

Abstract
Progression of breast cancer to metastatic bone disease is linked to deregulated expression of the transcription factor Runx2. Therefore, our goal was to evaluate the potential for clinical use of Runx2-targeting microRNAs (miRNAs) to reduce tumor growth and bone metastatic burden. Expression analysis of a panel of miRNAs regulating Runx2 revealed a reciprocal relationship between the abundance of Runx2 protein and two miRNAs, miR-135 and miR-203. These miRNAs are highly expressed in normal breast epithelial cells where Runx2 is not detected, and absent in metastatic breast cancer cells and tissue biopsies that express Runx2. Reconstituting metastatic MDA-MB-231-Luc cells with miR-135 and miR-203 reduced the abundance of Runx2 and expression of the metastasis-promoting Runx2 target genes IL-11, MMP-13, and PTHrP. Additionally, tumor cell viability was decreased and migration suppressed in vitro. Orthotopic implantation of MDA-MB-231-luc cells delivered with miR-135 or miR-203, followed by an intratumoral administration of the synthetic miRNAs reduced the tumor growth and spontaneous metastasis to bone. Furthermore, intratibial injection of these miRNA-delivered cells impaired tumor growth in the bone environment and inhibited bone resorption. Importantly, reconstitution of Runx2 in MDA-MB-231-luc cells delivered with miR-135 and miR-203 reversed the inhibitory effect of the miRNAs on tumor growth and metastasis. Thus, we have identified that aberrant expression of Runx2 in aggressive tumor cells is related to the loss of specific Runx2-targeting miRNAs and that a clinically relevant replacement strategy by delivery of synthetic miRNAs is a candidate therapeutic approach to prevent metastatic bone disease by this route.
Keywords: metastasis, breast cancer, osteolysis, miRNA, Runx2
Introduction
Breast cancer is among the most prevalent cancers worldwide. The vast majority of breast cancer-related deaths are due to metastatic tumor growth in distant organs, with bone being the most common site of metastasis (1). Approximately 70% of breast cancer patients suffer from bone metastases, which are associated with debilitating fragility fractures, severe pain, nerve compressions, and hypercalcemia, all resulting in an increased morbidity and mortality (2). The complex process of bone metastasis involves cancer cell dissemination from the primary tumor, extravasation into the blood stream, and occupation of the bone marrow space. Metastatic growth in bone is supported by the ‘vicious cycle‘, a multi-directional interaction between tumor cells, osteoblasts, and osteoclasts, leading to the development of osteolytic bone disease, a localized bone destruction caused by an accelerated resorption of bone by osteoclasts (3). Tumor cells express and secrete a variety of cytokines, including interleukins (e.g., IL-11, IL-20) and matrix metalloproteinases (e.g., MMP-9, MMP-13) that activate osteoclasts either directly or through osteoblasts (2). The prevailing tumor-secreted factor activating the osteolytic cycle is parathryroid hormone-related protein (PTHrP). PTHrP stimulates osteoblasts to produce RANKL and other cytokines that in turn activates osteoclasts and subsequent bone destruction. Bone resorption causes the release of TGF-β from the bone matrix, which in concert with other factors further stimulates tumor cell proliferation, thereby perpetuating the vicious cycle (4).
Tumor cell homing to and survival in bone is a result of a deregulated activation of osteomimetic genes (e.g., Runx2, Ibsp, Opn) and signaling pathways, including bone morphogenetic protein (BMP) and canonical Wnt-signaling (5, 6). Runx2, a runt-related transcription factor, has a pivotal role in breast and prostate cancer progression and metastasis (7). Runx2 is aberrantly expressed in breast and prostate cancer cells that metastasize to bone and promotes early and late stages of tumor progression, including the development of osteolytic lesions in the bone marrow environment through transcriptional activation of metastasis-related genes including IL-11, several MMPs, and PTHrP (8-13). Inhibition of Runx2 in metastatic cancer cell lines by RNA interference inhibits tumor cell activities and prevents metastatic bone disease, thus providing a basis for Runx2 as a potential therapeutic target against metastasis (14, 15). However, regulating transcription factors using conventional pharmacological approaches is challenging, while a potential novel avenue for the targeted control of the abundance of nuclear factors in a clinical context might be provided by the microRNA (miRNA) technology.
The small non-coding miRNAs function by binding to the 3’UTR of their target mRNAs and inhibit protein translation either by interfering with the mRNA stability and/or by blocking protein translation (16). MiRNAs are key regulators of various biological processes including bone homeostasis and bone pathologies (14, 15, 17), cancer progression and metastasis (18, 19). Therefore in vivo delivery of miRNAs or miRNA antagonists provides an attractive therapeutic tool to reverse bone tissue degeneration (16), or to prevent cancer-induced bone diseases (20). Very recently, miRNAs targeting osteoclast function have been shown to reduce bone metastatic disease (21, 22). Thus, increasing evidence suggests that miRNAs can be used as therapeutic targets, supporting the concept that the identification of miRNA-based mechanisms to repress Runx2 may provide a novel approach for the treatment of metastatic bone disease.
Here, we show that the diminished expression of specific miRNAs contributes to the elevation of Runx2 in bone metastatic breast cancer disease. Reconstituting highly metastatic MDA-MB-231 breast cancer cells with miR-135 and miR-203 by delivering synthetic miRNA mimics to the mammary fat pad in mice, led to an impaired tumor growth and metastasis in vivo. We further demonstrate that ectopic expression of miR-135 and miR-203 in metastatic cells suppressed both tumor growth in the bone environment and the development of metastatic lesions through direct downregulation of Runx2. In vitro studies revealed a suppressed tumor cell properties through multiple mechanisms, including downregulation of Runx2 target genes, along with pathway co-regulatory factors known to mediate metastasis. Importantly, our data provide compelling evidence that targeting Runx2 by a miRNA-based approach using synthetic miRNA mimics, can be used to reduce metastatic disease progression.
Materials and Methods
Tissue samples
Tissue biopsies derived from primary tumors and bone metastases of breast cancer patients were obtained from the archives of the University Medical Center Hamburg-Eppendorf, Germany, following institutional guidelines. Tissue samples were evaluated independently by two expert pathologists. All studies using human samples were carried out in accordance with the declaration of Helsinki and in agreement with the institutional regulations.
Immunohistochemistry
Human tissue biopsies, mouse bones, and lungs were fixed in 4% Formalin/PBS. Bones were decalcified in 4% Na-EDTA solution at pH 7.4 for two weeks. Tissues were dehydrated, embedded in paraffin and cut. Consecutive 4 μm thick sections were analyzed by immunohistochemistry using antibodies against Runx2 (MBL), Ki-67 (Dako), and HLA Class 1 ABC (Abcam), Pan-Cytokeratin (Abcam), and Smad-5 (Cell Signaling) with positive and negative controls following established protocols (23). Antigen retrieval was performed using citrate buffer at pH 6.0. Vectastain (Vector Laboratories) and DAB+ (Dako) systems were used for detection.
