

Abstract
Background & Aims
Pancreatic cancer (PC) frequently causes diabetes. We recently proposed adrenomedullin (AM) as a candidate mediator of pancreatic β-cell dysfunction in PC. How PC-derived AM reaches β-cells remote from the cancer to induce β-cell dysfunction is unknown. We tested a novel hypothesis that PC sheds AM-containing exosomes into circulation which are transported to β-cells and impair insulin secretion.
Methods
We characterized exosomes from conditioned media of PC-cell lines (n=5) and portal/peripheral venous blood of PC patients (n=20). Western blot analysis showed the presence of AM in PC-Exosomes. We determined the effect of AM-containing PC-Exosomes on insulin secretion from INS-1 β-cells and human islets, and showed how exosomes internalize into β-cells. We studied the interaction between β-cell AM receptors and AM present in PC-Exosomes. In addition, we studied the effect of AM on endoplasmic reticulum (ER) stress response genes and reactive oxygen/nitrogen species generation in β-cells.
Results
Exosomes were found to be the predominant extracellular vesicles secreted by PC into culture media and human plasma. PC-Exosomes contained AM and CA19-9, readily entered β-cells through caveolin-mediated endocytosis or macropinocytosis, and inhibited insulin secretion. AM in PC-Exosomes interacted with its receptor on β-cells. AM receptor blockade abrogated the inhibitory effect of exosomes on insulin secretion. β-cells exposed to AM or PC-Exosomes showed upregulation of ER stress genes and increased reactive oxygen/nitrogen species.
Conclusions
Pancreatic cancer causes paraneoplastic β-cell dysfunction by shedding AM+/CA19-9+ exosomes into circulation that inhibit insulin secretion, likely through AM-induced ER stress and failure of the UPR.
Keywords: Pancreatic cancer, Diabetes Mellitus, Paraneoplastic syndrome, Exosome, Adrenomedullin
Introduction
Nearly 85% of pancreatic cancer (PC) patients have hyperglycemia, and the majority (45%–67%) have DM (3, 4), which is frequently new onset (75%), i.e., less than 36 months in duration (3, 5, 6). The mechanism of PC-induced DM (PC-DM) remains unclear. Understanding the pathogenesis and mediators of PC-DM could lead to identification of novel biomarkers for early diagnosis of the cancer (2, 7).
Although the American Diabetes Association classifies PC-DM as a form of pancreatogenous DM (8), its mechanism remains unclear. PC-DM and type 2 DM share a number of features including risk factors (3), presence of marked insulin resistance, and high plasma insulin levels (4, 9–12). These features also distinguish PC-DM from chronic pancreatitis-related DM which is associated with decreased insulin levels secondary to low β-cell mass. Unlike type 2 DM, which is ameliorated by weight loss, PC-DM develops in the face of ongoing, often profound, weight loss (13). Finally, in PC, new-onset DM paradoxically resolves in over 50% of patients following pancreatico-duodenectomy (3). Amyloid deposits in islets, a histologic hallmark of type 2 DM and a major contributor to β-cell dysfunction and loss (14–16) in type 2 DM, are absent in PC-DM (17, 18). Thus, PC-DM appears to be distinct from both type 2 DM and other forms of pancreatogenous DM.
The very high prevalence of new-onset DM in PC suggests a PC-specific mediator that dysregulates glucose homeostasis (7). We have previously shown that PC cell line–conditioned media induce β-cell dysfunction in both the rat insulinoma (INS-1) β-cell line and mouse islets (19). In previous studies, glucose intolerance could be induced in mice homozygous for the scid mutation by repeated intraperitoneal injection of PC-conditioned media (20) or by xenografting human PC (19). The weight of epidemiologic, clinical, and laboratory evidence suggests that PC-DM is a paraneoplastic phenomenon caused by tumor-secreted factors (7).
In recent years, membrane-derived extracellular vesicles (EV) have emerged as important conduits for cell-to-cell communication (21). Both normal and malignant cells shed microparticles of varying sizes into the surrounding extracellular space (22). EVs contain components of the cells’ membrane and cytoplasm, which affect recipient cells by transferring their biologically active cargoes (22). EVs are produced by at least 3 distinct mechanisms: inward or reverse budding of multivesicular bodies (MVBs) leading to formation of nanoparticle-sized (30 to 100 nm) exosomes; outward membrane blebbing producing 100 to 1,000 nm sized microvesicles; and outward blebbing of apoptotic cell membranes producing large (500 to 2,000 nm) apoptotic bodies (23). The morphologic and functional characteristics of EVs shed by PC have not been previously delineated. Here, we show that the majority of EVs isolated from PC cell line–conditioned media and in the plasma of PC patients are exosomes. This finding led us to study the in vitro biological action of exosomes shed by PC on insulin secretion. We hypothesize that β-cell dysfunction in PC is mediated by exosomes carrying β-cell toxic cargo that are shed into blood circulation.
Microarray analysis of commercially available cell lines that caused β-cell dysfunction led us to identify adrenomedullin (AM) as a candidate mediator of PC-DM (19). AM is the most highly expressed gene in PC cell lines grown under harsh conditions of low glucose and low oxygen (24). Although this ubiquitous polypeptide inhibits insulin secretion, AM has no known physiologic role in glucose metabolism (25). We showed that inhibition of insulin secretion by conditioned media from PC cell lines is abrogated by inhibiting AM expression using short hairpin RNA (shRNA) (19). However, how AM is transported from PC to β-cells and the molecular mechanism of AM-induced β-cell dysfunction are unknown.
In this study, we show that the predominant EVs shed by PC, not only in conditioned media but also in peripheral and portal venous blood of patients, are exosomes. We show that PC-Exosomes (PC-Exo), containing AM and CA19-9, readily enter INS-1 β-cells and human islets by both caveolin-mediated endocytosis and macropinocytosis, and inhibit insulin secretion. We demonstrate that PC-exosomal AM interacts with AM receptors (ADMR; calcitonin receptor–like receptors [CRLRs]) on β-cells with the inhibitory effect of PC-Exo on insulin secretion being reversed by AM receptor blockade. Finally, we show that AM causes upregulation of genes associated with endoplasmic reticular (ER) stress (Bip, Chop) which promotes increased ROS/RNS production, due to failure of the unfolded protein response (UPR) leading to increased β-cell death. AM receptor blockade abolished these effects. Taken together, our studies provide insight into a novel molecular mechanism of paraneoplastic diabetes in PC.
