaviours after a blood meal, and Rund et al. (2013) identified well-defined cyclic expression patterns of Ors and odorant binding proteins, a family of soluble proteins found in insect olfactory organs. Given that the peaks in expression correspond with times of increased blood-feeding behaviour, such cyclic expression may be responsible for coordinating the olfactory system with An. gambiae's pre-dusk/dusk circadian niche. Researchers seeking targets for control measures against Aedes aegypti, the principal mosquito vector of dengue viruses, have examined fat body transcriptomes in blood-fed and non-blood-fed Ae. aegypti (Price et al., 2011), while other studies characterizing blood feeding have examined salivary gland and midgut tissues (Table S1).
Response to plant defence and pesticides
Although the ability of herbivores to cope with naturally occurring plant defences and the evolution of resistance to synthetic insecticides might seem like separate topics, recent research using RNA-Seq has revealed a surprising amount of overlap between them (Bass et al., 2013; Dermauw et al., 2013). For example, the spider mite Tetranychus urticae is polyphagous, and many strains have evolved resistance to all insecticides. To investigate whether the genes that allow T. urticae to feed on many chemically defended hosts are also involved in its ability to rapidly evolve pesticide resistance, Dermauw et al. (2013) compared transcriptomes from two different insecticide-resistant strains to those of a strain selected for five generations on tomato (a challenging host for T. urticae). They found >40% of the differentially regulated genes were shared by all three groups, and prominent amongst these were some ‘usual suspects’ commonly implicated in detoxification or transport of xenobiotics (e.g. cytochrome P450s, carboxyl/cholinesterases, and glutathione-S-transferases), although genes of unknown function had some of the most marked expression changes. A more striking result, however, was the detection of expression differences in some previously unexplored gene categories, namely binding proteins (e.g. lipocalins) and transport proteins (e.g. the major facilitator superfamily). While the role of these proteins in polyphagy and insecticide resistance is not yet known, their detection – which would have been impossible using a candidate gene based approach – illustrates the value of unbiased whole transcriptome comparisons.
Based on such results, it seems likely that many of the studies that have looked only at the transcriptional response to either naturally occurring plant compounds or only at the response to insecticides will in fact shed light on the reciprocal question. By characterizing differential gene regulation in pest insects in response to plant defences, new targets for pest management could emerge. Many recent studies have examined gene expression changes associated with feeding on well-defended host plants. Chi et al. (2009) found that feeding on defended soybean is energetically costly for the cowpea bruchid, Callosobruchus maculatus: the expression of structural, defence and stress-related genes was much lower on defended plants, suggesting that insects reallocate resources to deal with plant compounds at a cost to their ability to respond to other potential challenges. In the case of the whitefly, Bemisia tabaci, however, Alon et al. (2012) found that expression changes in response to phenylpropanoids (a large group of plant secondary defence metabolites) do not appear to be costly. They examined reproductive performance and transcriptome profiles after feeding on phenylpropanoid-enriched tobacco and concluded that the resulting elevated expression of genes involved in metabolism, protein synthesis and defence was not accompanied by reduced reproductive performance.
The response of the fall armyworm, Spodoptera frugiperda, to toxins present in maize was examined by Fescemyer et al. (2013). They found upregulation of genes involved in the production of midgut constituents (to counteract the peritrophic membrane depletion the maize toxins cause) and of genes involved in the production of digestive enzymes (to help clear the toxins more rapidly). Similarly, Asian rice gall midges, Orseolia oryzae, feeding on resistant vs. susceptible rice varieties upregulate genes for proteolysis and protein phosphorylation, possibly in an attempt to break down the toxic proteins more quickly (Sinha et al., 2012).
Some researchers have used the transcriptional response of herbivores to a novel or recently adopted host as a proxy for the evolution of host plant range, hypothesizing that the genes expressed might encompass those involved in historical transitions to new hosts (Liu et al., 2010; Matzkin, 2012; Whiteman et al., 2012; Celorio-Mancera et al., 2013; Luan et al., 2013). These studies involve the examination of experimentally manipulated insects using specific plants as hosts. For instance, (Matzkin, 2012) looked at host range evolution in the cactophilic fruit fly, Drosophila mojavensis, which exhibits population-level specialization on different cactus species. Matzkin found that >20% of the genome was differentially regulated in response to a challenging cactus species. Genes associated with mRNA binding and toxin response were upregulated, while genes associated with energy and carbohydrate metabolism and structural components were downregulated. In another study, Celorio-Mancera et al. (2013) examined the evolution of host range in the butterfly Polygonia c-album, a polyphagous, widely distributed butterfly. They compared the transcriptomes of larvae feeding on an established host plant (Urtica dioica) vs. one that represents a recent evolutionary shift onto a very divergent host (Ribes uva-crispa). Like Dermauw et al. (2013) they found unexpectedly high levels of differential regulation of previously unsuspected proteins, namely genes coding for transporter and membrane-binding proteins (including major facilitator superfamily and monocarboxylate types), in response to the newer host. In addition, they found upregulation of cuticular proteins, which they suggest may strengthen the insect gut and prevent toxic plant compounds from entering the insect body.
Sexual dimorphism
Sexual selection is the driving force responsible for a vast array of novel morphological and behavioural phenotypes found throughout insects (Wilkinson & Dodson, 1997; Emlen, 2008; Allen et al., 2011). The extent of sexual dimorphism ranges from minor differences in somatic structures to dramatic showy or exaggerated characters – such as beetle horns, butterfly wing patterning and cricket calls – involved in competition, sexual advertisement and mate selection. Despite the widespread study of sexual dimorphism, we still know little about how variation at the molecular level controls this morphological diversity. A fundamental aspect of the genetic basis of sexual differences is that males and females have virtually identical genomes. Other than the few genes located on the sex-limited copy of the heteromorphic sex chromosomes (e.g. the Y chromosome in XY sex determination systems), there are no genes that exist exclusively in only one sex. As a result, sexual dimorphism is shaped primarily by differential patterns of gene expression. Research examining the specifics of sexual differences in gene expression has flourished recently (for reviews, see Ellegren & Parsch, 2007; Parsch & Ellegren, 2013), motivated by the advent of next-generation genomic techniques and an interest in a number of different evolutionary phenomena, including: the resolution of sexually antagonistic selection pressures; the molecular evolution of reproductive proteins; the chromosomal distribution of sex-biased genes; and the origin and evolution of dosage compensation. The vast majority of this research has focused on Drosophila, but RNA-Seq offers the opportunity to explore these topics in other insect systems distinguished by dramatic sexually dimorphic phenotypes. Below we summarize the details associated with much of this research and highlight some recent studies on non-Drosophila systems.