Cell culture
The human mammary epithelial cell line (MCF-10A) and the breast cancer cell lines MCF-7 and MDA-MB-231-a (hereafter MDA-MB-231) were purchased from ATCC. The MDA-MB-231-b subclone was kindly provided by Dr. Theresa Guise (24). MCF-10A cells were cultured in MEGM medium (Lonza) supplemented with 100 ng/ml cholera toxin. MCF-7 cells were cultured in D-MEM high Glucose (Lonza) supplemented with 10% Fetal Bovine Serum (FBS, Atlanta) and 1% Penicillin/Streptomycin (Gibco). MDA-MB-231 cells were maintained in alpha-MEM (Lonza), 10% FBS and 1% Penicillin/Streptomycin. Both cell lines had similar responses to miRNA mimics and were validated at the Vermont Cancer Center DNA Analysis Facility by STR DNA fingerprinting using the Promega GenePrint® 10 System according to manufacturer's instructions (Promega #B9510). The STR profiles were compared to known ATCC fingerprints (ATCC.org), and to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima) (25). The STR profiles of all cell lines matched (>85%) known DNA fingerprints. To collect conditioned medium (CM), MDA-MB-231 cells were seeded at 80% confluence in complete medium. Cells were serum starved for 24 h in 2% FBS prior collection of the CM.
Transfections
Cells were plated in 6-well plates and transfected at 70-80% confluence with miRVana miRNA mimics (Ambion mirVana™ miRNA Mimic, Negative Control #1, miR-135, miR-203) and inhibitors (mirVana™ miRNA Inhibitor, Negative Control #1, miR-135, miR-203) at a final concentration of 50 nM in OPTI-MEM using Oligofectamine Reagent (Invitrogen) according to manufacturer’s instructions. Fresh culture medium containing 2x FBS was added after 4 h. Cells were used for functional studies or harvested for RNA and protein analyses as described. Adenoviruses expressing Runx2 and the control vector have been reported previously (10). The pCMV5 empty vector and pCMV5-Smad5 plasmids were purchased from Addgene and transfected with lipofectamine according to manufacturer’s instructions.
For the 3’ UTR assays, human distal, proximal, and full-length Runx2 3’UTR and Smad5 3’UTR were cloned into a pMIR-Reporter Luciferase Plasmid (Applied Biosystems) to obtain Luc-Runx2-3’UTR and Luc-Smad5-3’UTR reporter plasmids. Cells were transfected with miRNA control or miRNA mimic oligonucleotides using oligofectamine as described above. On the following day, cells were transfected with 500 ng of 3’UTR plasmids each along with 50 ng Renilla luciferase plasmid (Promega) using Xtremegene9 (Roche) following manufacturer’s instructions. Luciferase assays were performed using the Dual Luciferase Reporter Gene Assay System (Promega) according to instructions provided by the manufacturer.
Total RNA extraction and gene expression analysis
Total RNA was isolated using Trizol reagent (Invitrogen) and purified with Zymo RNA purification kit (Zymo) according to manufacturer’s instructions. cDNA was synthesized from 1 μg of total RNA using Supercript RT kit (Invitrogen) following the protocol provided by manufacturer. Quantitative real-time PCR (qRT-PCR) was performed with the 7300 sequence detection system (Applied Biosystems/Roche) using SYBR® Green Master Mix (Applied Biosystems). After normalization to Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA, relative expression levels and fold induction of each target gene were calculated using the comparative CT (ΔΔCT) method.
Real-time PCR gene array
RNA was extracted and purified from MDA-MB-231-luc cells transfected with miR-C, miR-135 or miR-203 as described above. cDNA was synthesized using the RT2 First Strand Kit (SABiosciences). Real-time PCR was then performed by the Human Tumor Metastasis RT2 Profiler PCR array (SABiosciences) using the SABiosciences' SYBR® Green qPCR Mastermix, and analyzed according to manufacturer’s instructions.
MiRNA extraction and expression analysis
Total RNA was isolated from cells and purified as described above. RNA extraction from paraffin embedded tissue samples was performed using the High Pure FFPE RNA Micro-kit (Roche) according to manufacturer’s instructions (23). The QuantimiR-kit (SBI System Biosciences) was used to add a polyA tail to the small RNAs for cDNA synthesis (SBI System Biosciences) according to manufacturer’s guidelines. Relative miRNA expression was determined by SYBR Green detection (Applied Biosystems) using a universal reverse primer and a specific forward primer designed for each miRNA of interest (26). U6 expression was used as internal control. The relative miRNA expression was calculated using the ΔΔCT method.
Western blot analysis
Cells were lysed in RIPA buffer (Boston Bioproducts) supplemented with protease inhibitors (Complete, EDTA-free, Roche Diagnostics) and MG132 (Calbiochem). Lysates were separated on a 12% acrylamide gel and subjected to western blot analysis. Immunoblots were incubated over night at 4°C with the following primary antibodies: anti-RUNX2 (mouse monoclonal, MBL 1:2000), anti-Smad5 (Rabbit polyclonal, Cell Signaling Technologies, 1:1000), anti-p-Smad5 (Rabbit polyclonal, Cell Signaling Technologies, 1:1000). Anti-Tubulin (mouse monoclonal, Sigma, 1:5000) and anti-Actin antibodies (goat polyclonal, Santa Cruz Biotechnology, 1:2000) were used as loading controls. Peroxidase-labeled anti-mouse or anti-goat secondary antibodies (Santa Cruz, 1:10 000) were used to visualize bands using the enhanced chemiluminescence (ECL) kit (PerkinElmer) on BioMax film (Kodak).
Proliferation assay
Cell proliferation was determined by the CellTiter MTS assay (Promega). Cells were seeded in 96-well plates at a density of 3000 cells/well before performing the MTS assay according to manufacturer’s instructions.
Migration assays
For performing the wound-healing assay, MCF-7 and MDA-MB-231 cells were plated in 6-well plates, transfected with miRNA mimics or inhibitors as described and grown to 90-100% confluence. A wound was then applied with a pipette tip and closure of the wound was monitored by live cell imaging or by obtaining images at indicated time points using an Improvision LiveCell Spinning Disk microscope (Perkin Elmer). Migration velocity, track length, and displacement index were quantified using the Volocity6 program (Perkin Elmer).
A trans-well system was used for chemotactic migration assays. Cells were transfected as described, enzymatically released and re-plated in a migration chamber (BD Biosciences) in serum-free medium. Cells were allowed to migrate through 8 μM pores towards medium containing 10% FBS for the indicated period of time. Cells that had migrated through the membrane were stained with the Hemacolor-kit (Merck Millipore) and counted.
Osteoclast differentiation assay
Bone marrow macrophages (BMM) were obtained from wild type mice by flushing the bone marrow, collecting the non-adherent cells after 4 h in culture and expanding the non-adherent cells in the presence of MCS-F (Peprotek 100 ng/ml) for 3 days. Osteoclast differentiation was stimulated by supplementing the CM with 25 ng MCS-F and 50 ng/ml RANK-L (Peprotek). Cultures were terminated by day 4 and stained for Tartrate-resistant acid phosphatase (TRAP) activity. TRAP positive cells with a minimum of three nuclei were considered as osteoclasts and counted using the Osteomeasure system (Osteometrics).