Materials and Methods
Studies pertaining to human subjects, including the study of exosomes in human plasma, were approved by the Mayo Clinic Institutional Review Board.
Cell culture
INS-1 cells were cultured in RPMI medium containing 10% fetal bovine serum, 10 mmol/L HEPES, 2 mmol/L L-glutamine, 1 mmol/L sodium pyruvate, and 50 µmol/L β-mercaptoethanol. Primary PC patient–derived xenograft cell lines developed in the Mukhopadhyay laboratory were cultured in DMEM/F-12 medium supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic solution (Life Technologies) on plates coated with rat tail collagen type I. Whole human islets purchased from Prodo Laboratories were cultured for 48 hours in islet complete medium (Prodo Laboratories) before studies on glucose responsiveness.
Isolation of exosomes
Exosomes were isolated by differential centrifugation of conditioned media collected from HUVEC, HPDE, PANC-1, primary patient-derived xenograft cell lines, or human plasma. Cells were grown in their respective media to 70% to 80% confluency. The medium was then replaced with medium containing fetal bovine serum deprived of microparticles by differential centrifugation (60 minutes at 100,000 × g). After a 72-hour incubation, the conditioned media were initially cleared of cellular debris/dead cells with 2 sequential spins at 3,000 rpm for 10 minutes at 4°C. The resulting supernatants were then spun at 100,000 × g for 70 minutes at 4°C. The exosome pellet was washed with 1× phosphate-buffered saline (PBS) solution and centrifuged again at 100,000 × g for 70 minutes. The final exosome pellet was resuspended in 1x PBS. To isolate exosomes from peripheral or portal blood, samples were centrifuged at 3,000 rpm for 10 minutes to separate the plasma from the red blood cells. The collected plasma was centrifuged again at 3,000 rpm for 10 minutes and then isolated with high-speed centrifugation, as described above. The resulting exosome pellet was washed and resuspended as described. Protein quantification of exosome preparations was determined by the bicinchoninic acid assay (Pierce).
NanoTracker analysis
Isolated exosome fractions from PC cell lines and PC patient blood plasma were analyzed on the NanoSight NS300. Fractions were diluted accordingly and 5, 60 second movies were taken for each sample. Analysis of the data was done using the software supplied with the machine. Graphical analysis shows particle size distribution of the microparticles in the fractions, and a concentration was reported as particles per milliliter.
Western blot analysis
Exosomes were isolated by the procedure mentioned above from HUVEC, HPDE, PANC-1, 4 primary PC cell lines, and peripheral and portal venous PC blood samples. After isolation, exosomes were quantified by bicinchoninic acid assay (Pierce), and tergitol-type NP-40 (Boston BioProducts) was added to 20 µg of exosomes from each preparation. Samples were run on a western blot according to standard protocols, and probed with CA19-9 antibody (Abcam), or adrenomedullin antibody (Phoenix Pharmaceuticals) and either Alix (Cell Signaling Technology) or TSG101 (Abcam) to detect exosomes in the fractions.
For insulin and pro-insulin western blotting, INS-1 cells were grown in a 6-well plate to ~70% confluency. The media was replaced with media containing varying concentrations of AM peptide and incubated for 48 hours. Cells were glucose stimulated for 4 to 6 hours and cell lysates were isolated using Tergitol-type NP-40 (Boston BioProducts). 20 µg of protein was loaded and blots were probed with the insulin antibody (Santa Cruz Biotechnology Inc.) or pro-insulin antibody (Novus Biologicals) with β-actin (Sigma-Aldrich) serving as the loading control.
Electron microscopic imaging of exosomes
Exosomes suspended in water from the PANC-1 cell line were resuspended in Trump fixative solution (4% formaldehyde: 1% glutaraldehyde in 0.1M sodium phosphate buffer [pH 7.2]). Multiple rinses occurred in this sequence: 0.1M sodium phosphate buffer (pH 7.2), 1% osmium tetroxide in 0.1M sodium phosphate buffer, distilled water, 2% uranyl acetate, distilled water, ethanol, and 100% acetone. Exosomes were then placed on a transmission electron microscopy grid, and images were taken using a Philips Tecnai T12 transmission electron microscope.
Exosome internalization and confocal analysis
Exosomes were isolated from the PANC-1 cell line and dyed with green fluorescent linker PKH67 (Sigma-Aldrich) according to the manufacturer’s protocol. Fifty micrograms of PKH67-dyed exosomes were incubated with cultured human islets in a 35-mm culture dish (culture conditions as described above). Confocal microscopy images were obtained after 24 and 48 hours using a Zeiss LSM 780 confocal microscope.
Insulin secretion assay
INS-1 or human islet medium was replaced with microparticle-free medium containing no glucose. Fifty micrograms of exosomes were incubated with the cells for 24 hours to allow uptake. Cells were glucose stimulated with 15 mM glucose (INS-1) or 16.7 mM glucose (islets) for 4 to 6 hours. For the dose response curve, varying amounts of PC-Exo were incubated with INS-1 cells for 24 hours. Cells were stimulated with 15 mM glucose for 4 to 6 hours. The supernatants were collected and assayed using a rat insulin enzyme-linked immunosorbent assay kit (Crystal Chem), and insulin values were normalized to total protein content. Protein samples were quantified using the bicinchoninic acid assay (Pierce).
Duolink™ Assay System
INS-1 cells were treated with varying amounts of PKH67-dyed (as per manufacturer’s protocol, Sigma-Aldrich) PC-Exo or PC-Exo with 10 nM AM inhibitor (AM 22–52, AnaSpec, Inc) for 24 hours. Cells were washed with 1x PBS and fixed with 4% paraformaldehyde. Duolink in situ kits were purchased from Olink Biosciences. Antibodies for AM (Phoenix Pharmaceuticals) and CRLR (Santa Cruz Biotechnology, Inc) were used for assessing AM/ADMR interactions. Antibodies for Bip (Abcam) and pro-insulin (Novus Biologicals) were used to detect Bip/pro-insulin in the presence of increasing PC-Exo. Conjugation of proximity ligation assay (PLA) probes, rolling circle amplification, and detection procedures were done according to the manufacturer’s instructions. Cells were mounted using DAPI-containing mounting media (Vector Labs). Images were taken on a Zeiss LSM 780 confocal microscope.