Genetic basis of sexual dimorphism
Sexually dimorphic traits arise because different selective pressures affect male and female phenotypes (Lande, 1980). Differences in gene expression between the sexes (termed sex-biased or sex-specific gene expression) evolve to resolve the conflict between male- and female-specific adaptive peaks so that each sex can approach its own adaptive optima (Parsch & Ellegren, 2013); therefore, cataloguing genes that are over-expressed in one sex relative to the other in sexually dimorphic tissue can help identify candidate genes and pathways responsible for sexually dimorphic development. Studies in Drosophila have shown that sex-biased gene expression is pervasive and evolutionarily dynamic (Meiklejohn et al., 2003; Ranz et al., 2003; Zhang et al., 2007; Assis et al., 2012). Depending on the study, up to 50% of the genes examined exhibit differential expression between males and females. Substantial differences in the patterns of sex-biased expression are also found between closely related species, particularly for genes serving male reproductive functions (Meiklejohn et al., 2003; Ranz et al., 2003; Zhang et al., 2007; Assis et al., 2012). These studies, however, have focused primarily on whole-body gene expression where much of the sex-biased expression results from reproductive tissues. Few studies, even in Drosophila, have examined sex-biased expression in specific somatic tissues (but see Chang et al., 2011), and fewer still in a somatic tissue with respect to a specific sexually dimorphic phenotype in an attempt to associate expression differences with phenotypic variation.
In non-Drosophila insects, we are aware of only two studies comparing male and female expression levels at a genomic scale in specific (not whole-body) somatic tissue. Baker et al. (2011) examined sex-biased gene expression in several tissues of the mosquito, An. gambiae, and found that, depending on the tissue, 5–15% of the genome was differentially expressed between males and females. A more directed approach was taken by Wilkinson et al. (2013) in an attempt to understand the expression differences underlying extreme sexual dimorphism in the eyestalks of stalk-eyed flies (Diopsidae). Using microarrays designed from an EST library (Baker et al., 2009), they identified nearly 900 genes that were sex-biased in the developing eyestalk tissue but not in the adult head. As might be expected because males possess a larger eyestalk, male-biased genes were over-represented for genes involved in growth and mitochondrial function. Results from both of these studies suggest that transcriptome analysis provides a valuable tool for understanding the overall architecture of sexual dimorphism, but, because sex-biased expression is so widespread, such analyses are unlikely to directly identify candidate genes. It is also important to note that the causal loci directly responsible for sexually dimorphism may not exhibit any expression differences between the sexes, but rather exert their influence through cis-regulatory effects or alternative splicing of pre-mRNA. It will be critical for future studies to identify essential upstream regulators of sexual dimorphism and exaggerated traits, such as doublesex and Insulin-like receptor (Emlen et al., 2012; Gotoh et al., 2014), and to examine the transcriptional impact of these genes through functional manipulation (e.g. RNAi).
Evolution of reproductive proteins
Genes involved in mating and reproduction, particularly for males, are among the most rapidly evolving loci in the genome, and often show evidence of positive selection (Haerty et al., 2007; Singh & Artieri, 2010). Because of their adaptive significance, reproductive proteins have been the subject of many evolutionary studies, and transcriptome analyses offer an efficient way of examining their molecular evolution. While few non-Drosophila insect studies have directly measured sex-biased gene expression on a genomic scale, examination of transcript diversity in a sexually dimorphic tissue for one sex is much more common. These studies include gene expression analysis of the sex pheromone glands in the moth Heliothis virescens (Vogel et al., 2010), the developing horn tissue in the beetle Onthophagus taurus (Choi et al., 2010), and the male reproductive organs of several insects – two Heliconius species (Walters & Harrison, 2010, 2011), Ceratitis capitata (Scolari et al., 2012), Lutzomyia longipalis (Azevedo et al., 2012), Teleopsis dalmanni (Baker et al., 2012; Reinhardt et al., 2014), Teleogryllus oceanicus (Bailey et al., 2013) and two Gryllus species (Andres et al., 2013). Protein sequences identified through transcriptome surveys can be compared with protein sequences from other species to test various hypotheses of molecular evolution. For instance, Walters and Harrison (2011) recently used a transcriptome analysis of seminal fluid proteins in Heliconius butterflies to examine the correlation between mating system and the rate of protein evolution. Contrary to expectation, they found that rates of protein evolution were not accelerated in species with promiscuous vs. monogamous female mating behaviour.
Another critical feature of transcriptome studies is that they facilitate the identification of lineage-specific genes that have no direct homologue in other insect groups. Novel genes can arise in many ways (e.g. gene duplication, lateral gene transfer, de novo gene creation from noncoding regions of DNA) and may rapidly acquire adaptive functional roles (Ranz & Parsch, 2012; Chen et al., 2013). Studies in Drosophila have shown that novel genes represent a substantial component of male-biased gene expression and play a critical role in spermatogenesis. A recent study in stalk-eyed flies (Baker et al., 2012) highlights the ability of RNA-Seq to uncover novel genetic diversity associated with reproductive proteins. More recently, testes transcriptomes from additional stalk-eyed fly species have been analysed, and numerous lineage-specific novel genes have been found (see Fig. 3 for an example). The gene family represented in Fig. 3 comprises homologues of the D. melanogaster gene, Nucleosome assembly protein 1 (Nap1), an important histone chaperone gene that is expressed in the testes (Kimura, 2013). In Drosophila and the mosquito species, Nap1 is present as a single homologue, while stalk-eyed fly species exhibit as many as nine paralogues of Nap1. Overall, the tree suggests there have been at least 14 duplications of the Nap1 gene within the family, and it appears these new genes function primarily in spermatogenesis. Of the seven paralogous copies in T. dalmanni, the one with the closest homology to the D. melanogaster Nap1 protein is expressed ubiquitously throughout the body, while the other six copies exhibit testes-specific gene expression (Fig. 3). DNA undergoes dramatic condensation during spermatogenesis involving the replacement of histones with protamines (White-Cooper, 2010), so it is possible these new Nap1 proteins play some role in this process. Overall, the use of transcriptomes for gene discovery has some inherent limitations because variation in expression levels across taxa will impact their representation in any gene family analysis. Even with this caveat, transcriptome surveys are an incredibly powerful tool for exploring the genetic diversity that is unique, and therefore of particular relevance, to non-model organism groups.
Figure 3.