Orthotopic cancer cell transplantation and intratumoral miRNA delivery to mammary tumor
All animal studies were conducted in accordance with approved Institutional Animal Care and Use Committee protocols following the ethical guidelines of the NIH Guide for the Care and Use of Laboratory Animals. Severe combined immunodeficient (SCID) mice were obtained from the Jackson Laboratory.
MDA-MB-231 cells stably expressing luciferase (MDA-MB-231-luc) were transfected with miR-C, miR-135, or miR-203 oligonucleotides as described. After 24 h, cells were counted and suspended in matrigel (500,000 cells/50 μl). Female SCID mice (six-weeks old) were anesthetized by intraperitoneal (i.p.) injection of Ketamine/Xylazine (Ketamine: 100 mg/kg body weight; Xylazine: 10mg/kg body weight). The cell/matrigel suspension (50 μl) was injected into the mammary fat pad using a small syringe. After two weeks, miRNA mimics or miR-C (0.5 nmol/mouse) were complexed with 50 μl volume RNALancerII in vivo delivery reagent (BIOO Scientific) and injected into the tumor.
Intratibial injection of miRNA-transfected breast cancer cells
Six week-old female NOD-SCID mice were used for intratibial injections. Prior to injection, mice were anesthetized, the knee of the right hind limb was flexed and a short incision was made over the knee. A C-arm fluoroscope was used to visualize the needle entry into the tibia to allow a precise injection into the metaphysis. A 30-gauge needle on a tuberculin syringe was then inserted into the intramedullary space of the tibia, followed by the injection of 100,000 MDA-MB-231-luc cells transfected with either miR-C, miR-135, or miR-203 and suspended in 100 μl of sterile PBS.
Monitoring tumor growth and osteolysis
Bioluminescence imaging (Xenogen) was used to follow the tumor growth at the orthotopic transplantation site and in the bone. Mice were anesthetized using isoflurane and D-luciferin was injected (i.p.) at a dose of 150 mg/kg body weight. The animals were placed on the warm (37°C) IVIS imaging stage and bioluminescent images were acquired for each mouse using the IVIS Imaging System. Analyses were performed using the LivingImage software (Xenogen) by measuring the photon flux within a region of interest drawn around the bioluminescence signals. Blank regions of interest were also measured for each scan and substracted from each tumor photon flux for normalization.
Statistical analysis
Normal distribution of the data was tested using the D'Agostino-Pearson omnibus test. Comparisons between groups were analyzed using one-way ANOVA with Tukey’s post-hoc tests.
Results
Reciprocal expression between Runx2 and miRNAs targeting Runx2 in breast cancer
Runx2 promotes metastatic bone disease in mouse models of breast cancer (7), but its activity is attenuated by miRNAs, as evidenced by studies using mesenchymal cells (26). To determine the relationship between Runx2 expression and miRNAs in human breast cancer metastasis, we first examined the presence of Runx2 in bone metastases of breast cancer patients using immunohistochemistry, and determined a robust abundance of Runx2 in the nuclei of cytokeratin and SMAD5 positive metastatic breast cancer cells (Fig. 1A) but not in the primary breast tumors (Supplementary Fig. S1). Furthermore, Runx2 is highly expressed in metastatic MDA-MB-231-luc breast cancer cells that grow aggressively in bone, yet Runx2 abundance is comparatively modest in the normal epithelial (MCF-10A) and non-metastatic MCF-7 breast cancer cell lines (Fig. 1B). However, despite the strikingly increased expression of Runx2 protein in MDA-MB-231-luc cells compared to MCF-7 cells, the difference in Runx2 mRNA expression was only two-fold between both cell lines (Supplementary Fig. S2A), suggesting that Runx2 protein is regulated through both transcription-dependent and alternate mechanisms, such as control of protein translation.
Figure 1.
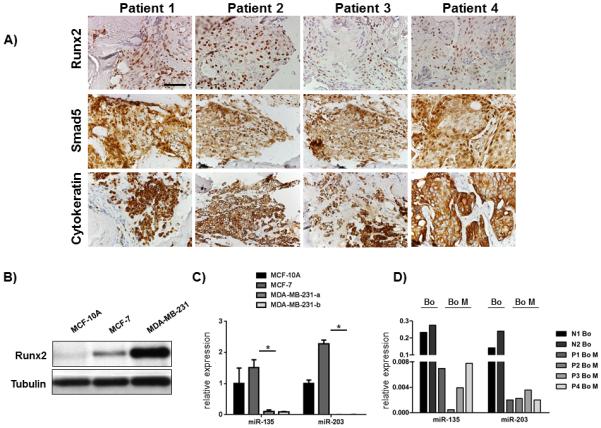
Runx2 expression in bone metastases from breast cancer patients inversely correlates with Runx2 targeting miRNAs. A, Runx2, Smad5 and pan-cytokeratin proteins were detected by immunohistochemistry in four representative tissue biopsies obtained from bone metastases of breast cancer patients and B, protein by western blot analysis in cell lysates from normal epithelial MCF-10A cells, non-metastatic MCF-7 cells, and metastatic MDA-MB-231 cell lines. C, expression of miRNAs that target Runx2 was analyzed in MCF-10A cells, MCF-7 cells, and in two highly metastatic MDA-MB-231 cell lines by qRT-PCR. U6 expression was used as internal control. D, miRNAs were isolated from paraffin-embedded tissue specimens, reverse transcribed, and expression of miR-135 and miR-203 was quantified by qRT-PCR. U6 was used as an internal control. Scale bar 50 μm. Data are represented as means ± SD of each group. *significant difference (p < 0.05), Tukey’s post-hoc test.
At least 12 different miRNAs have been documented to regulate Runx2 (26). To determine which specific miRNAs contribute to the elevation of Runx2 protein in the context of breast cancer, we investigated the expression profile of miRNAs that regulate Runx2 in osteoblasts. We found that elevated Runx2 in metastatic cells inversely correlated with the expression of most of the Runx2 targeting miRNAs (Fig. 1C, Supplementary Fig. S2B). Among the 12 miRNAs whose expression was quantified, eight miRNAs (miR-23a, miR-133a, miR-135, miR-203, miR-204, miR-205, miR-217, and miR-338) were downregulated in metastatic cells compared to non-metastatic breast cancer cells. Additionally, miR-30c and miR-137 were expressed at a lower level in metastatic cells in comparison to normal epithelial cells. Notably, expression of miR-135 and miR-203 was depleted (Fig. 1C) in MDA-MB-231-luc cells, as well as in bone metastases of breast cancer patients compared to healthy bone (Fig. 1D, Supplementary Fig. S2C). Collectively, these data demonstrate that Runx2 is maintained at physiological levels by specific miRNAs, and that the absence of these miRNAs may contribute to the elevation of Runx2 protein in bone metastases.