Pharmacological inhibition and quantification of exosome internalization
INS-1 cells were grown in 4-well chamber slides (800,000 cells/well) with optimal treatment concentrations and timings adapted from Bhattacharya et al (26)(Supplementary Table 1). After treatment, cells were washed three times with 1x PBS, and MV free media was added to the cells with the addition of 5 µl PKH67-dyed PANC-1 exosomes. Exosomes were incubated with cells for 5 hours to allow internalization, then washed with 1x PBS 2 times, fixed with 4% PFA, and mounted with DAPI-containing mounting media (Vector Labs). Images were taken using a Zeiss LSM 780 confocal microscope. Quantification of green fluorescence (exosome) internalization was performed using KS400 Image Analysis software (Zeiss). For each image, the nuclei were counted and the total green fluorescence for the entire image was divided by the nuclei (cell) count to obtain green fluorescence per cell.
Real-time polymerase chain reaction
After treating INS-1 cells with varying concentrations of AM peptide (Phoenix Pharmaceuticals) or PC-Exo (50 µg) for 24 or 48 hours, total RNA was isolated using the RNeasy kit (Qiagen), and complement DNA (cDNA) was reverse transcribed from 1 µg of RNA using the iScript cDNA kit (BioRad). RT-PCR analysis was performed using the ABI 7500 Real Time PCR System and the SYBR Green Master Mix (Applied Biosystems). Primers for insulin and ER stress markers (Bip, Chop, and Xbp-1s) were adapted from Lipson et al (27).
Annexin V staining
INS-1 cells were plated in a 6-well plate (4×106 cells/well) in microparticle-free medium. Fifty micrograms of PC-Exo were incubated with the cells for 48 hours. Cells were trypsinized, collected, and resuspended in 1× binding buffer supplied with the Annexin V-FITC Apoptosis kit (BioVision, Inc). Annexin V–propidium iodide staining was completed according to the manufacturer’s protocol. Flow cytometry analysis was completed to detect early and late apoptotic cells.
Cellular ROS/superoxide detection
INS-1 cells were seeded in 4 well chamber slides (800,000 cells/well). Cells were pretreated with 1 or 20 pM adrenomedullin peptide for 24 hours, then glucose stimulated for 4 hours with 15 mM glucose. Cellular ROS/superoxide detection assay kit (Abcam) was used for ROS/RNS detection. Treatment of cells for detection was adapted by this protocol including for the positive and negative controls. Cells were then washed in 1x wash buffer (supplied with kit), and mounted with DAPI-containing mounting media. Images were obtained using a Zeiss LSM 780 confocal microscope.
Statistical analysis
Statistical comparisons between 2 groups were performed using a 2-sample t test. Comparisons across more than 2 groups were performed using analysis of variance with a Dunnett posttest to compare all columns to the control. All analyses were conducted using GraphPad software. P values less than .05 were considered statistically significant.
Results
Extracellular vesicles in PC are predominantly exosomes
We isolated all microparticles (microvesicles and exosomes) shed by PC using differential centrifugation. With the NanoSight NS300 system (Malvern Instruments Ltd) we determined that the predominant microparticles in PC cell lines and patient plasma were of exosomal size (modes, ~100 nm) (Fig. 1A; Supplementary Fig. S1). However, this did not hold true for all PC cell lines, as exemplified in Supplementary Fig. S1b, where the majority of the microparticles were of microvesicular size. In addition, scanning electron microscopy revealed that microparticles from PANC-1 conditioned medium are of exosomal size and morphology, thereby confirming the NanoTracker results (Fig. 1E).
Figure 1. PC sheds CA19-9 and AM-containing exosomes that readily enter β-cells.

(A) NanoTracker analysis (average of 5, 60 second movies) of size distribution of microparticles isolated from conditioned medium of a PC patient–derived cell line (left), PANC-1 cell line (center), and plasma from a PC patient (right). Red bars indicate ±1 standard error of the mean. (B) Western blot for CA19-9 in 15 µg of exosomes from HUVEC and HPDE (controls), and PC patient plasma (PC1-5). (C–D) Western blot probing for AM in 20 µg of exosomes. (C) PC-Exo isolated from PANC-1 and HPDE cell lines and PC1-PC4 (PC patient–derived xenograft cell lines). (D) PC-Exo isolated from peripheral venous blood (pev) or portal venous blood (pov) of PC patients. (E) Electron microscopy of exosomes isolated from PANC-1 cell line. Scale bar, 200 µm. (F) Confocal microscopy after co-incubation for 48 hours of PKH67-dyed exosomes from the PANC-1 cell with human islet cells. Scale bar, 50 µm.
PC-Exosomes readily enter human islets
Human islets were incubated with PKH67-dyed exosomes isolated from the PANC-1 cell line. Using confocal microscopy, we identified PKH67-dyed exosome internalization into islets within 48 hours of co-incubation (Fig. 1F).
Exosomes from PC-conditioned media and plasma of PC patients contain CA19-9 and AM
Western blot analysis revealed the presence of CA19-9 in all PC patient exosome samples (n=11) with TSG101 serving as a marker of exosomes (Fig. 1B; Fig. 5 representative samples). In contrast, exosomes isolated from normal pancreatic cell lines HPDE and HUVEC showed no CA19-9 and TSG101 expression (Fig. 1B). This suggests that the content and amount of normal exosomes being produced differs in comparison to tumor-derived exosomes and that isolated exosome fractions from PC patient plasma are in fact derived from the PC tumor. We next looked for the presence of AM through western blotting in exosomes shed by human pancreatic ductal epithelial (HPDE; control cell line), PANC-1, and 4 PC patient–derived xenograft cell lines. All revealed the presence of AM, however, exosomes from the HPDE cell line may be produced less because of the lack of TSG101 expression found in 20 µg of exosomes (Fig. 1C). Western blotting of exosomes isolated from peripheral (n=18) and portal venous (n=2) plasma of 20 PC patients showed the presence of AM (Fig. 1D; representative samples shown). Although exosome bioavailability and half-life studies were not conducted to determine the rate of PC-Exo uptake in peripheral organs, this result suggests that a steady production of PC-Exo is secreted from the tumor into circulation which contains metabolically active cargoes.
Figure 5. AM and PC-Exosomes increase expression of ER stress markers.