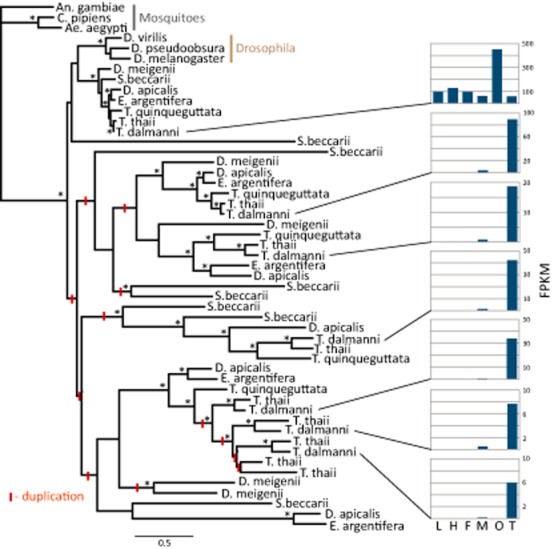
Phylogeny of the Nap1 gene family depicting abundant gene expansion within stalk-eyed flies. Contig sequences homologous to Drosophila Nap1 were extracted from an assembly of a testes transcriptome for each stalk-eyed fly species except Teleopsis dalmanni whose sequence data was derived from a multi-tissue assembly (GenBank accession numbers: KM821168-KM821212). The maximum likelihood tree was generated in PhyML (Guindon and Gascuel 2003) using an LG + G model with 100 bootstrap replicates. Branches with bootstrap values >90 are indicated by an asterisk. Red marks on the branches denote putative duplication events. Gene expression values (FPKM) for each T. dalmanni paralogue are presented to the right of the tree. Tissue abbreviations: L,larva; H, adult head; F, female carcass (whole body with reproductive tissues removed); M, male carcass, O, ovaries; T, testes. Stalk-eyed fly generic abbreviations: D. meigenii, Diasemopsis meigenii; D. apicalis, Diopsis apicalis; E., Eurydiopsis; S., Sphyracephala; T., Teleopsis.
Evolution of sex chromosomes
Perhaps the most widespread use of sex transcriptome analysis involves addressing evolutionary questions regarding the origin and composition of sex chromosomes. Most insect species have a genetic sex determination system with one sex possessing heteromorphic sex chromosomes comprised of one gene-rich chromosome and one degenerate, heterochromatic, gene-poor chromosome (for reviews of the evolutionary processes driving this state see: Charlesworth, 1991; Charlesworth et al., 2005; Bachtrog, 2006). Which sex carries the heteromorphic pair varies among taxa (e.g. XY males/XX females in Diptera, ZZ males/ZW females in Lepidoptera), but in either case, the result is a dose imbalance for sex-limited genes in the heterogametic sex. To offset the potential fitness costs of having a single gene copy, dosage compensation mechanisms that balance gene expression levels across the genome are expected to evolve.
The abundant variation found among insects in both sex-determination systems and sex chromosome composition (Baker & Wilkinson, 2010; Gempe & Beye, 2011; Sahara et al., 2012; Vicoso & Bachtrog, 2013) has motivated a number of studies using RNA-Seq to examine the evolution of dosage compensation by comparing the expression levels of autosomal and sex-linked genes (Vicoso & Charlesworth, 2009; Prince et al., 2010; Harrison et al., 2012; Wilkinson et al., 2013). These studies have confirmed the observation that hypertranscription of the heterogametic sex chromosome occurs in XY systems (Prince et al., 2010; Wilkinson et al., 2013), but not ZW systems (Zha et al., 2009; Harrison et al., 2012). However, the mechanism of dosage compensation appears to differ between insect groups: beetles exhibit hypertranscription of the X chromosome in both sexes (Prince et al., 2010), while, in stalk-eyed flies, hypertranscription of the X chromosome appears to occur only in males (Wilkinson et al., 2013). In addition, ambiguity remains regarding the absence of dosage compensation in ZW species (Walters & Hardcastle, 2011). Transcriptome analyses in several more insect groups will be needed to map the evolutionary origin of dosage compensation systems and to understand the specific genetic mechanisms controlling dosage effects.
The existence of heteromorphic chromosomes affects the evolution of sex-biased gene expression well beyond the effects of dosage compensation. Sex-linked genes in species with heteromorphic chromosomes are found more often in the homogametic sex (e.g. the X chromosome spends 2/3 of its time in females in XY systems), a condition that has strong implications for the resolution of sexually antagonistic selective pressures (Rice, 1992; Bonduriansky & Chenoweth, 2009). Sex chromosomes are predicted to be a preferred location for sex-biased genes, and early transcriptome surveys of sex-biased gene expression in Drosophila revealed dramatic differences in the distribution of sex-biased genes, with the X chromosome significantly over-represented for female-biased genes (Parisi et al., 2003; Ranz et al., 2003; Sturgill et al., 2007). This research sparked an interest in the composition of sex chromosomes, and every study to date has found some form of differential representation of sex-biased genes on the sex chromosomes. For XY species, there is evidence of over-representation of female-biased genes on the X chromosome (Prince et al., 2010; Wilkinson et al., 2013) and under-representation of male-biased genes on the X chromosome (Magnusson et al., 2012). In contrast, the Z chromosome in the silk moth, Bombyx mori, is enriched for genes that function in the testes and is the source of excessive gene movement of female-biased genes onto autosomes (Arunkumar et al., 2009; Wang et al., 2012), while pea aphids (Acyrthosiphon pisum), which have an XO sex-determination system, have a highly masculinized X chromosome that is consistent with predictions based on the unusual mode of X chromosome inheritance in this species (Jaquiery et al., 2013). Given the rapid turnover in sex chromosome identity within insects (Kaiser & Bachtrog, 2010; Pease & Hahn, 2012; Vicoso & Bachtrog, 2013), the effect of chromosomal location on the pattern of sex-biased gene expression is likely to provide a major evolutionary force shaping the phenotypic differences between the sexes.
Conclusion
The democratization of RNA-Seq technology in terms of affordability, accessibility and interpretability, has paved the way to exciting progress in understanding the transcriptional basis of phenotypic diversity in non-model insects. Over the past few years, the number of published studies using this approach has more than doubled, allowing researchers to address previously intractable questions. RNA-Seq has been especially useful in cases where extreme phenotypic divergence occurs between individuals with similar or identical genomes, such as sexual dimorphism and social polyphenism.
RNA-Seq has been used in a variety of ways in non-model insects. At its most fundamental level, RNA-Seq allows for the identification, annotation and comparison of the sets of genes expressed in different species, individuals or phenotypes. Such annotation-based studies have led to the identification of gene families that are evolving especially rapidly, and thus are plausible candidates for explaining the evolution of divergent phenotypes. Beyond cataloging the genes present in a transcriptome, RNA-Seq has further been used to compare the expression of genes across species, under various environmental conditions, and at different developmental stages. A third, and very recent, application of RNA-Seq is for examination of alternative pre-messenger RNA splicing. Alternate splice sites give rise to different protein isoforms, and these can radically alter protein function. In humans, recent studies suggest that >90% of all genes encode two or more proteins via alternative splicing of pre-messenger RNA (Wang et al., 2008), and in cases where both genetic sequence and gene expression levels are identical, it may be the case that alternative splicing is responsible for phenotypic divergence. While detailed surveys of the prevalence and impact of alternative splicing on insect phenotypes are still lacking, work on the Dscam gene, which plays a role in axonal guidance and can generate 38 016 distinct mRNA isoforms in D. melanogaster (Schmucker et al., 2000), suggests that hypervariable splicing of this gene is common throughout the Arthopoda (Armitage et al., 2012). In non-model insects, preliminary results from the tarnished plant bug (Lygus lineolaris) suggest that alternative splicing may contribute to the divergence of odorant binding proteins (Hull et al., 2014). Because odorant binding proteins mediate odorant detection and discrimination, alternative splicing of the genes coding for these proteins could affect oviposition, host plant use, mate preference and many other evolutionarily important behaviours.
