

Abstract
Pathophysiological hypoxia, which fosters the glioma stem-like cell (GSC) phenotype, is present in high-grade gliomas and has been linked to tumor development, invasiveness, and resistance to chemotherapy and radiation. Oncolytic virotherapy with engineered herpes simplex virus-1 (HSV-1) is a promising therapy for glioblastoma; however efficacy of γ134.5-deleted HSVs, which have been used in clinical trials, was diminished in hypoxia. We investigated the ability of a chimeric HCMV/HSV-1 virus, which expresses the human CMV PKR evasion gene IRS1 and is in preparation for clinical trials, to infect and kill adult and pediatric patient-derived glioblastoma xenografts in hypoxia and normoxia. Infectivity, cytotoxicity and viral recovery were significantly greater with the chimeric virus compared to the γ134.5-deleted virus, regardless of oxygen tension. The chimeric virus infected and killed CD133+ GSCs similarly to wild-type HSV-1. Increased activation of mitogen-activated protein kinase (MAPK) p38 and its substrate heat shock protein 27 (Hsp27) was seen after viral infection in normoxia compared to hypoxia. Hsp27 knockdown or p38 inhibition reduced virus recovery indicating that the p38 pathway plays a role in the reduced efficacy of the γ134.5-deleted virus in hypoxia. Together these findings demonstrate chimeric HCMV/HSV-1 efficiently targets both CD133+ GSCs and glioma cells in hypoxia.
Keywords: Hypoxia, herpes simplex virus, oncolytic, glioma stem cells, CD133, chimeric HCMV/HSV-1, p38 MAPK, Hsp27
Introduction
High-grade gliomas are the most common brain malignancy in adults and represent approximately 10% of childhood brain tumors.1 Outcomes for adult and pediatric glioblastoma multiforme (GBM) are poor with 2-year survival rates less than 20% despite multimodality therapy including surgery, chemotherapy, and radiotherapy.2,3 Novel therapies are desperately needed, and oncolytic engineered herpes simplex virotherapy (oHSV) is one such therapy that offers a promising approach by targeting glioma cells while sparing normal cells. Normal cells are unharmed through deletions of various dispensable genes such as the neurovirulence gene γ134.5, which enables the wild-type virus to overcome a host cell's defense mechanism to prevent virus replication.4 Oncolytic HSVs with γ134.5 deleted cannot evade protein kinase R (PKR)-mediated translational arrest which occurs in normal cells in response to HSV double-stranded RNA and prevents viral replication, rendering the virus safe for normal cells. Mutant γ134.5-deleted viruses may replicate in tumor cells with defective signaling pathways that result in an attenuated PKR response. Non-essential genes can be replaced with therapeutic foreign genes that augment the oncolytic effect, such as the human cytolomegalovirus (HCMV) IRS1 gene. The IRS1 gene product restores a critical function that is lost with the γ134.5-deletion by facilitating late viral protein synthesis and viral replication without contributing to virulence for normal cells.5,6 First generation γ134.5-deleted viruses have been used safely in phase I human adult high-grade glioma trials with evidence of virus replication in tumors and several radiologic responses seen, however many responses were modest.7,8
One recently discovered limitation of the first generation γ134.5-deleted (ICP34.5-) HSV-1, which may explain some of the moderate clinical responses, is the reduced efficacy of the virus under hypoxic conditions.9 Pathophysiological hypoxia is a hallmark of high-grade gliomas with hypoxic gradients varying from 10%-2.5% (mild to moderate) to as low as 0.1% (severe), which can be found in necrotic areas of tumors.10,11 This hypoxia has been associated with tumor initiation, invasiveness, angiogenesis, loss of apoptotic potential and resistance to radiation and chemotherapy, all characteristics which have been ascribed to glioma stem-like cells (GSCs).12 Furthermore, hypoxia has been shown to maintain and drive the GSC phenotype with the number of GSCs, as measured by the most commonly used GSC marker CD133, increasing dramatically in hypoxia.13,14 Accordingly, the reduced efficacy of the γ134.5-deleted virus in hypoxia has important implications for clinical application of oHSV because hypoxia plays a critical role in shaping tumor behavior and therapeutic resistance. An effective oHSV must be able to replicate well under hypoxic conditions to eradicate the resistant GSC population.