miR-135 and miR-203 markedly suppress breast cancer cell migration and inhibit cancer cell proliferation in vitro
Based on the relatively high abundance of miR-135 and miR-203 in non-metastatic breast cancer cells, and a strong downregulation in metastatic cells and tissue biopsies (Fig. 1), we postulated that miR-135 and miR-203 may influence tumorigenic properties of breast cancer cells. To test this hypothesis, we ectopically expressed miR mimics or inhibitors as well as corresponding non-targeting control miRNAs (miR-C, antimiR-C) in breast cancer cell lines and examined their oncogenic properties. Reconstitution of MDA-MB-231-luc cells with miR-135 and miR-203 markedly suppressed breast cancer cell migration in a wound-healing assay as determined by a decreased track length, displacement, and migration velocity using live cell imaging (Fig. 2A). Consistent with these findings, inhibition of the endogenous miR-135 and miR-203 in MCF-7 cells augmented the migratory capacity of these cells (Fig. 2B). Similarly, the chemotactic migration was significantly suppressed in a trans-well system by exogenous miR-135 and miR-203 expression (Fig. 2C, Supplementary Fig. S3). Consistent with our observations that miR-135 and miR-203 regulate cell migration, gene expression analysis revealed the downregulation of multiple genes involved in cell motility and adhesion, including ROCK1, CD44, and PTK2 in miR-135 and miR-203 administrated cells compared to control (Fig. 2D). Moreover, delivery of miR-135 and miR-203 resulted in an inhibition of cell viability of MDA-MB-231-luc cells in vitro as measured by an MTS assay (Fig. 2E) and as demonstrated by an expression analysis of genes related to cell proliferation such as CCL7 and CXCL12 (Supplementary Fig. S4).
Figure 2.
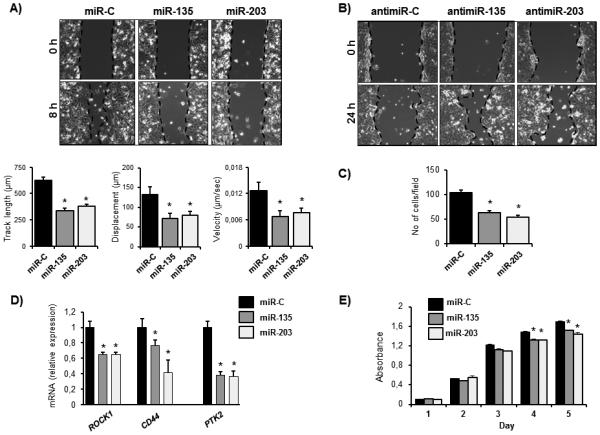
miR-135 and miR-203 suppress breast cancer cell migration and proliferation in vitro. A, MDA-MB-231-luc cells were transfected with miR-135 and miR-203 mimics or scrambled negative control miRNA (miR-C) and cell migration in wound healing assays was evaluated by live cell imaging. Migration velocity, track length, and displacement index were calculated using a velocity program, B, miRNA inhibitors (antimiR-135 and antimiR-203) and respective control antimiRNA were transfected in MCF-7 cells. Migration was evaluated in a wound-healing assay. C, miR-135, miR-203, or miR-C expressing MDA-MB-231-luc cells were subjected to a trans-well assay to evaluate chemotactic migration. D, gene expression was evaluated in miR-C miR-135 and miR-203 expressing MDA-MB-231-luc cells by qRT-PCR. Values were normalized to GAPDH. E, cell proliferation of MDA-MB-231-luc cells ectopically expressing miR-135, miR-203, or miR-C was investigated by an MTS assay. Data are represented as means ± SEM A) or ± SD (C, D, E) of each group. *significant difference compared to miR-C control (p< 0.05), Tukey’s post-hoc test.
miR-135 and miR-203 reduce tumor growth and inhibit spontaneous metastasis
To recapitulate the in vitro findings and to examine the role of these miRNAs in tumorigenesis in vivo, miR-135 and miR-203 mimics along with a control miRNA (miR-C) were delivered in MDA-MB-231 cells stably expressing the luciferase gene (MDA-MB-231-Luc) and subsequently implanted orthotopically into the mammary fat pad of six-weeks old female immune-deficient SCID mice (Fig. 3). Additional intratumoral injection of synthetic miRNA oligos was performed one week after implantation and miRNA level in tumors was evaluated two weeks after the initial transplantation to assure a successful delivery (Supplementary Fig. S5). Ectopic expression of miR-135 and miR-203 significantly reduced the primary tumor size as determined by bioluminescence imaging (Fig. 3A). Since miR-135 and miR-203 decreased the migration capacity of the cells in vitro, we hypothesized that they may also inhibit metastasis in vivo. Indeed, delivery of miR-135 and miR-203 followed by a direct intratumoral injection of synthetic miRNA-mimic molecules suppressed spontaneous metastasis to bone seven weeks after orthotopic transplantation (Fig. 3B). Importantly, metastasis to the lung was also reduced in the presence of ectopic miR-135 and miR-203 (Fig. 3C).
Figure 3.
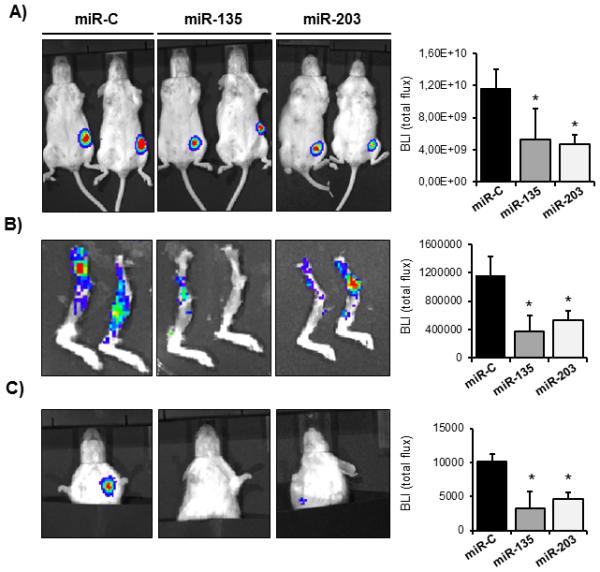
miR-135 and miR-203 impair tumor growth and spontaneous metastasis to bone and lung. A, tumor growth in the mammary fat pad in vivo was determined by quantifying the bioluminescence signal after transplanting MDA-MB-231-luc cells ectopically expressing miR-135 and miR-203, followed by intratumoral injection of synthetic miRNAs. Tumor growth was quantified by the bioluminescence signal using the LivingImage software. B, spontaneous metastasis from the orthotopic site to bones and C, to the lung was visualized by bioluminescence. Ectopic expression of miR-135 and miR-203 significantly impaired spontaneous metastasis. Bioluminescence intensity was quantified using the LivingImage software and data are represented as means ± SD of each group. *significant difference (p< 0.05) between indicated miRNA and miR-C, Tukey’s post-hoc test. Representative mice from n= 6-12 in each group are shown.