One of the most striking results of the increasingly large number of de novo (rather than candidate-based) RNA-Seq studies published to date is the realization that many genes of unknown function are being expressed in virtually every species that has been examined. Because transcriptome annotation necessarily relies on sequence similarity to previously identified genes, the finding that 50% (or more) of the differentially expressed (or alternatively spliced) genes in one's study are unannotated means that it is difficult to know what role, if any, these genes have in explaining the phenotype of interest. Now that the most vexing aspects of RNA isolation, library preparation, sequencing, assembly and analysis are fairly well worked out, the next great challenge for those who study non-model organisms is to simplify the process of figuring out just what all those genes are for.
Acknowledgments
We thank an anonymous reviewer for constructive comments. We thank the Korein Foundation and the Lewis and Dorothy Cullman Foundation for their continued generous support. The Sackler Institute for Comparative Genomics at the American Museum of Natural History provided a vibrant community for discussion of the issues addressed here. Apurva Narechania and Josie Reinhardt provided valuable assistance with transcriptome assemblies and bioinformatic analysis. S.S. would like to thank the German Academic Exchange Service (DAAD) and the Biosystematics Group at Wageningen University for support. S.O. is supported by NSF DBI-1307844. Funding for R.B.'s research on stalk-eyed flies was provided by NSF DEB-0951816.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1. Recent transcriptome studies in non-model insects.
References
- Allen CE, Zwaan BJ, Brakefield PM. Evolution of sexual dimorphism in the Lepidoptera. Annu Rev Entomol. 2011;56:445–464. doi: 10.1146/annurev-ento-120709-144828. [DOI] [PubMed] [Google Scholar]
- Alon M, Elbaz M, Ben-Zvi MM, Feldmesser E, Vainstein A, Morin S. Insights into the transcriptomics of polyphagy: bemisia tabaci adaptability to phenylpropanoids involves coordinated expression of defense and metabolic genes. Insect Biochem Mol Biol. 2012;42:251–263. doi: 10.1016/j.ibmb.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Amdam GV, Norberg K, Fondrk MK, Page RE. Reproductive ground plan may mediate colony-level selection effects on individual foraging behavior in honey bees. Proc Natl Acad Sci U S A. 2004;101:11350–11355. doi: 10.1073/pnas.0403073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres JA, Larson EL, Bogdanowicz SM, Harrison RG. Patterns of transcriptome divergence in the male accessory gland of two closely related species of field crickets. Genetics. 2013;193:501–513. doi: 10.1534/genetics.112.142299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage SAO, Freiburg RY, Kurtz J, Bravo IG. The evolution of Dscam genes across the arthropods. BMC Evol Biol. 2012;12:53–67. doi: 10.1186/1471-2148-12-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunkumar KP, Mita K, Nagaraju J. The silkworm Z chromosome is enriched in testis-specific genes. Genetics. 2009;182:493–501. doi: 10.1534/genetics.108.099994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assis R, Zhou Q, Bachtrog D. Sex-biased transcriptome evolution in Drosophila. Genome Biol Evol. 2012;4:1189–1200. doi: 10.1093/gbe/evs093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo RV, Dias DB, Bretas JA, Mazzoni CJ, Souza NA, Albano RM, et al. The transcriptome of Lutzomyia longipalpis (Diptera: Psychodidae) male reproductive organs. PLoS One. 2012;7:e34495. doi: 10.1371/journal.pone.0034495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D. A dynamic view of sex chromosome evolution. Curr Opin Genet Dev. 2006;16:578–585. doi: 10.1016/j.gde.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Badisco L, Ott SR, Rogers SM, Matheson T, Knapen D, Vergauwen L, et al. Microarray-based transcriptomic analysis of differences between long-term gregarious and solitarious desert locusts. PLoS One. 2011;6:e28110. doi: 10.1371/journal.pone.0028110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek JH, Oh JH, Kim YH, Lee SH. Comparative transcriptome analysis of the venom sac and gland of social wasp Vespa tropica and solitary wasp Rhynchium brunneum. J Asia-Pac Entomology. 2013;16:497–502. [Google Scholar]
- Bailey NW, Veltsos P, Tan YF, Millar AH, Ritchie MG, Simmons LW. Tissue-specific transcriptomics in the field cricket Teleogryllus oceanicus. Genes Genomes Genet. 2013;3:225–230. doi: 10.1534/g3.112.004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D, Nolan T, Fischer B, Pinder A, Crisanti A, Russell S. A comprehensive gene expression atlas of sex- and tissue-specificity in the malaria vector, Anopheles gambiae. BMC Genomics. 2011;12:296–307. doi: 10.1186/1471-2164-12-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RH, Wilkinson GS. Comparative genomic hybridization (CGH) reveals a neo-X chromosome and biased gene movement in stalk-eyed flies (Genus Teleopsis. PLoS Genet. 2010;6:e1001121. doi: 10.1371/journal.pgen.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RH, Morgan J, Wang X, Boore JL, Wilkinson GS. Genomic analysis of a sexually-selected character: EST sequencing and microarray analysis of eye-antennal imaginal discs in the stalk-eyed fly Teleopsis dalmanni (Diopsidae) BMC Genomics. 2009;10:1–20. doi: 10.1186/1471-2164-10-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RH, Narechania A, Johns PM, Wilkinson GS. Gene duplication, tissue-specific gene expression and sexual conflict in stalk-eyed flies (Diopsidae) Philos Trans R Soc B Biol Sci. 2012;367:2357–2375. doi: 10.1098/rstb.2011.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao YY, Qu LY, Zhao D, Chen LB, Jin HY, Xu LM, et al. The genome- and transcriptome-wide analysis of innate immunity in the brown planthopper, Nilaparvata lugens. BMC Genomics. 2013;14:160–182. doi: 10.1186/1471-2164-14-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass C, Zimmer CT, Riveron JM, Wilding CS, Wondji CS, Kaussmann M, et al. Gene amplification and microsatellite polymorphism underlie a recent insect host shift. Proc Natl Acad Sci U S A. 2013;110:19460–19465. doi: 10.1073/pnas.1314122110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birol I, Jackman S, Nielsen C, Qian J, Varhol R, Stazyk G, et al. De novo transcriptome assembly with ABySS. Bioinformatics. 2009;25:2872–2877. doi: 10.1093/bioinformatics/btp367. [DOI] [PubMed] [Google Scholar]