Therefore, we sought to determine if the second-generation, chimeric HCMV/HSV-1 oncolytic virus C134, which has proven safe in normal brain cells and prolonged survival in two in vivo experimental murine brain tumor models, has an improved ability to infect, replicate in, and kill pediatric and adult GBM xenograft lines (xenolines) in hypoxia compared to the first-generation ICP34.5- virus and to establish a mechanism for the reduced efficacy of the γ134.5-deleted virus in hypoxia.6 C154 is a green fluorescence protein (GFP) expressing version of C134, which is being prepared under current GMP procedures and qualified by the NExT Program (RAID) for clinical application. We explored the role of the p38 mitogen-activated protein kinase (MAPK) signaling pathway, including its known substrate heat shock protein 27 (Hsp27), which together enhance expression of late viral genes, in the moderation of the γ134.5-deleted virus in hypoxia.15,16 We hypothesized that reduced activation of p38 MAPK and Hsp27 in hypoxia would result in reduced efficacy of the ICP34.5- virus. Since the C154 chimeric virus contains the HCMV IRS1 gene, which facilitates late viral protein synthesis, we anticipated that C154 would be less affected by hypoxia. Importantly, our data demonstrate that the chimeric HCMV/HSV-1 is superior to the γ134.5-deleted virus in hypoxia and can target both CD133+ GSCs and glioma cells similar to wild-type HSV-1. Furthermore, the p38 pathway plays an important role in the reduced efficacy of γ134.5-deleted viruses in hypoxia.
Results
Viral Infectivity
We previously showed that the ability of γ134.5-deleted viruses to infect and kill GBM xenolines was diminished in hypoxia.9 To determine if the chimeric virus had improved infectability, GBM-XD456, GBM-X12 and GBM-X6 cells that were maintained in Neurobasal medium either in normoxia or hypoxia (1% oxygen) were infected with the recombinant GFP-tagged C154 virus at 1, 3.3 and 10 multiplicities of infection (MOI) for 30 hours. One percent oxygen was chosen to recapitulate the severe hypoxia observed in high-grade gliomas by Evans et al.11 Using this assay, we previously demonstrated that the γ134.5-deleted virus infected a similar percentage of cells between 1-5% oxygen levels.17 Thirty hours was established as the optimal time point in our previous study, which examined multiple time points, because it allowed the infection to be well underway but was prior to cells lysis.9 The proportion of cells infected was determined by FACS analysis. Similar to our previous observations about the sensitivity of these GBM xenolines to the γ134.5-deleted virus, GBM-X12 was the most sensitive in both normoxia and hypoxia while GBM-X6 was the most resistant to infection with the chimeric virus. The chimeric virus infected a significantly greater number of cells than the γ134.5-deleted virus in both hypoxia and normoxia in all three xenolines at 10 MOI (Table 1). GBM-X6 cells experienced the greatest increase in infection with C154 infecting 2.7-fold and 5.9-fold more cells than the γ134.5-deleted virus in normoxia and hypoxia respectively. While the chimeric virus infected significantly fewer GBM-XD456 cells (p=0.008) and GBM-X12 cells (p=0.001) in hypoxia compared to normoxia, the relative decrease in infection (25% for GBM-XD456 and 15% for GBM-X12) was less than that observed for the γ134.5-deleted virus which infected 36% fewer GBM-XD456 cells and 26% fewer GBM-X12 cells in hypoxia. Unlike the ICP34.5- virus, the chimeric virus infected more GBM-X6 cells in hypoxia (58.8 ± 0.4%) than in normoxia (54.3 ± 1.4%; p=0.006) after 30 hours. These data demonstrate that the chimeric HCMV/HSV-1 is superior at infecting adult and pediatric GBM xenoline cells in both normoxia and hypoxia and is not limited to the same degree in hypoxia as the γ134.5-deleted virus.
Table 1. Percentage (mean + SD) of GBM xenoline cells infected with γ134.5-deleted virus (ICP34.5-) and chimeric C154 virus at 10 MOI 30 hours post-infection in normoxia or 1% hypoxia as measured by GFP expression.