We then evaluated the possible role of miRNAs in regulating tumor growth in bone and subsequent development of osteolytic lesions by injecting the miRNA reconstituted breast cancer cells directly into the tibiae of six weeks old female mice. As expected, cells transfected with control miRNAs grew aggressively in the bone and developed osteolytic lesions within four weeks (Fig. 4A, B). Although cells reconstituted with miR-135 and miR-203 were able to grow in the bone, the tumor sizes were significantly reduced compared to control mice (Fig. 4A, right panel). Notably, miR-135 and miR-203 protected against breast cancer-induced osteolytic bone disease as evaluated by X-ray, μCT, and histological analyses of TRAP-positive osteoclasts in the tumor-bone interface (Fig. 4B, C). This observation was confirmed by the ~40 % reduced number of actively proliferating Ki-67-positive cells in bone metastases in the presence of miR-135 and miR-203 (Fig. 4D). Of further potential clinical relevance, miR-135 and miR-203 also reduced secondary metastasis to the lung, demonstrated by a significantly decreased number of HLA-positive metastatic nodules and a decreased proliferation of metastatic cells (Fig. 4E).
Figure 4.
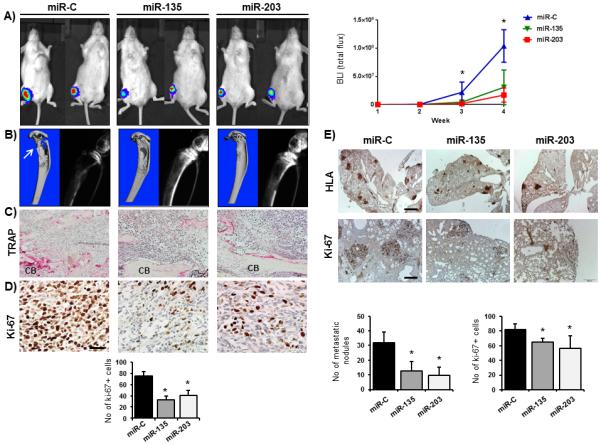
miR-135 and miR-203 suppress the tumor-induced osteolytic lesions. A, MDA-MB-231-luc cells were transfected with synthetic miR-C, miR-135 and miR-203 oligonucleotides prior to injection of the cells into the tibiae as a model of osteolytic bone disease. Tumor growth was quantified by the bioluminescence signal intensity using the LivingImage software. B, bone resorption was visualized by μCT imaging (left) and radiographs (right). C, TRAP stain for osteoclast activity of tumor-bearing tibiae at the tumor-cortical bone (CB) interface. Scale bar 100 μm. D, tumor-bearing bones were stained with Ki-67 to visualize and quantify proliferating tumor cells. Scale bar 50 μm. E, lung metastases were analyzed by HLA and Ki-67 immunohistochemistry after ectopic expression of miR-C, miR-135 and miR-203. Scale bar 200 μm (upper panel) and 1 mm (lower panel). *significant difference (p<0.05) between indicated miRNA and miR-C, Tukey’s post-hoc test. Representative mice from n= 6-12 in each group are shown.
miR-135 and miR-203 suppress multiple Runx2 target genes related to osteolysis
We next sought to determine the underlying molecular mechanisms by which miR-135 and miR-203 mediate their effect on tumor progression and bone metastasis. To identify the targets of miR-135 and miR-203, we utilized the in silico target prediction tools Target Scan, Pictar and miR Walk, which allows a comparison between multiple target prediction algorithms. We first evaluated the activity of the miR-135 and miR-203 on the Runx2 3’UTR in the context of human breast cancer. The human RUNX2 3’UTR contains three putative binding sites for miR-135, which are all located in the proximal 3’UTR. However, the putative miR-203 binding site is located in the distal 3’UTR (Supplementary Fig. S6). Consistently, ectopic miR-135 suppressed both the proximal and the full-length 3’UTR activity, while having no effect on the distal 3’UTR activity. In contrast, miR-203 significantly inhibited the distal and the full-length RUNX2 3’UTR activity (Fig. 5A). The net result for both miRNAs is that administration of exogenous miR-135 and miR-203 decreased Runx2 protein levels, suggesting a functional role in controlling protein translation (Fig. 5B). In addition to the reporter assay and corroborating the in vitro findings, Runx2 expression was significantly reduced in tumors obtained from the miR-135 and miR-203-injected mice compared to controls (Fig. 5C). Similarly, while in the control mice Runx2 protein was detected in tumors at the metastatic sites, its expression was greatly diminished in the miR-135 and miR-203 administrated tumors in both the lung and in the bone (Fig. 5D). As a consequence of the impaired Runx2 expression, several Runx2 target genes implicated in metastasis, including IL-11, MMP-13, and PTHrP were downregulated in the presence of miR-135 and miR-203 compared to the miR-C-treated cells (Fig. 5E, upper panel). A similar expression pattern was found in the primary tumors (Fig. 5E, lower panel), further supporting the notion that the effect of these miRNAs in tumorigenesis and metastasis is mediated through the inhibition of Runx2 and its metastasis-promoting downstream targets.
Figure 5.
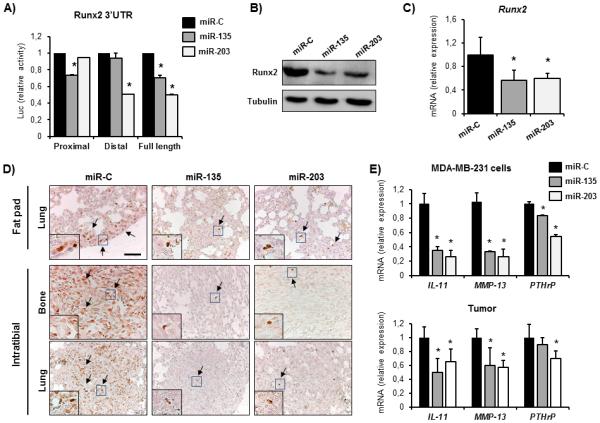
miR-135 and miR-203 directly target Runx2 in metastatic breast cancer cells. A, Luc-Runx2-3’UTR reporter functional assay. MiR-C, miR-135 and miR-203 were delivered in MDA-MB-231-luc cells and Firefly Luciferase activity was normalized to co-transfected Renilla luciferase and presented as fold change to miR-C. B, endogenous Runx2 protein was detected in miR-C, miR-135- and miR-203 transfected MDA-MB-231-luc cells by western blot analysis. C, Runx2 mRNA expression in primary tumors delivered miR-C, miR-135 and miR-203 was quantified by qRT-PCR. D, Runx2 protein in lung metastases after orthotopic (top) or intratibial tumor transplantation (lower) was analyzed by immunohistochemistry. E, expression of Runx2 target genes mediating osteolysis including IL-11, MMP-13 and PTHrP was analyzed in miR-C, miR-135 and miR-203 transfected cells (top) and in orthotopically transplanted mammary tumors (lower) by qRT-PCR. GAPDH was used as control. *significant difference (p < 0.05) between indicated miRNA and miR-C, Tukey’s post-hoc test. Scale bars 50 μm.