- Bonduriansky R, Chenoweth SF. Intralocus sexual conflict. Trends Ecol Evol. 2009;24:280–288. doi: 10.1016/j.tree.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Boomsma JJ. Lifetime monogamy and the evolution of eusociality. Philosophical Trans R Soc B-Biol Sci. 2009;364:3191–3207. doi: 10.1098/rstb.2009.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradnam KR, Fass JN, Alexandrov A, Baranay P, Bechner M, Birol I, et al. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. GigaScience. 2013;2:1–31. doi: 10.1186/2047-217X-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalan A, Hutter S, Parsch J. Population and sex differences in Drosophila melanogaster brain gene expression. BMC Genomics. 2012;13:654–666. doi: 10.1186/1471-2164-13-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celorio-Mancera MD, Wheat CW, Vogel H, Soderlind L, Janz N, Nylin S. Mechanisms of macroevolution: polyphagous plasticity in butterfly larvae revealed by RNA-Seq. Mol Ecol. 2013;22:4884–4895. doi: 10.1111/mec.12440. [DOI] [PubMed] [Google Scholar]
- Chang PL, Dunham JP, Nuzhdin SV, Arbeitman MN. Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing. BMC Genomics. 2011;12:364–384. doi: 10.1186/1471-2164-12-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The evolution of sex chromosomes. Science. 1991;251:1030–1033. doi: 10.1126/science.1998119. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B, Marais G. Steps in the evolution of heteromorphic sex chromosomes. Heredity. 2005;95:118–128. doi: 10.1038/sj.hdy.6800697. [DOI] [PubMed] [Google Scholar]
- Chen S, Yang P, Jiang F, Wei Y, Ma Z, Kang L. De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PLoS One. 2010;5:e15633. doi: 10.1371/journal.pone.0015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Krinsky BH, Long M. New genes as drivers of phenotypic evolution. Nat Rev Genet. 2013;14:645–660. doi: 10.1038/nrg3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi YH, Salzman RA, Balfe S, Ahn JE, Sun W, Moon J, et al. Cowpea bruchid midgut transcriptome response to a soybean cystatin: costs and benefits of counter-defense. Insect Mol Biol. 2009;18:97–110. doi: 10.1111/j.1365-2583.2008.00854.x. [DOI] [PubMed] [Google Scholar]
- Choi JH, Kijimoto T, Snell-Rood E, Tae H, Yang Y, Moczek AP, et al. Gene discovery in the horned beetle Onthophagus taurus. BMC Genomics. 2010;11:703–722. doi: 10.1186/1471-2164-11-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty THF, Toth AL, Robinson GE. Nutrition and division of labor: effects on foraging and brain gene expression in the paper wasp Polistes metricus. Mol Ecol. 2011;20:5337–5347. doi: 10.1111/j.1365-294X.2011.05344.x. [DOI] [PubMed] [Google Scholar]
- Dermauw W, Wybouw N, Rombauts S, Menten B, Vontas J, Grbic M, et al. A link between host plant adaptation and pesticide resistance in the polyphagous spider mite Tetranychus urticae. Proc Natl Acad Sci U S A. 2013;110:E113–E122. doi: 10.1073/pnas.1213214110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning LT, Dennis AB, Park D, Sinclair BJ, Newcomb RD, Buckley TR. Identification of cold-responsive genes in a New Zealand alpine stick insect using RNA-Seq. Comp Biochem Phys D-Genomics Proteomics. 2013;8:24–31. doi: 10.1016/j.cbd.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 2007;8:689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- Emlen DJ. The Evolution of Animal Weapons. Annu Rev Ecol Evol Systs. 2008;39:387–413. [Google Scholar]
- Emlen DJ, Warren IA, Johns A, Dworkin I, Lavine LC. A mechanism of extreme growth and reliable signaling in sexually selected ornaments and weapons. Science. 2012;337:860–864. doi: 10.1126/science.1224286. [DOI] [PubMed] [Google Scholar]
- Evans JD, Wheeler DE. Gene expression and the evolution of insect polyphenisms. Bioessays. 2001;23:62–68. doi: 10.1002/1521-1878(200101)23:1<62::AID-BIES1008>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Ewen-Campen B, Shaner N, Panfilio KA, Suzuki Y, Roth S, Extavour CG. The maternal and early embryonic transcriptome of the milkweed bug Oncopeltus fasciatus. BMC Genomics. 2011;12:61–83. doi: 10.1186/1471-2164-12-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira PG, Patalano S, Chauhan R, Ffrench-Constant R, Gabaldon T, Guigo R, et al. Transcriptome analyses of primitively eusocial wasps reveal novel insights into the evolution of sociality and the origin of alternative phenotypes. Genome Biol. 2013;14:R20. doi: 10.1186/gb-2013-14-2-r20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fescemyer HW, Sandoya GV, Gill TA, Ozkan S, Marden JH, Luthe DS. Maize toxin degrades peritrophic matrix proteins and stimulates compensatory transcriptome responses in fall armyworm midgut. Insect Biochem Mol Biol. 2013;43:280–291. doi: 10.1016/j.ibmb.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Gempe T, Beye M. Function and evolution of sex determination mechanisms, genes and pathways in insects. Bioessays. 2011;33:52–60. doi: 10.1002/bies.201000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser-Schmitt A, Catalan A, Parsch J. Adaptive divergence of a transcriptional enhancer between populations of Drosophila melanogaster. Philosophical Trans R Soc B Biol Sci. 2013;368:20130024. doi: 10.1098/rstb.2013.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong ZJ, Wu YQ, Miao J, Duan Y, Jiang YL, Li T. Global transcriptome analysis of orange wheat blossom midge, Sitodiplosis mosellana (Gehin) (Diptera: Cecidomyiidae) to identify candidate transcripts regulating diapause. PLoS One. 2013;8:e71564. doi: 10.1371/journal.pone.0071564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh H, Miyakawa H, Ishikawa A, Ishikawa Y, Sugime Y, Emlen DJ, et al. Developmental link between sex and nutrition; doublesex regulates sex-specific mandible growth via juvenile hormone signaling in stag beetles. PLoS Genet. 2014;10:e1004098. doi: 10.1371/journal.pgen.1004098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg JK, Xia J, Zhou X, Thatcher SR, Gu X, Ament SA, et al. Behavioral plasticity in honey bees is associated with differences in brain microRNA transcriptome. Genes Brain Behav. 2012;11:660–670. doi: 10.1111/j.1601-183X.2012.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang XH, Ma ZY, Xue LA, Han JY, Yu D, et al. CSP and takeout genes modulate the switch between attraction and repulsion during behavioral phase change in the migratory locust. PLoS Genet. 2011;7:e1001291. doi: 10.1371/journal.pgen.1001291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerty W, Jagadeeshan S, Kulathinal RJ, Wong A, Ram KR, Sirot LK, et al. Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics. 2007;177:1321–1335. doi: 10.1534/genetics.107.078865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13:469–483. doi: 10.1038/nrg3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PW, Mank JE, Wedell N. Incomplete sex chromosome dosage compensation in the Indian meal moth, Plodia interpunctella, based on de novo transcriptome assembly. Genome Biol Evol. 2012;4:1118–1126. doi: 10.1093/gbe/evs086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Sun P, Zhou X, Lei C. Characterization of head transcriptome and analysis of gene expression involved in caste differentiation and aggression in Odontotermes formosanus (Shiraki) PLoS One. 2012;7:e50383. doi: 10.1371/journal.pone.0050383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull JJ, Perera OP, Snodgrass GL. Cloning and expression profiling of odorant-binding proteins in the tarnished plant bug, Lygus lineolaris. Insect Mol Biol. 2014;23:78–97. doi: 10.1111/imb.12064. [DOI] [PubMed] [Google Scholar]