(A–C) mRNA expression of ER stress markers Bip (A), Chop (B), and Xbp-1s (C) in INS-1 cells after treatment with varying concentrations of AM peptide, or peptide + AM inhibitor (AM 22–52). (D–E) mRNA expression of ER stress markers Bip (D) and Chop (E) in INS-1 cells after treatment with 50 µg of PC-Exo (+Exo) from a PC patient–derived cell line compared with control culture medium (-Exo). (F) Percentage of apoptotic cells determined by annexin V staining of INS-1 cells treated with 50 µg of PC-Exo (+Exo) compared to control (untreated) cells (-Exo). (G) Western blot for pro-insulin protein levels with the addition of AM peptide (1, 20, or 40 pM AM) vs. untreated INS-1 cells. * P<.05; ** P<.001; *** P<.0001.
PC-Exosomes inhibit insulin secretion
In a recent study, we reported that conditioned media from various PC cell lines decreased insulin secretion in both a rat INS-1 β-cell line and mouse islets (19). We hypothesized that the inhibitory effect of conditioned media resided in exosomes. The addition of PC-Exo from a patient-derived PC cell line showed a decrease in insulin secretion in both INS-1 cells and human islets (Fig. 2A–B). In addition, PC-Exo isolated from the PANC-1 cell line and 2 PC patient plasma samples showed a similar effect of decreasing insulin secretion from β-cells (Fig. 2C–E). AM receptor abrogation blocked the insulin inhibitory effect of PC-Exo with values exceeding past the baseline due to the blocking of endogenous (free) AM to ADMRs (Fig. 2A–E). In addition, exosome-depleted media had no effect on insulin secretion, confirming that the inhibitory effect is caused by exosomes and not any other soluble factor(s) secreted into the media (Fig. 2A–C). Additionally, this insulin inhibitory effect was seen in PC-Exo isolated from additional PC cell lines and plasma of PC patients (Supplementary Fig. S2). Lastly, varying concentrations of PC-Exo isolated from PC patient plasma were incubated with INS-1 cells for 48 hours to determine insulin secretion in a dose-dependent manner. Indeed, increasing the amount of PC-Exo decreased insulin secretion from the β-cell line (Fig. 2F).
Figure 2. PC-Exosomes decrease insulin secretion with the effect abrogated by ADMR blockade.

Glucose-stimulated insulin secretion in INS-1 cells (A, C–E) and human islets (B) co-incubated with PC-Exo (+Exo) compared to control culture medium (-Exo), PC-Exo with ADMR inhibitor (+Exo + AM inhib) and PC-conditioned medium depleted of exosomes (Exo-free medium). Exosomes were isolated from a PC patient–derived cell line (A–B), PANC-1 (C), and plasma from 2 PC patients (D–E). (F) Insulin secretion dose response curve with increasing amounts of PC-Exo in INS-1 cells. * P<.05; ** P<.001; *** P<.0001.
PC-Exosomal AM binds to cell surface receptors on β-cells to inhibit insulin secretion
To assess the interaction between exosomal AM and its receptor ADMR, the Duolink Assay System (Olink Bioscience) was used. This method allows for determination of protein-protein interactions by using specialized fluorescent PLA probes that attach to the primary antibodies for AM and the ADMR. ADMRs require activation of both co-receptors CRLR and either RAMP 2 or 3, therefore we chose to use a primary antibody for CRLR since this co-receptor is consistently activated each time there is an AM/ADMR interaction. Rolling circle amplification amplifies this region, and punctate dots indicate ligand-receptor interactions. INS-1 cells were incubated for 24 hours with varying amounts of PKH67-dyed PC-Exo. Confocal microscopy was used to visualize AM/ADMR interactions with increasing PC-Exo as indicated by the diffuse cytoplasmic red dots (Fig. 3A). Additionally, quantification of the interactions revealed an overall increase in AM/ADMR interactions with increasing addition of PC-Exo (data not shown). Conversely, ADMR inhibition with AM 22–52 in the presence of PC-Exo decreased AM/ADMR interactions, as indicated by less punctate red staining (Fig. 3B). These data indicate that PC-Exo containing AM are able to shuttle the peptide efficiently to recipient cells and that competitively inhibiting AM coming from PC-Exo results in less AM/ADMR interaction.
Figure 3. PC-Exosomes deliver AM to INS-1 cells and increase AM/ADMR interaction with the effect abrogated by ADMR blockade.
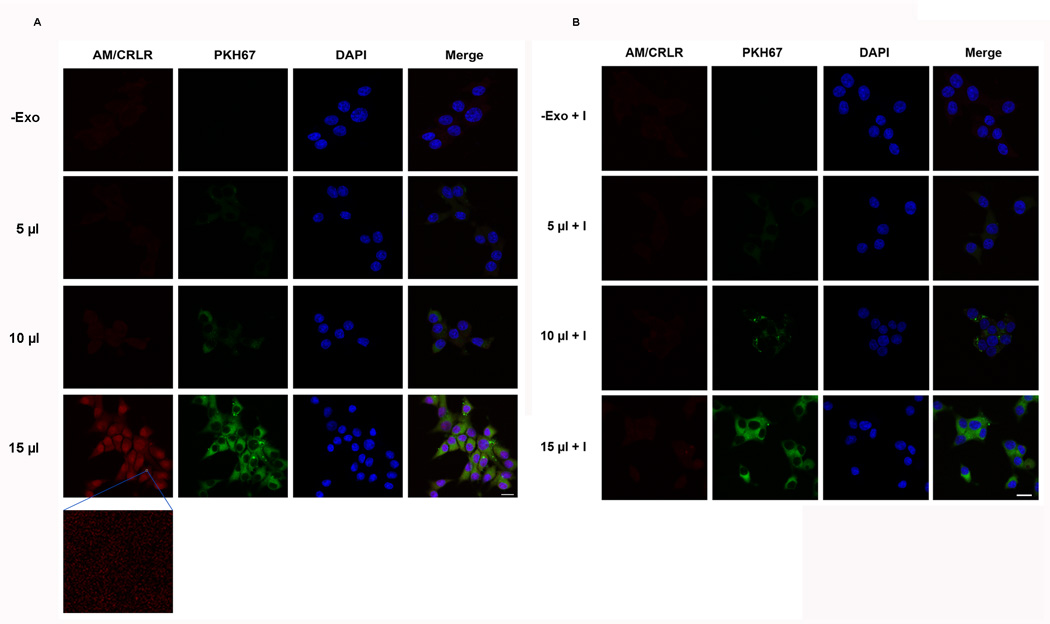
(A–B) The Duolink Assay System was used to assess the interaction of AM/ADMR in INS-1 cells treated with increasing amounts (0, 5, 10, or 15 µl) of PKH67-dyed PC-Exo in the absence (A) and presence (B) of AM 22–52, an ADMR antagonist. Scale bar, 10 µm.