miR-135 functions through the BMP-Runx2-axis
We demonstrated that miR-135 induces a significant decrease of Runx2 protein yet only a 35% decrease in RUNX2 3’UTR activity. Thus, we postulated that miR-135 might exert its action through additional mechanisms upstream of Runx2. Notably, miR-135 is predicted to target several components of the BMP-signaling pathway (receptors BMPIR, BMPIIR, and downstream mediators SMAD5 and SMAD4). Since BMP-signaling is known to activate Runx2 and reported to facilitate breast cancer cell migration and metastasis to bone (5), we determined the effect of miR-135 on the BMP signaling activity. First, we evaluated the SMAD5 expression in a clinical context and detected high levels of SMAD5 in bone metastases biopsies obtained from breast cancer patients while the expression was only mildly detected in primary tumors (Fig. 1A, Supplementary Fig. S1). Western blot analysis and SMAD5 3’UTR -reporter assays confirmed that miR-135 directly targets Smad5 (Fig. 6A, B). Consistently, we found a significantly reduced expression of the BMP-2 downstream target gene Id2 in the miR-135 expressing tumors compared to the controls (Fig. 6C). Consequently, exogenous miR-135 impaired the BMP-2-induced phosphorylation of Smad 1,5,8 and the expression of the BMP target gene Id2 in vitro (Fig. 6D, E). The BMP-Runx2 axis has been shown to support osteoclast differentiation and to favor the development of osteolytic disease over osteosclerotic lesions in prostate cancer (27, 28). To investigate whether the BMP-Runx2 pathway has a similar function also in breast cancer, we overexpressed Smad5 in MDA-MB-231-luc cells, which were transfected with miR-C or miR-135 and used the CM to determine osteoclast differentiation (Fig. 6F). Interestingly, CM from MDA-MB-231-luc cells delivered with miR-135 decreased the number of multinucleated osteoclasts, which was partially rescued by overexpression of Smad5. These data suggest that miR-135 reduces the tumorigenic and osteolytic properties at least in part via regulation of the BMP pathway and by subsequent reduction of Runx2 expression.
Figure 6.
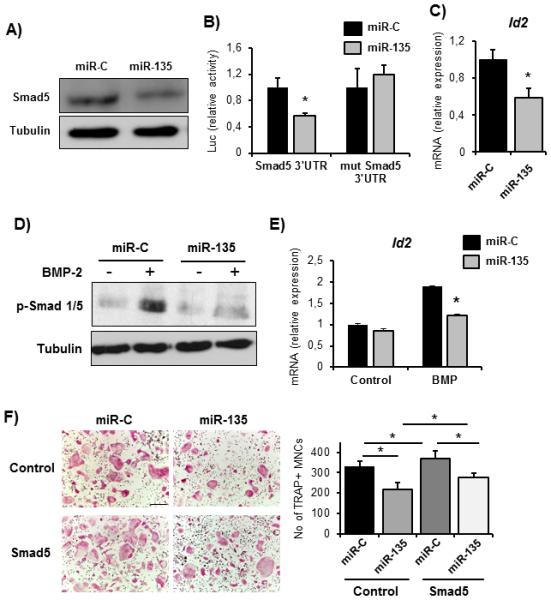
miR-135 targets the BMP-pathway. A, direct targeting of Smad5 by miR-135 in MDA-MB-231-luc cells was confirmed by Smad5 western blot analysis and B, Luc-Smad5-3’UTR reporter assay. C, expression of the BMP target gene Id2 was analyzed in miR-C and miR-135 expressing tumors by qRT-PCR. D, BMP-signaling in MDA-MB-231-luc cells was stimulated by BMP-2 treatment and pathway activity was determined by phospho-Smad1/5/8 immunoblotting and by E, Id2 mRNA expression analysis. F, osteoclast differentiation was stimulated with conditioned medium (CM) collected from MDA-MB-231-luc cells transfected with miR-C or miR-135 and control vector or Smad5. TRAP-positive multinucleated cells (MNCs) were counted using the Osteomeasure system. *significant difference (p < 0.05), Tukey’s post-hoc test. Scale bar 200 μm.
Reconstitution of Runx2 restores miRNA-mediated suppression of tumorigenesis and metastasis
To test if miR-135 and miR-203 indeed exert their effects through Runx2, we reconstituted Runx2 in MDA-MB-231-luc cells miR-135 and miR-203 and evaluated their tumorigenic properties both in vitro and in vivo. As previously reported for prostate cancer cells (28), overexpression of Runx2 promoted migration of MDA-MB-231-luc cells (Fig. 7A). Importantly, reconstitution with Runx2 restored the migration capacity of cells delivered with miR-135 and miR-203. In order to determine the in vivo relevance of the miR-Runx2 axis we performed orthotopic transplantations MDA-MB-231-luc cells delivered with miR-135 and miR-203 cells with and without overexpression of Runx2. Remarkably, reconstitution of breast cancer cells with Runx2 rescued the miRNA-mediated reduction of tumor growth and spontaneous metastasis to the bone and the lung (Fig. 7B, Supplementary Fig. S7). These data suggest that miR-135 and miR-203 inhibit cell migration in vitro as well as tumor growth and metastasis in vivo at least in part through reducing the abundance of Runx2.
Figure 7.
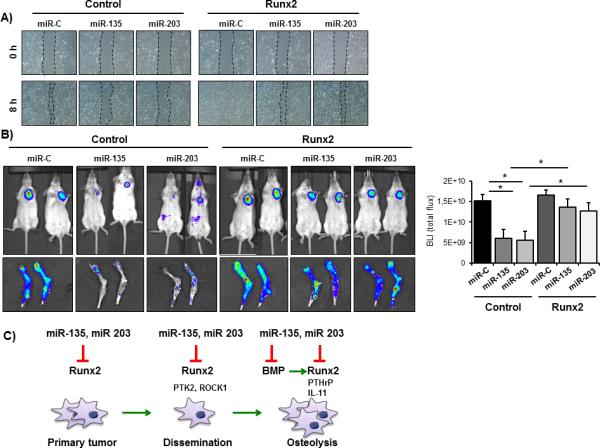
Reconstitution of breast cancer cells with Runx2 restores miRNA-mediated suppression of tumor growth and metastasis. A, MDA-MB-231-luc cells expressing control vector or Runx2 were transfected with miR-C, miR-135 or miR-203 and transplanted orthotopically into SCID mice. Tumor growth was evaluated by bioluminescence and quantified using the LivingImage Software. B, spontaneous metastasis to bone was examined by bioluminescence after sacrificing the mice. C, model for miRNA-mediated inhibition of Runx2 and its downstream effectors in the progression of breast cancer metastasis. *significant difference (p < 0.05) between indicated miRNA and miR-C, Tukey’s post-hoc test.
In summary, our findings demonstrate the effectiveness of targeting the abnormally and highly expressed transcription factor Runx2 by delivery of miRNA mimics to rescue the loss of anti-metastatic miRNAs. Both specific miRNAs examined were effective in downregulating Runx2 and its target genes as well as additional signaling pathways that reduce tumor growth, cell migration and metastatic bone disease as presented in a schematic (Fig. 7C).