- Husseneder C, McGregor C, Lang RP, Collier R, Delatte J. Transcriptome profiling of female alates and egg-laying queens of the Formosan subterranean termite. Comp Biochem Physiol D-Genomics Proteomics. 2012;7:14–27. doi: 10.1016/j.cbd.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Jaquiery J, Rispe C, Roze D, Legeai F, Le Trionnaire G, Stoeckel S, et al. Masculinization of the x chromosome in the pea aphid. PLoS Genet. 2013;9:e1003690. doi: 10.1371/journal.pgen.1003690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BR, Linksvayer TA. Deconstructing the superorganism: social physiology, groundplans, and sociogenomics. Quart Rev Biol. 2010;85:57–79. doi: 10.1086/650290. [DOI] [PubMed] [Google Scholar]
- Johnson BR, Tsutsui ND. Taxonomically restricted genes are associated with the evolution of sociality in the honey bee. BMC Genomics. 2011;12:164–174. doi: 10.1186/1471-2164-12-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BR, Atallah J, Plachetzki DC. The importance of tissue specificity for RNA-seq: highlighting the errors of composite structure extractions. BMC Genomics. 2013;14:586–594. doi: 10.1186/1471-2164-14-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser VB, Bachtrog D. Evolution of sex chromosomes in insects. Annu Rev Genet. 2010;44:91–112. doi: 10.1146/annurev-genet-102209-163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S. The Nap family proteins, CG5017/Hanabi and Nap1, are essential for Drosophila spermiogenesis. FEBS Lett. 2013;587:922–929. doi: 10.1016/j.febslet.2013.02.019. [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- Korb J, Weil T, Hoffmann K, Foster KR, Rehli M. A gene necessary for reproductive suppression in termites. Science. 2009;324:758–758. doi: 10.1126/science.1170660. [DOI] [PubMed] [Google Scholar]
- Lande R. Sexual dimorphism, sexual selection, and adaptation in polygenic characters. Evolution. 1980;34:292–305. doi: 10.1111/j.1558-5646.1980.tb04817.x. [DOI] [PubMed] [Google Scholar]
- Le Conte Y, Alaux C, Martin JF, Harbo JR, Harris JW, Dantec C, et al. Social immunity in honeybees (Apis mellifera): transcriptome analysis of varroa-hygienic behaviour. Insect Mol Biol. 2011;20:399–408. doi: 10.1111/j.1365-2583.2011.01074.x. [DOI] [PubMed] [Google Scholar]
- Li LT, Zhu YB, Ma JF, Li ZY, Dong ZP. An analysis of the Athetis lepigone transcriptome from four developmental stages. PLoS ONE. 2013;8:e73911. doi: 10.1371/journal.pone.0073911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang ZS, Trang N, Mattila HR, Rodriguez-Zas SL, Seeley TD, Robinson GE. Molecular determinants of scouting behavior in honey bees. Science. 2012;335:1225–1228. doi: 10.1126/science.1213962. [DOI] [PubMed] [Google Scholar]
- Linksvayer TA, Wade MJ. Genes with social effects are expected to harbor more sequence variation within and between species. Evolution. 2009;63:1685–1696. doi: 10.1111/j.1558-5646.2009.00670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Li M, Li J-M, Huang C-J, Zhou X-P, Xu F-C, et al. Viral infection of tobacco plants improves performance of Bemisia tabaci but more so for an invasive than for an indigenous biotype of the whitefly. J Zhejiang Univ Sci B. 2010;11:30–40. doi: 10.1631/jzus.B0900213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan JB, Wang YL, Wang J, Wang XW, Liu SS. Detoxification activity and energy cost is attenuated in whiteflies feeding on Tomato yellow leaf curl China virus-infected tobacco plants. Insect Mol Biol. 2013;22:597–607. doi: 10.1111/imb.12048. [DOI] [PubMed] [Google Scholar]
- Ma J, Wang J, Mao XF, Wang Y. Differential expression of two antifreeze proteins in the desert beetle Anatolica polita (Coleoptera: Tenebriondae): seasonal variation and environmental effects. Cryoletters. 2012;33:337–348. [PubMed] [Google Scholar]
- Magnusson K, Lycett G, Mendes A, Lynd A, Papathanos P-A, Crisanti A, et al. Demasculinization of the Anopheles gambiae X chromosome. BMC Evol Biol. 2012;12:69–82. doi: 10.1186/1471-2148-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannello F, Ligi D, Magnani M. Deciphering the single-cell omic: innovative application for translational medicine. Expert Rev Proteomics. 2012;9:635–648. doi: 10.1586/epr.12.61. [DOI] [PubMed] [Google Scholar]
- Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- Matzkin LM. Population transcriptomics of cactus host shifts in Drosophila mojavensis. Mol Ecol. 2012;21:2428–2439. doi: 10.1111/j.1365-294X.2012.05549.x. [DOI] [PubMed] [Google Scholar]
- Maza E, Frasse P, Senin P, Bouzayen M, Zouine M. Comparison of normalization methods for differential gene expression analysis in RNA-Seq experiments: a matter of relative size of studied transcriptomes. Communicative Integr Biol. 2013;6:e25849. doi: 10.4161/cib.25849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Parsch J, Ranz JM, Hartl DL. Rapid evolution of male-biased gene expression in Drosophila. Proc Natl Acad Sci U S A. 2003;100:9894–9899. doi: 10.1073/pnas.1630690100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ometto L, Shoemaker D, Ross KG, Keller L. Evolution of gene expression in fire ants: the effects of developmental stage, caste, and species. Mol Biol Evol. 2011;28:1381–1392. doi: 10.1093/molbev/msq322. [DOI] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM. Single-molecule direct RNA sequencing without cDNA synthesis. Wiley Interdisciplinary Rev-RNA. 2011;2:565–570. doi: 10.1002/wrna.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, et al. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science. 2003;299:697–700. doi: 10.1126/science.1079190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsch J, Ellegren H. The evolutionary causes and consequences of sex-biased gene expression. Nat Rev Genet. 2013;14:83–87. doi: 10.1038/nrg3376. [DOI] [PubMed] [Google Scholar]
- Patel A, Fondrk MK, Kaftanoglu O, Emore C, Hunt G, Frederick K, et al. The making of a queen: TOR pathway Is a key player in diphenic caste development. PLoS One. 2007;2:e509. doi: 10.1371/journal.pone.0000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease JB, Hahn MW. Sex chromosomes evolved from independent ancestral linkage groups in winged insects. Mol Biol Evol. 2012;29:1645–1653. doi: 10.1093/molbev/mss010. [DOI] [PubMed] [Google Scholar]