Exosome internalization is mediated through caveolin-dependent endocytosis and macropinocytosis
The results from the Duolink Assay suggest that AM/ADMR interactions occur intracellularly as indicated by the diffuse red cytoplasmic dots (Fig. 3). We sought to gain an understanding of the mechanism of exosome entry and subsequent AM delivery to the cell. Pharmacological inhibition of various endocytic mechanisms was used in order to determine the exact method of exosome internalization. Chlorpromazine (clathrin inhibitor), Nystatin (caveolin inhibitor), Amiloride (inhibitor of macropinocytosis), and Nocodazole (inhibitor of endosomal trafficking) were tested in INS-1 cells using the concentrations and incubation timings listed in Supplementary Table 1. Exosome internalization was significantly inhibited with Amiloride and Nystatin treatments as indicated by less green fluorescence (Fig. 4A, D). Quantification of green fluorescence/cell showed a 42.5% decrease in internalized exosomes with the addition of 100 µM Amiloride, and 20.5% and 59.5% decreases in exosome internalization with 25 µg/ml and 50 µg/ml Nystatin, respectively (Fig. 4C, F). Conversely, varying treatment concentrations of Chlorpromazine and Nocodazole did not have an inhibitory effect on internalizing PC-Exo (Supplementary Fig. S3). Taken together, these data show that PC-Exo internalize into INS-1 cells through both caveolin-dependent endocytosis and macropinocytosis.
Figure 4. Pharmacological inhibition of the endocytic pathway blocks exosome uptake into INS-1 cells.

(A) INS-1 cells were treated with varying concentrations of Amiloride (25, 50, and 100 µM) for 15 minutes. PANC-1 exosomes were dyed with PKH67 and incubated with the cells after Amiloride treatment for 5 hours to allow exosome uptake. Scale bars represent 10 µm. (B) Zoom images (10.5X) of exosome internalization into INS-1 cells in the presence or absence of 100 µM Amiloride. Scale bar represents 5 µm. (C) Quantification of green fluorescence/cell in all treatment groups. (D) INS-1 cells were treated with 25 µg/ml or 50 µg/ml Nystatin for 30 minutes. PKH67-dyed exosomes were incubated with the cells for 5 hours to allow internalization. Scale bars represent 10 µm. (E) Zoom images (10.5X) of cells treated with 50 µg/ml Nystatin compared to control (untreated) cells show decreased PKH67-dyed exosome internalization. Scale bar represents 5 µm. (F) Green fluorescence quantification of internalized exosomes in the presence of absence of Nystatin. ***, P<.0001.
AM deregulates the UPR pathway leading to β-cell dysfunction
As in many other target cells, AM has been reported to cause an increase in cAMP levels in islets (29) which is normally associated with increased insulin secretion (30–32). This is mediated through increases in insulin exocytosis as well as insulin production (33). Indeed we found a modest increase in insulin mRNA in response to AM (Supplementary Fig. S4). ER stress is a physiologic β-cell response to increased insulin production, while an efficient unfolded protein response (UPR) safely resolves ER stress. However, the question arises whether the paradoxical inhibition of insulin secretion by AM, despite an increase in cAMP, could be due to an independent AM-induced unresolved ER stress resulting from a failed unfolded protein response (UPR).
Classical characteristics of a failed UPR response are increased Bip (ER chaperone protein) coupling to pro-insulin in the ER, and activation of Chop (inducer of apoptosis) (34). To assess whether the presence of AM in the cell can activate these ER stress markers, we looked at Bip and Chop gene expressions in the presence of AM. INS-1 cells were treated with varying concentrations of the AM peptide for 48 hours and then glucose stimulated for 4 to 6 hours. RT-PCR analysis showed upregulation of ER stress genes Bip and Chop, with only modest upregulation of Xbp-1s (ER stress marker involved in the normal UPR) with the addition of increasing concentrations of AM (Fig. 5A–C). The addition of the AM inhibitor (AM 22–52) with varying concentrations of AM peptide caused mRNA levels of all three genes to subside to endogenous levels (Fig. 5A–C). The modest increase in Xbp-1s suggests that normal UPR function may be activated slightly in the presence of AM, however the evident upregulation of Bip and Chop in the presence of AM suggests failure of this response. In addition, 50 µg of PC-Exo increased both markers (Fig. 5D, E). Since Chop mRNA levels were elevated, annexin V staining was done to determine cellular apoptosis. Fifty micrograms of PC-Exo were incubated with INS-1 cells for 48 hours; a higher percentage of cells were observed undergoing early apoptosis compared with the control (untreated) cells (Fig. 5F).
Another defining characteristic of a failed UPR in β-cells is the increased coupling of pro-insulin to Bip in the ER (34). Pro-insulin protein levels were assessed in INS-1 cells with the addition of varying concentrations of AM. Our results showed increased pro-insulin in the presence of increasing AM (Fig. 5G). In addition, the Duolink Assay System (as noted above) was utilized to assess the Bip/pro-insulin interactions in situ with increasing amounts of PC-Exo. Indeed, INS-1 cells incubated with increasing amounts of AM-containing PC-Exo showed increasing Bip/pro-insulin interactions as indicated by increased red punctate dots (Fig. 6A).
Figure 6. AM-containing PC-Exosomes can increase Bip/pro-insulin interactions in β-cells.

(A)The Duolink Assay System was used to assess Bip/pro-insulin interactions with increasing amounts (0, 5, 10, or 15 µl) of PKH67-dyed PC-Exo. Scale bar represents 10 µm. (B) The potential mechanism for exosomal AM release and signaling differs from the conventional mechanism shown in 1) in which AM binds to cell surface AM receptors (ADMRs) and activates the cAMP-dependent pathway. Instead PC-Exo internalize through either caveolin-mediated endocytosis or macropinocytosis thus fusing to the early endosome which is also the site of endocytosed ADMRs (2). The early endosome/multivesicular body could be another site of AM/ADMR interaction. Once the pathway is activated adenyl cyclase activation of cAMP releases the catalytic subunits of PKA, which translocate to the nucleus and subsequently activates insulin gene transcription. Excessive exosomal AM activates ER stress proteins that regulate degradation of misfolded or unfolded ER proteins. However, overproduction of insulin due to excessive AM pathway activation eventually leads to failure of the UPR marked by increase Bip/proinsulin coupling in the ER, increased ROS/RNS production, and an increase in Chop, an inducer of apoptosis, leading to a decrease in insulin secretion due to β-cell death.