Discussion
In this study we identified miR-135 and miR-203 as suppressors of breast cancer metastasis and osteolytic bone disease in vivo through direct targeting of the bone-related transcription factor Runx2. Expression analyses revealed a downregulation of miR-135 and miR-203 in bone metastases that have a high level of Runx2, revealing that the pathophysiological increase in Runx2 protein is due to the lack of these specific miRNAs that are missing in breast cancer metastases. Indeed, reconstituting miR-135 and miR-203 in metastatic cells impaired their oncogenic properties in vitro and more importantly it impaired tumor growth at the orthotopic site in vivo. Furthermore, miRNA replacement with synthetic oligonucleotides suppressed both spontaneous metastasis to bone and tumor growth in the bone environment. These data strongly suggest that miRNA delivery is a viable therapeutic strategy to disable the metastatic disease progression (Fig. 7C).
The concept that miRNAs are linked to the biological activities of Runx2 in different cancer types is corroborated by our recent finding that the activity of Runx2 in osteosarcoma is directly linked to loss of miR-34, another key Runx2-targeting miRNA (23). In addition, miR-203 has been identified as a regulator of prostate cancer progression and metastasis through inhibition of Runx2, strongly supporting our current findings in breast cancer progression (29, 30). In breast cancer, both Runx2-targeting miRNAs miR-135 and miR-203 suppress primary tumor growth and impair the events leading to the osteolytic disease, reflecting the oncogenic function of Runx2 in the initiation of metastasis, disease progression, and bone destruction. With regard to the early events in breast cancer progression, si-RNA-mediated silencing of Runx2 has been shown to inhibit tumor growth in vivo (10), whereas Runx2 overexpression induces epithelial-mesenchymal transition (EMT) (31). Consistent with these properties, miR-203 inhibits EMT (32-34) and may do so through knockdown of Runx2. At later stages, Runx2 promotes cell motility and invasion in vitro (13), tumor growth in the bone environment, and formation of osteolytic lesions in vivo (9). Consequently, depletion of Runx2 protects from the vicious cycle of bone metastasis, demonstrating the feasibility of developing Runx2 as a therapeutic target to prevent the late metastatic event in primary tumors and subsequent osteolytic bone disease. However, since Runx2 is a nuclear factor, translating these findings into clinical applications is challenging. Here, we propose a miRNA-mediated attenuation of Runx2 as a novel therapeutic approach to prevent the devastating bone metastatic disease in breast cancer (31).
A wealth of studies has contributed to the elucidation of miRNAs involved in primary breast cancer growth, metastasis initiation, and progression. However, the role of miRNAs in bone metastasis has just begun to emerge (19). Our xenograft models provide evidence that a miRNA-based therapy can be used to disrupt the metastatic cascade by targeting key transcription factors, which are difficult to target by conventional pharmacological approaches. Osteolytic bone disease is driven by accelerated osteoclast activity, which eventually destroys the bone. Current therapies to treat bone metastases and to reduce tumor burden in the bone environment are largely based on anti-resorptives, which inhibit the osteoclast activity via antibody-based blocking of osteoblast-derived RANKL (i.e., Denosumab) or by inducing osteoclast apoptosis using bisphosphonates. Very recently, Ell et al. reported a novel strategy to impair the osteolytic bone disease by miRNA-mediated targeting of osteoclasts (21). This approach elegantly demonstrates the remarkable potential of miRNAs in the therapeutic interventions for bone pathologies. Here, we propose to target the tumor cells, which induce both osteoblast-dependent and independent activation of osteoclast differentiation and function and subsequent resorption of the bone matrix. It can be considered that the role of miR-135 and miR-203 in the control of breast cancer progression and osteolytic disease is partially due to decreased cancer cell proliferation and tumor growth in either the primary or a secondary site. This finding would reduce the risk of potential metastasis and tumor growth, which is still a beneficial effect of the miRNA targeting tumors to inhibit disease progression. However, the authors believe there is a direct role for miR-135 and miR-203 in reducing the metastatic potential of breast cancer cells. This is evidenced by marked downregulation of several metastasis-associated genes (including PTK2 and IL-11) as well as decreased migration capacity in the presence of miR-135 and miR-203, shown by our own and previously published results (29,30). Furthermore, the decreased osteolytic capacity is mediated through downregulation of the bone-resorbing secreted factor PTHrP (intrinsic to metastatic breast cancer cells) and with subsequent impairment of osteoclast activity. In conclusion, our experimental approaches demonstrate that miRNAs can be used to therapeutically target breast cancer at an early disease stage, in addition to what is generally considered the incurable bone metastatic disease at later stages.
Together, our findings open new avenues for the development of translational applications of miRNAs in bone pathological conditions, including metastatic bone disease. Currently, miRNA signatures are used as prognostic, diagnostic, and predictive miRNA biomarkers. Furthermore, miRNA replacement therapy aimed at restoring the abundance of a miRNA that is expressed at a low level is currently being developed with miR-34, which already entered Phase I clinical trials (34). The ability of a single miRNA to target a broad range of molecular regulators in a context-dependent manner is both an advantage and a restriction of the miRNA replacement therapy. While targeting multiple genes or signaling pathways simultaneously can be beneficial, it may also increase the risk of adverse effects for instance by suppressing signaling networks that maintain tissue homeostasis. Strategies might be developed to improve the targeted delivery of miRNAs for instance by modifying the oligonucleotide chemistry. Thus, although given systemically, miRNAs could target the cancer cells specifically in any organ tissue. In the future, miRNA therapeutics might enter the clinics to provide novel opportunities to target key molecules, including transcription factors that drive the disease progression.
Supplementary Material
Acknowledgements
We thank all members of our laboratories, especially Ying Zhang, Ronglin Xie, Jason Dobson, Jitesh Pratap, Terri Messier, Shirwin Pockwinse and Saskia Schröder for assistance with experimentation, critical comments, technical advice, sharing of reagents, and/or general support. We thank the team of the Microscopy Imaging Facility at the University Medical Center Hamburg-Eppendorf (Umif) for expert advice. The MDA-MB-231-b subclone was kindly provided by Dr. Theresa Guise (Indiana University School of Medicine). Dr. Matthias Priemel (University Medical Center Hamburg-Eppendorf) contributed to the acquisition of the surgical specimen. We are grateful to the Vermont Cancer Center DNA Analysis Facility for data pertaining to cell line authentication.
Grant Support
This work was supported, in whole or in part, by grants of the National Institutes of Health NCI P01 CA082834 (GSS, JBL), R01 AR039588 (GSS, JLS, JBL), R37 DE012528 (JBL, JLS), R01 AR049069 (AJvW), and the Deutsche Forschungsgemeinschaft HE 5208/2-1 (EH), and by post-doctoral fellowships from EMBO and Alexander von Humboldt Foundation (HT). The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
The authors have no conflicts of interest.