- Picelli S, Björklund Å, Faridani O, Winberg G, Sagasser S, Sandberg R. Improved Smart-Seq for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Poelchau MF, Reynolds JA, Denlinger DL, Elsik CG, Armbruster PA. Transcriptome sequencing as a platform to elucidate molecular components of the diapause response in the Asian tiger mosquito Aedes albopictus. Physiol Entomol. 2013a;38:173–181. doi: 10.1111/phen.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelchau MF, Reynolds JA, Elsik CG, Denlinger DL, Armbruster PA. RNA-Seq reveals early distinctions and late convergence of gene expression between diapause and quiescence in the Asian tiger mosquito, Aedes albopictus. J Exp Biol. 2013b;216:4082–4090. doi: 10.1242/jeb.089508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DP, Nagarajan V, Churbanov A, Houde P, Milligan B, Drake LL, et al. The fat body transcriptomes of the yellow fever mosquito Aedes aegypti, pre- and post- blood meal. PLoS One. 2011;6:e22573. doi: 10.1371/journal.pone.0022573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince EG, Kirkland D, Demuth JP. Hyperexpression of the X chromosome in both sexes results in extensive female bias of X-linked genes in the flour beetle. Genome Biol Evol. 2010;2:336–346. doi: 10.1093/gbe/evq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu LM, Mao XF, Hou F, Ma J. A novel function; Thermal protective properties of an antifreeze protein from the summer desert beetle Microdera punctipennis. Cryobiology. 2013;66:60–68. doi: 10.1016/j.cryobiol.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Ranz JM, Parsch J. Newly evolved genes: moving from comparative genomics to functional studies in model systems. How important is genetic novelty for species adaptation and diversification? Bioessays. 2012;34:477–483. doi: 10.1002/bies.201100177. [DOI] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science. 2003;300:1742–1745. doi: 10.1126/science.1085881. [DOI] [PubMed] [Google Scholar]
- Reinhardt JA, Brand CL, Paczolt KA, Johns PM, Baker RH, Wilkinson GS. Meiotic drive impacts expression and evolution of x-linked genes in stalk-eyed flies. PLoS Genet. 2014;10:e1004362. doi: 10.1371/journal.pgen.1004362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR. Sexually antagonistic genes: experimental evidence. Science. 1992;256:1436–1439. doi: 10.1126/science.1604317. [DOI] [PubMed] [Google Scholar]
- Riesgo A, Andrade SC, Sharma P, Novo M, Pérez-Porro A, Vahtera V, et al. Comparative description of ten transcriptomes of newly sequenced invertebrates and efficiency estimation of genomic sampling in non-model taxa. Front Zool. 2012;9:33–57. doi: 10.1186/1742-9994-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinker DC, Pitts RJ, Zhou X, Suh E, Rokas A, Zwiebel LJ. Blood meal-induced changes to antennal transcriptome profiles reveal shifts in odor sensitivities in Anopheles gambiae. Proc Natl Acad Sci U S A. 2013;110:8260–8265. doi: 10.1073/pnas.1302562110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessingh P, Bouaichi A, Simpson SJ. Effects of sensory stimuli on the behavioural phase state of the desert locust, Schistocerca gregaria. J Insect Physiol. 1998;44:883–893. doi: 10.1016/s0022-1910(98)00070-5. [DOI] [PubMed] [Google Scholar]
- Rund SS, Bonar NA, Champion MM, Ghazi JP, Houk CM, Leming MT, et al. Daily rhythms in antennal protein and olfactory sensitivity in the malaria mosquito Anopheles gambiae. Scientific Rep. 2013;3:2494. doi: 10.1038/srep02494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackton TB, Werren JH, Clark AG. Characterizing the infection-induced transcriptome of Nasonia vitripennis reveals a preponderance of taxonomically-restricted immune genes. PLoS One. 2013;8:e83984. doi: 10.1371/journal.pone.0083984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara K, Yoshido A, Traut W. Sex chromosome evolution in moths and butterflies. Chromosome Res. 2012;20:83–94. doi: 10.1007/s10577-011-9262-z. [DOI] [PubMed] [Google Scholar]
- Saminadin-Peter SS, Kemkemer C, Pavlidis P, Parsch J. Selective sweep of a cis-regulatory sequence in a non-African population of Drosophila melanogaster. Mol Biol Evol. 2012;29:1167–1174. doi: 10.1093/molbev/msr284. [DOI] [PubMed] [Google Scholar]
- Schmucker D, Clemens JC, Shu H, Worby CA, Xiao J, Muda M, et al. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell. 2000;101:671–684. doi: 10.1016/s0092-8674(00)80878-8. [DOI] [PubMed] [Google Scholar]
- Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scolari F, Gomulski LM, Ribeiro JM, Siciliano P, Meraldi A, Falchetto M, et al. Transcriptional profiles of mating-responsive genes from testes and male accessory glands of the Mediterranean fruit fly, Ceratitis capitata. PLoS One. 2012;7:e46812. doi: 10.1371/journal.pone.0046812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R, Raychoudhury R, Cai Y, Sun Y, Lietze V-U, Boucias DG, et al. Differential impacts of juvenile hormone, soldier head extract and alternate caste phenotypes on host and symbiont transcriptome composition in the gut of the termite Reticulitermes flavipes. BMC Genomics. 2013;14:1–18. doi: 10.1186/1471-2164-14-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuel RW, Dixon SE. The early establishment of dimorphism in the female honeybee, Apis mellifera L. Insectes Sociaux. 1960;7:265–282. [Google Scholar]
- Simola DF, Wissler L, Donahue G, Waterhouse RM, Helmkampf M, Roux J, et al. Social insect genomes exhibit dramatic evolution in gene composition and regulation while preserving regulatory features linked to sociality. Genome Res. 2013;23:1235–1247. doi: 10.1101/gr.155408.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson SJ, Sword GA, Lo N. Polyphenism in Insects. Curr Biol. 2011;21:R738–R749. doi: 10.1016/j.cub.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Singh RS, Artieri CG. Male sex drive and the maintenance of sex: evidence from Drosophila. J Hered. 2010;101(Suppl 1):S100–S106. doi: 10.1093/jhered/esq006. [DOI] [PubMed] [Google Scholar]
- Sinha DK, Nagaraju J, Tomar A, Bentur JS, Nair S. Pyrosequencing-based transcriptome analysis of the Asian rice gall midge reveals differential response during compatible and incompatible interaction. Int J Mol Sci. 2012;13:13079–13103. doi: 10.3390/ijms131013079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CR, Toth AL, Suarez AV, Robinson GE. Genetic and genomic analyses of the division of labour in insect societies. Nat Rev Genet. 2008;9:735–748. doi: 10.1038/nrg2429. [DOI] [PubMed] [Google Scholar]