Overwhelming ER stress can also induce oxidative stress (34). We therefore examined whether AM can invoke reactive oxygen/nitrogen species (ROS/RNS) production in INS-1 cells. With the addition of 1 and 20 pM AM ROS (green filter) and superoxide (orange filter) production increased compared to the control (untreated) cells (Supplementary Fig. S5). This collective body of evidence suggests that AM and PC-Exo containing AM is capable of deregulating the UPR pathway which inevitably leads to marked β-cell dysfunction.
Discussion
Compelling clinical, epidemiologic, and experimental evidence strongly supports the notion that new-onset DM in PC is a paraneoplastic phenomenon caused by tumor secreted products (7). We observed that PC sheds nanoparticle-sized exosomes that are abundantly present in PC-conditioned media and in portal and peripheral venous blood of patients with PC. There is growing evidence that exosomes are important mediators of cell-cell communication (22). Their ability to influence adjacent cells and stroma is now increasingly recognized as a critical cellular function. We observed that the PC-Exosomes derived from in vitro and in vivo sources were morphologically and functionally indistinguishable and exosomes from both sources readily entered β-cells and inhibited insulin secretion. We also show that the inhibitory effect of PC cell line conditioned media on insulin secretion is mediated by exosomes as exosome-free media had no effect on insulin secretion. Thus, our studies not only show that exosomes are the predominant extracellular vesicles shed by PC, both into culture media and plasma, but also demonstrate their ability to dysregulate insulin secretion.
We have previously proposed adrenomedullin as a candidate mediator of β-cell dysfunction in PC as it is overexpressed in PC and inhibits insulin secretion (19). We have also reported increased plasma AM concentrations in PC-DM patients (22.9 ± 10.7 fmol/L) compared to patients with PC but normal fasting glucose (18.3 ± 7.0 fmol/L), noncancer patients with new-onset type 2 diabetes (14.8 ± 10.7 fmol/L), and noncancer subjects with normal fasting glucose levels (12.9 ± 6.6 fmol/L)(19). However, because plasma concentrations were only modestly elevated this raises the question of whether AM is acting as a hormone. Our present work suggests that AM is carried from PC to β-cells as exosomal cargo. After Kitamura et al first reported isolating AM from human phaeochromocytoma in 1993 (35), AM was proposed to be a circulating hormone regulating systemic and pulmonary blood pressure (36). In healthy individuals, circulating AM levels are in the low picomolar range. Physiologic increases in AM levels are seen during pregnancy, with the source being the placenta (37). Pathologic conditions associated with increased AM levels include diabetes, severe hypertension, chronic renal failure, heart failure, pulmonary hypertension, subarachnoid hemorrhage, sepsis, hyperthyroidism, and during cardiac surgery.
It has been debated whether these elevations simply reflect excess tissue AM production or whether excess circulating AM has systemic biologic effects. However, infusion of AM into humans (38) at a rate that quadrupled physiologic levels of AM had no effect on heart rate and blood pressure. Extremely high doses that achieved a concentration over 40 times normal circulating AM levels did significantly decrease blood pressure (38). In another study, high dose AM infusion affected hemodynamic parameters, but not low-dose AM infusion (39). Thus, the circulating concentration of AM necessary to affect blood pressure greatly exceeds that observed in healthy volunteers and in patients with a range of physiologic (notably pregnancy) and pathologic (notably cancer, heart failure, vasculopathies) conditions.
In relation to our work, neither low-dose nor high dose AM infusion had any effect on blood glucose (38). Thus, the elevation of plasma AM is unlikely to be the cause of the biological effects of AM in PC. We are not aware of any mechanism for intra-pancreatic transport of molecules through the length of the pancreas to explain global β-cell dysfunction leading to DM in PC. Therefore, we studied a novel hypothesis that AM was being transported from PC to islets distant from the cancer in exosomes shed into portal circulation and eventually reaching the islets through systemic circulation.
In PC-DM, there are both β-cell dysfunction and increased insulin resistance that resolves with tumor resection (6, 9, 12, 19, 40). Insulin levels in PC are high due to insulin resistance. Our data suggest that β-cell dysfunction leads to inadequate insulin response to insulin resistance. We believe it is this dual hit that promotes a consistent destabilization of glucose homeostasis in PC, resulting in hyperglycemia (12). Thus far, the mechanism and mediators of insulin resistance in PC remain unknown, and the role of PC-Exo, AM, and other factors contributing to insulin resistance needs further study.
Through use of the Duolink Assay System we showed that AM contained in PC-Exo can interact with ADMRs (CRLR) intracellularly with the absence of PC-Exo showing little to no interaction in INS-1 cells (Fig. 3). Currently, the exact mechanism of how AM in PC-Exo interacts with ADMRs is not known. We hypothesize that PC-Exo internalize along with ADMRs in the early endosomes as this location is a potential site of AM release from the exosomes which leaves the peptide in close proximity to ADMRs internalized into early endosomes (Fig. 6B). Further studies are necessary to confirm this exact mechanism.
We show that exosomes inhibit insulin secretion and AM receptor blockade abrogates the effect of exosomes on insulin secretion. Based on our studies, we propose a mechanism for β-cell dysfunction in PC-DM in that the effect of AM and exosomal AM is mediated via upregulation and eventual failure of the unfolded protein response (UPR) due to hyperstimulation of the cAMP-dependent pathway. We found hallmark features of a failed UPR, including increased mRNA expression of ER stress markers Bip and Chop (Fig. 5). We observed increased association of Bip with pro-insulin in the presence of increasing amounts of PC-Exo, another classic feature of a failed UPR response in β-cells (Fig. 6A). We also showed increased β-cell apoptosis after exposure to AM, further confirming Chop-mediated apoptosis (Fig. 5F). The upregulation of Bip and Chop was abolished by AM receptor blockade, suggesting that the effects seen were due to AM-ADMR interactions and not a non-specific toxic effect of AM on β-cells. We conclude from these data that AM leads to excessive activation and subsequent failure of the UPR leading to eventual β-cell death (Fig. 6B). Further work will be needed to understand the molecular mechanism of the failed UPR in response to exosomal AM.