References
- 1.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411–25. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–64. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 3.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–93. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 4.Juarez P, Guise TA. TGF-beta in cancer and bone: implications for treatment of bone metastases. Bone. 2011;48:23–9. doi: 10.1016/j.bone.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Katsuno Y, Hanyu A, Kanda H, Ishikawa Y, Akiyama F, Iwase T, et al. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene. 2008;27:6322–33. doi: 10.1038/onc.2008.232. [DOI] [PubMed] [Google Scholar]
- 6.Sethi N, Kang Y. Dysregulation of developmental pathways in bone metastasis. Bone. 2011;48:16–22. doi: 10.1016/j.bone.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Pratap J, Lian JB, Stein GS. Metastatic bone disease: role of transcription factors and future targets. Bone. 2011;48:30–6. doi: 10.1016/j.bone.2010.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pratap J, Javed A, Languino LR, van Wijnen AJ, Stein JL, Stein GS, et al. The Runx2 osteogenic transcription factor regulates matrix metalloproteinase 9 in bone metastatic cancer cells and controls cell invasion. Mol Cell Biol. 2005;25:8581–91. doi: 10.1128/MCB.25.19.8581-8591.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pratap J, Wixted JJ, Gaur T, Zaidi SK, Dobson J, Gokul KD, et al. Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Res. 2008;68:7795–802. doi: 10.1158/0008-5472.CAN-08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pratap J, Imbalzano KM, Underwood JM, Cohet N, Gokul K, Akech J, et al. Ectopic runx2 expression in mammary epithelial cells disrupts formation of normal acini structure: implications for breast cancer progression. Cancer Res. 2009;69:6807–14. doi: 10.1158/0008-5472.CAN-09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akech J, Wixted JJ, Bedard K, van der Deen M, Hussain S, Guise TA, et al. Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene. 2010;29:811–21. doi: 10.1038/onc.2009.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das K, Leong DT, Gupta A, Shen L, Putti T, Stein GS, et al. Positive association between nuclear Runx2 and oestrogen-progesterone receptor gene expression characterises a biological subtype of breast cancer. Eur J Cancer. 2009;45:2239–48. doi: 10.1016/j.ejca.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leong DT, Lim J, Goh X, Pratap J, Pereira BP, Kwok HS, et al. Cancer-related ectopic expression of the bone-related transcription factor RUNX2 in non-osseous metastatic tumor cells is linked to cell proliferation and motility. Breast Cancer Res. 2010;12:R89. doi: 10.1186/bcr2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lian JB, Stein GS, van Wijnen AJ, Stein JL, Hassan MQ, Gaur T, et al. MicroRNA control of bone formation and homeostasis. Nat Rev Endocrinol. 2012;8:212–27. doi: 10.1038/nrendo.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taipaleenmaki H, Bjerre Hokland L, Chen L, Kauppinen S, Kassem M. Mechanisms in endocrinology: micro-RNAs: targets for enhancing osteoblast differentiation and bone formation. Eur J Endocrinol. 2012;166:359–71. doi: 10.1530/EJE-11-0646. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Guo B, Li Q, Peng J, Yang Z, Wang A, et al. miR-214 targets ATF4 to inhibit bone formation. Nat Med. 2013;19:93–100. doi: 10.1038/nm.3026. [DOI] [PubMed] [Google Scholar]
- 17.van Wijnen AJ, van de Peppel J, van Leeuwen JP, Lian JB, Stein GS, Westendorf JJ, et al. MicroRNA functions in osteogenesis and dysfunctions in osteoporosis. Curr Osteoporos Rep. 2013;11:72–82. doi: 10.1007/s11914-013-0143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4:143–59. doi: 10.1002/emmm.201100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Browne G, Taipaleenmäki H, Stein GS, Stein JL, Lian JB. MicroRNAs in the control of metastatic bone disease. Trends Endocrinol Metab. 2014 doi: 10.1016/j.tem.2014.03.014. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeshita F, Patrawala L, Osaki M, Takahashi RU, Yamamoto Y, Kosaka N, et al. Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther. 2010;18:181–7. doi: 10.1038/mt.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ell B, Mercatali L, Ibrahim T, Campbell N, Schwarzenbach H, Pantel K, et al. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell. 2013;24:542–56. doi: 10.1016/j.ccr.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krzeszinski JY, Wei W, Huynh H, Jin Z, Wang X, Chang TC, et al. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature. 2014;512:431–5. doi: 10.1038/nature13375. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.van der Deen M, Taipaleenmaki H, Zhang Y, Teplyuk NM, Gupta A, Cinghu S, et al. MicroRNA-34c inversely couples the biological functions of the runt-related transcription factor RUNX2 and the tumor suppressor p53 in osteosarcoma. J Biol Chem. 2013;288:21307–19. doi: 10.1074/jbc.M112.445890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romano P, Manniello A, Aresu O, Armento M, Cesaro M, Parodi B. Cell Line Data Base: structure and recent improvements towards molecular authentication of human cell lines. Nucleic Acids Res. 2009;37:D925–32. doi: 10.1093/nar/gkn730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Xie RL, Croce CM, Stein JL, Lian JB, van Wijnen AJ, et al. A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc Natl Acad Sci U S A. 2011;108:9863–8. doi: 10.1073/pnas.1018493108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta A, Cao W, Chellaiah MA. Integrin alphavbeta3 and CD44 pathways in metastatic prostate cancer cells support osteoclastogenesis via a Runx2/Smad 5/receptor activator of NF-kappaB ligand signaling axis. Mol Cancer. 2012;11:66. doi: 10.1186/1476-4598-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H, Pan Y, Zheng L, Choe C, Lindgren B, Jensen ED, et al. FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res. 2011;71:3257–67. doi: 10.1158/0008-5472.CAN-10-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viticchie G, Lena AM, Latina A, Formosa A, Gregersen LH, Lund AH, et al. MiR-203 controls proliferation, migration and invasive potential of prostate cancer cell lines. Cell Cycle. 2011;10:1121–31. doi: 10.4161/cc.10.7.15180. [DOI] [PubMed] [Google Scholar]
- 30.Saini S, Majid S, Yamamura S, Tabatabai L, Suh SO, Shahryari V, et al. Regulatory Role of mir-203 in Prostate Cancer Progression and Metastasis. Clin Cancer Res. 2011;17:5287–98. doi: 10.1158/1078-0432.CCR-10-2619. [DOI] [PubMed] [Google Scholar]
- 31.Niu DF, Kondo T, Nakazawa T, Oishi N, Kawasaki T, Mochizuki K, et al. Transcription factor Runx2 is a regulator of epithelial-mesenchymal transition and invasion in thyroid carcinomas. Lab Invest. 2012;92:1181–90. doi: 10.1038/labinvest.2012.84. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Zhang B, Li W, Fu L, Fu L, Zhu Z, et al. Epigenetic Silencing of miR-203 Upregulates SNAI2 and Contributes to the Invasiveness of Malignant Breast Cancer Cells. Genes Cancer. 2011;2:782–91. doi: 10.1177/1947601911429743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding X, Park SI, McCauley LK, Wang CY. Signaling between transforming growth factor beta (TGF-beta) and transcription factor SNAI2 represses expression of microRNA miR-203 to promote epithelial-mesenchymal transition and tumor metastasis. J Biol Chem. 2013;288:10241–53. doi: 10.1074/jbc.M112.443655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouchie A. First microRNA mimic enters clinic. Nat Biotechnol. 2013;31:577. doi: 10.1038/nbt0713-577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.