- Sturgill D, Zhang Y, Parisi M, Oliver B. Demasculinization of X chromosomes in the Drosophila genus. Nature. 2007;450:238–241. doi: 10.1038/nature06330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21:2213–2223. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarver MR, Coy MR, Scharf ME. Cyp15F1: a novel cytochrome P450 gene linked to juvenile hormone-dependent caste differentiation in the termite Reticulitermes flavipes. Arch Insect Biochem Physiol. 2012;80:92–108. doi: 10.1002/arch.21030. [DOI] [PubMed] [Google Scholar]
- Teets NM, Denlinger DL. Autophagy in Antarctica Combating dehydration stress in the world's southernmost insect. Autophagy. 2013;9:629–631. doi: 10.4161/auto.23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teets NM, Peyton JT, Colinet H, Renault D, Kelley JL, Kawarasaki Y, et al. Gene expression changes governing extreme dehydration tolerance in an Antarctic insect. Proc Natl Acad Sci U S A. 2012;109:20744–20749. doi: 10.1073/pnas.1218661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans MJTN, Roelofs D, Nota B, Ylstra B, Holmstrup M. Sugar sweet springtails: on the transcriptional response of Folsomia candida (Collembola) to desiccation stress. Insect Mol Biol. 2009;18:737–746. doi: 10.1111/j.1365-2583.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- Toth AL, Robinson GE. Evo-devo and the evolution of social behavior. Trends Genet. 2007;23:334–341. doi: 10.1016/j.tig.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Toth AL, Varala K, Henshaw MT, Rodriguez-Zas SL, Hudson ME, Robinson GE. Brain transcriptomic analysis in paper wasps identifies genes associated with behaviour across social insect lineages. Proc R Soc B Biol Sci. 2010;277:2139–2148. doi: 10.1098/rspb.2010.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uvarov BP. Grasshoppers and Locusts: A Handbook of General Acridology. Cambridge, UK: Anti-Locust Research Centre (Cambridge University Press); 1966. Vol. 1. [Google Scholar]
- Van Verk MC, Hickman R, Pieterse CM, Van Wees SC. RNA-Seq: revelation of the messengers. Trends Plant Sci. 2013;18:175–179. doi: 10.1016/j.tplants.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Vicoso B, Bachtrog D. Reversal of an ancient sex chromosome to an autosome in Drosophila. Nature. 2013;499:332–335. doi: 10.1038/nature12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. The deficit of male-biased genes on the D. melanogaster X chromosome Is expression-dependent: a consequence of dosage compensation? J Mol Evol. 2009;68:576–583. doi: 10.1007/s00239-009-9235-4. [DOI] [PubMed] [Google Scholar]
- Vilcinskas A, Stoecker K, Schmidtberg H, Rohrich CR, Vogel H. Invasive harlequin ladybird carries biological weapons against native competitors. Science. 2013;340:862–863. doi: 10.1126/science.1234032. [DOI] [PubMed] [Google Scholar]
- Vogel H, Heidel AJ, Heckel DG, Groot AT. Transcriptome analysis of the sex pheromone gland of the noctuid moth Heliothis virescens. BMC Genomics. 2010;11:29–50. doi: 10.1186/1471-2164-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters JR, Hardcastle TJ. Getting a full dose? Reconsidering sex chromosome dosage compensation in the silkworm, Bombyx mori. Genome Biol Evol. 2011;3:491–504. doi: 10.1093/gbe/evr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters JR, Harrison RG. Combined EST and proteomic analysis identifies rapidly evolving seminal fluid proteins in Heliconius butterflies. Mol Biol Evol. 2010;27:2000–2013. doi: 10.1093/molbev/msq092. [DOI] [PubMed] [Google Scholar]
- Walters JR, Harrison RG. Decoupling of rapid and adaptive evolution among seminal fluid proteins in Heliconius butterflies with divergent mating systems. Evolution. 2011;65:2855–2871. doi: 10.1111/j.1558-5646.2011.01351.x. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Long M, Vibranovski MD. Retrogenes moved out of the z chromosome in the silkworm. J Mol Evol. 2012;74:113–126. doi: 10.1007/s00239-012-9499-y. [DOI] [PubMed] [Google Scholar]
- Wang X, Fang X, Yang P, Jiang X, Jiang F, Zhao D, et al. The locust genome provides insight into swarm formation and long-distance flight. Nat Commun. 2014;5:1–9. doi: 10.1038/ncomms3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White-Cooper H. Molecular mechanisms of gene regulation during Drosophila spermatogenesis. Reproduction. 2010;139:11–21. doi: 10.1530/REP-09-0083. [DOI] [PubMed] [Google Scholar]
- Whiteman NK, Gloss AD, Sackton TB, Groen SC, Humphrey PT, Lapoint RT, et al. Genes involved in the evolution of herbivory by a leaf-mining drosophilid fly. Genome Biol Evol. 2012;4:900–916. doi: 10.1093/gbe/evs063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson GS, Dodson G. Function and evolution of antlers and eye stalks in flies. In: Choe J, Crespi B, editors. The Evolution of Mating Systems in Insects and Arachnids. Cambridge, UK: Cambridge University Press; 1997. pp. 310–328. eds) [Google Scholar]
- Wilkinson GS, Johns PM, Metheny JD, Baker RH. Sex-biased gene expression during head development in a sexually dimorphic stalk-eyed fly. PLoS One. 2013;8:e59826. doi: 10.1371/journal.pone.0059826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EO. The Insect Societies. Cambridge, MA, USA: Belknap Press of Harvard University Press; 1971. [Google Scholar]
- Wolf JB. Principles of transcriptome analysis and gene expression quantification: an RNA-seq tutorial. Mol Ecol Resour. 2013;13:559–572. doi: 10.1111/1755-0998.12109. [DOI] [PubMed] [Google Scholar]
- Woodard SH, Fischman BJ, Venkat A, Hudson ME, Varala K, Cameron SA, et al. Genes involved in convergent evolution of eusociality in bees. Proc Natl Acad Sci U S A. 2011;108:7472–7477. doi: 10.1073/pnas.1103457108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014;30:1660–1666. doi: 10.1093/bioinformatics/btu077. [DOI] [PubMed] [Google Scholar]
- Zeng V, Ewen-Campen B, Horch HW, Roth S, Mito T, Extavour CG. Developmental gene discovery in a hemimetabolous insect: de novo assembly and annotation of a transcriptome for the cricket Gryllus bimaculatus. PLoS ONE. 2013;8:e61479. doi: 10.1371/journal.pone.0061479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha X, Xia Q, Duan J, Wang C, He N, Xiang Z. Dosage analysis of Z chromosome genes using microarray in silkworm, Bombyx mori. Insect Biochem Mol Biol. 2009;39:315–321. doi: 10.1016/j.ibmb.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature. 2007;450:233–238. doi: 10.1038/nature06323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Peng T, He W, Zhang H. High-throughput sequencing to reveal genes involved in reproduction and development in Bactrocera dorsalis (Diptera: Tephritidae) PLoS ONE. 2012;7:e36463. doi: 10.1371/journal.pone.0036463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Recent transcriptome studies in non-model insects.