Our studies thus far have focused specifically on the role that AM plays on β-cell dysfunction in PC-DM. However, it is possible that AM is not the only mediator of β-cell dysfunction, but rather it may be working in concert with other key factors, such as tumor necrosis factor α, to promote β-cell dysfunction (42). Therefore, further studies will investigate whether these factors are contained in PC-Exo and their potential interaction with AM. Understanding all the key modulators that cause exosome mediated β-cell dysfunction, and the interplay between them, will give us a more in depth understanding of the pathogenesis of PC-DM.
Our long-term programmatic goal is to develop a rational, evidence-based strategy to screen for sporadic PC. Our work has shown that new-onset DM is a harbinger of PC (43). We reported that subjects with new-onset DM are 8 times more likely than the general population to be diagnosed with PC within 3 years of meeting the criteria for DM (44). Based on these observations, we are conducting the first clinical trial of screening for PC in new-onset DM (NCT02001337). However, what is urgently needed in the field is a reliable marker to distinguish PC-DM from the more common type 2 DM.
Supplementary Material
Translational Relevance.
Pancreatic cancer is the 4th leading cause of cancer death in the United States and is projected to move to 2nd position by the year 2015. Pancreatic cancer has a dismal prognosis (~5% 5 year survival) mainly because 85% are diagnosed when the cancer is already at an advanced stage. New development of diabetes is the only harbinger of pancreatic cancer before cancer symptoms occur as it develops in nearly 50% of pancreatic cancer patients and average of 12 months prior to the development of cancer symptoms. Our previous work strongly suggests that the new diabetes associated with pancreatic cancer is caused by cancer-secreted factors. In this current study we looked at pancreatic cancer exosomes as mediators of β cell dysfunction which can help to understand the mechanism and mediators of pancreatic cancer-induced diabetes leading to early diagnosis of the cancer.
Acknowledgements
This work was partly supported by CA78383 and CA150190 to DM; CA100685 to STC; P50 CA102701 to GP and STC; 1T32 CA148073 to GS; and T32 CA148073 and Mayo Graduate School to NJ. The authors thank the Pancreatic Cancer SPORE; James Tarara and Kristin Mantz of the Mayo Clinic Core Microscopy facilities for assistance in confocal microscopy; and Drs. Gregory Gores and Petra Hirsova for suggestions and assistance on using the NanoSight NS300.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Reference
- 1.Matrisian LMRA. PANCAN. The alarming rise of pancreatic cancer deaths in the United States: Why we need to stem the tide. Pancreatic Cancer Action network. 2013 [Google Scholar]
- 2.Pannala R, Basu A, Petersen GM, Chari ST. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol. 2009;10:88–95. doi: 10.1016/S1470-2045(08)70337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pannala R, Leirness JB, Bamlet WR, Basu A, Petersen GM, Chari ST. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology. 2008;134:981–987. doi: 10.1053/j.gastro.2008.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Permert J, Ihse I, Jorfeldt L, von Schenck H, Arnqvist HJ, Larsson J. Pancreatic cancer is associated with impaired glucose metabolism. Eur J Surg. 1993;159:101–107. [PubMed] [Google Scholar]
- 5.Chari ST, Klee GG, Miller LJ, Raimondo M, DiMagno EP. Islet amyloid polypeptide is not a satisfactory marker for detecting pancreatic cancer. Gastroenterology. 2001;121:640–645. doi: 10.1053/gast.2001.27210. [DOI] [PubMed] [Google Scholar]
- 6.Chari ST, Leibson CL, Rabe KG, Timmons LJ, Ransom J, de Andrade M, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology. 2008;134:95–101. doi: 10.1053/j.gastro.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sah RP, Nagpal SJ, Mukhopadhyay D, Chari ST. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nature reviews Gastroenterology & hepatology. 2013 doi: 10.1038/nrgastro.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35(Suppl 1):S64–S71. doi: 10.2337/dc12-s064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chari ST, Zapiach M, Yadav D, Rizza RA. Beta-cell function and insulin resistance evaluated by HOMA in pancreatic cancer subjects with varying degrees of glucose intolerance. Pancreatology. 2005;5:229–233. doi: 10.1159/000085276. [DOI] [PubMed] [Google Scholar]
- 10.Permert J, Ihse I, Jorfeldt L, von Schenck H, Arnquist HJ, Larsson J. Improved glucose metabolism after subtotal pancreatectomy for pancreatic cancer. Br J Surg. 1993;80:1047–1050. doi: 10.1002/bjs.1800800841. [DOI] [PubMed] [Google Scholar]
- 11.Permert J, Larsson J, Westermark GT, Herrington MK, Christmanson L, Pour PM, et al. Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N Engl J Med. 1994;330:313–318. doi: 10.1056/NEJM199402033300503. [DOI] [PubMed] [Google Scholar]
- 12.Pannala RLJBW, Basu A, Petersen GM, Chari ST. Very high prevalence of new-onset diabetes and impaired fasting glucose in pancreatic cancer - Results of a case-control study. Gastroenterology. 2007:132. [Google Scholar]
- 13.Hart PA, Kamada P, Rabe KG, Srinivasan S, Basu A, Aggarwal G, et al. Weight loss precedes cancer-specific symptoms in pancreatic cancer-associated diabetes mellitus. Pancreas. 2011;40:768–772. doi: 10.1097/MPA.0b013e318220816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butler AE, Janson J, Soeller WC, Butler PC. Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes. 2003;52:2304–2314. doi: 10.2337/diabetes.52.9.2304. [DOI] [PubMed] [Google Scholar]
- 15.Hoppener JW, Ahren B, Lips CJ. Islet amyloid and type 2 diabetes mellitus. The New England journal of medicine. 2000;343:411–419. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- 16.Hull RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab. 2004;89:3629–3643. doi: 10.1210/jc.2004-0405. [DOI] [PubMed] [Google Scholar]
- 17.Saruc M, Iki K, Pour PM. Morphometric studies in human pancreatic cancer argues against the etiological role of type 2 diabetes in pancreatic cancer. Histology and histopathology. 2010;25:423–432. doi: 10.14670/HH-25.423. [DOI] [PubMed] [Google Scholar]
- 18.Li D. Diabetes and pancreatic cancer. Mol Carcinog. 51:64–74. doi: 10.1002/mc.20771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aggarwal G, Ramachandran V, Javeed N, Arumugam T, Dutta S, Klee GG, et al. Adrenomedullin is up-regulated in patients with pancreatic cancer and causes insulin resistance in beta cells and mice. Gastroenterology. 2012;143:1510–1517. doi: 10.1053/j.gastro.2012.08.044. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basso D, Brigato L, Veronesi A, Panozzo MP, Amadori A, Plebani M. The pancreatic cancer cell line MIA PaCa2 produces one or more factors able to induce hyperglycemia in SCID mice. Anticancer Res. 1995;15:2585–2588. [PubMed] [Google Scholar]
- 21.Andaloussi SE, Mager I, Breakefield XO, Wood M. Extracellular vesicles: biology and emerging therapeutic opportunities. Nature Reviews Drug Discovery. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 22.Shifrin DA, Jr, Demory Beckler M, Coffey RJ, Tyska MJ. Extracellular vesicles: communication, coercion, and conditioning. Mol Biol Cell. 24:1253–1259. doi: 10.1091/mbc.E12-08-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. Journal of proteomics. 2010;73:1907–1920. doi: 10.1016/j.jprot.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Natsuizaka M, Ozasa M, Darmanin S, Miyamoto M, Kondo S, Kamada S, et al. Synergistic up-regulation of Hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. Exp Cell Res. 2007;313:3337–3348. doi: 10.1016/j.yexcr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Lopez J, Cuesta N. Adrenomedullin as a pancreatic hormone. Microsc Res Tech. 2002;57:61–75. doi: 10.1002/jemt.10049. [DOI] [PubMed] [Google Scholar]
- 26.Bhattacharya S, Roxbury D, Gong X, Mukhopadhyay D, Jagota A. DNA conjugated SWCNTs enter endothelial cells via Rac1 mediated macropinocytosis. Nano letters. 2012;12:1826–1830. doi: 10.1021/nl204058u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Bonner-Weir S, O'Brien TD. Islets in type 2 diabetes: in honor of Dr. Robert C. Turner. Diabetes. 2008;57:2899–2904. doi: 10.2337/db07-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez A, Weaver C, Lopez J, Bhathena SJ, Elsasser TH, Miller MJ, et al. Regulation of insulin secretion and blood glucose metabolism by adrenomedullin. Endocrinology. 1996;137:2626–2632. doi: 10.1210/endo.137.6.8641217. [DOI] [PubMed] [Google Scholar]
- 30.Lemaire K, Schuit F. Integrating insulin secretion and ER stress in pancreatic beta-cells. Nat Cell Biol. 14:979–981. doi: 10.1038/ncb2594. [DOI] [PubMed] [Google Scholar]
- 31.Takhshid MA, Poyner DR, Chabot JG, Fournier A, Ma W, Zheng WH, et al. Characterization and effects on cAMP accumulation of adrenomedullin and calcitonin gene-related peptide (CGRP) receptors in dissociated rat spinal cord cell culture. Br J Pharmacol. 2006;148:459–468. doi: 10.1038/sj.bjp.0706750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, et al. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54:233–246. doi: 10.1124/pr.54.2.233. [DOI] [PubMed] [Google Scholar]
- 33.Knoch KP, Meisterfeld R, Kersting S, Bergert H, Altkruger A, Wegbrod C, et al. cAMP-dependent phosphorylation of PTB1 promotes the expression of insulin secretory granule proteins in beta cells. Cell Metab. 2006;3:123–134. doi: 10.1016/j.cmet.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitamura K, Kangawa K, Kawamoto M, Ichiki Y, Nakamura S, Matsuo H, et al. Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun. 1993;192:553–560. doi: 10.1006/bbrc.1993.1451. [DOI] [PubMed] [Google Scholar]
- 36.Ishimitsu T, Nishikimi T, Saito Y, Kitamura K, Eto T, Kangawa K, et al. Plasma levels of adrenomedullin, a newly identified hypotensive peptide, in patients with hypertension and renal failure. J Clin Invest. 1994;94:2158–2161. doi: 10.1172/JCI117573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Senna AA, Zedan M, el-Salam GE, el-Mashad AI. Study of plasma adrenomedullin level in normal pregnancy and preclampsia. Medscape J Med. 2008;10:29. [PMC free article] [PubMed] [Google Scholar]
- 38.Miyao Y, Nishikimi T, Goto Y, Miyazaki S, Daikoku S, Morii I, et al. Increased plasma adrenomedullin levels in patients with acute myocardial infarction in proportion to the clinical severity. Heart. 1998;79:39–44. doi: 10.1136/hrt.79.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piquard F, Charloux A, Mettauer B, Epailly E, Lonsdorfer E, Popescu S, et al. Exercise-induced increase in circulating adrenomedullin is related to mean blood pressure in heart transplant recipients. J Clin Endocrinol Metab. 2000;85:2828–2831. doi: 10.1210/jcem.85.8.6734. [DOI] [PubMed] [Google Scholar]
- 40.Chari ST, Leibson CL, Rabe KG, Ransom J, de Andrade M, Petersen GM. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology. 2005;129:504–511. doi: 10.1053/j.gastro.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramachandran V, Arumugam T, Hwang RF, Greenson JK, Simeone DM, Logsdon CD. Adrenomedullin is expressed in pancreatic cancer and stimulates cell proliferation and invasion in an autocrine manner via the adrenomedullin receptor, ADMR. Cancer Res. 2007;67:2666–2675. doi: 10.1158/0008-5472.CAN-06-3362. [DOI] [PubMed] [Google Scholar]
- 42.Jatoi A, Sideras K, Nguyen PL. Tumor Necrosis Factor-alpha as a Treatment Target for the Cancer Anorexia/Weight Loss Syndrome. Support Cancer Ther. 2004;1:237–242. doi: 10.3816/SCT.2004.n.016. [DOI] [PubMed] [Google Scholar]
- 43.Muniraj T, Chari ST. Diabetes and pancreatic cancer. Minerva gastroenterologica e dietologica. 2012;58:331–345. [PMC free article] [PubMed] [Google Scholar]
- 44.Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.