| GBM-XD456 | GBM-X6 | GBM-X12 | ||||
|---|---|---|---|---|---|---|
| Virus | Normoxia | Hypoxia | Normoxia | Hypoxia | Normoxia | Hypoxia |
| ICP34.5- | 41.5 ± 0.9 | 26.4 ± 0.9 | 20.4 ± 1.3 | 10.0 ± 0.2 | 65.5 ± 2.0 | 48.2 ± 0.8 |
| C154 | 65.4 ± 3.4 | 48.9 ± 1.2 | 54.3 ± 1.4 | 58.8 ± 0.4 | 86.3 ± 1.6 | 73.6 ± 2.1 |
| P value | 0.0003 | <0.0001 | <0.0001 | <0.0001 | 0.0003 | 0.0006 |
We next compared the abilities of the chimeric virus, the γ134.5-deleted virus and a wild type HSV-1 (M2001) to infect CD133+ GSCs from GBM-X6, the xenoline that was most resistant to the γ134.5-deleted virus, in normoxia and hypoxia. By 30 hours post-infection, the C154 virus infected a significantly greater number of CD133+ cells in both hypoxia and normoxia at each MOI (1, 3.3, and 10) compared to the ICP34.5- virus (Figure 1). At an MOI of 3.3, the chimeric virus infected 4-fold more CD133+ cells than the γ134.5-deleted virus in normoxia and 7-fold more CD133+ cells in hypoxia. Despite GBM-X6 being the most resistant xenoline, C154 was able to infect over half (55.9 ± 4.6%) of the CD133+ cells in hypoxia at 10 MOI within 30 hours compared to only 8.2 ± 0.9% for the γ134.5-deleted virus. At a low MOI of 1, the chimeric virus infected fewer cells than wild-type HSV-1; however at an MOI of 3.3 and 10, the chimeric virus was as effective at infecting cells as wild type virus. At 10 MOI, C154 infected 46.9 ± 4.6% of CD133+ cells in normoxia, and the wild-type virus infected 44.8 ± 5.1% (p=.62); in hypoxia, C154 infected 55.9 ± 4.6% of CD133+ cells versus 48. 9 ± 3.4% for the wild-type virus (p=.10). Importantly, in all three xenolines, the chimeric virus was able to infect as many CD133+ cells in hypoxia as in normoxia at various MOI (Figure 2). Taken together, these data show that the chimeric HCMV/HSV-1 infects CD133+ GSCs similar to wild-type HSV-1 except at low MOI. Furthermore, the chimeric virus is not limited to the same degree as the γ134.5-deleted virus in hypoxia, and its ability to infect GSCs is not limited by hypoxia like the γ134.5-deleted virus.
Figure 1.

Percentage (mean + SD) of CD133+ GBM-X6 cells infected by ICP34.5- HSV-1, chimeric C154, and wild-type HSV-1 (M2001) at various MOI in normoxia or 1% hypoxia at 30 hours as measured by GFP and APC expression by FACS analysis.
Figure 2.

Percentage (mean + SD) of CD133+ GBM-XD456, GBM-X6, and GBM-X12 cells infected by chimeric C154 at various MOI in normoxia or 1% hypoxia at 30 hours as measured by GFP and CD133-APC expression by FACS analysis.
Cytotoxicity and Viral Recovery
To confirm the infections were productive, we next determined the cytotoxic ability of the chimeric virus in normoxia and 1% hypoxia 3 days after infection, and we measured virus recovery 48 hours post-infection. Consistent with the infectivity data, GBM-X12 was the most sensitive to killing with the lowest dose (plaque forming units (PFU/cell)) required to kill 50% of the cells (LD50) followed by GBM-XD456, and GBM-X6 was the most resistant (Table 2). In all xenolines in both normoxia and hypoxia, significantly lower C154 doses were required to kill 50% of the cells compared to the γ134.5-deleted virus. Similar to the infectivity data, the C154 LD50 was higher in GBM-XD456 and GBM-X12 in hypoxia compared to normoxia whereas the C154 LD50 was lower in hypoxia than normoxia in GBM-X6. Importantly, significantly more GBM-X6 cells were killed by the chimeric virus in contrast to the γ134.5-deleted virus at multiple dose levels in both normoxia and hypoxia, and the chimeric virus killed similarly to wild-type HSV-1 (M2001) (Figure 3).
Table 2. Virus dose (PFU/cell) (mean + SD) required to kill 50% of cells (LD50) in normoxia or 1% hypoxia as measured by the alamarBlue® assay after a 3 day incubation period.
| GBM-XD456 | GBM-X6 | GBM-X12 | ||||
|---|---|---|---|---|---|---|
| Virus | Normoxia | Hypoxia | Normoxia | Hypoxia | Normoxia | Hypoxia |
| ICP34.5- | 7.0 ± 2.0 | 10.7 ± 0.8 | 44.8 ± 3.0 | 47.1 ± 2.9 | 1.9 ± 0.1 | 3.2 ± 0.2 |
| C154 | 2.1 ± 0.6 | 3.9 ± 0.4 | 18.1 ± 2.7 | 15.8 ± 4.4 | 0.4 ± 0.1 | 1.0 ± 0.2 |
| P value | 0.015 | 0.0002 | 0.0003 | 0.0005 | 0.009 | 0.004 |
Figure 3.
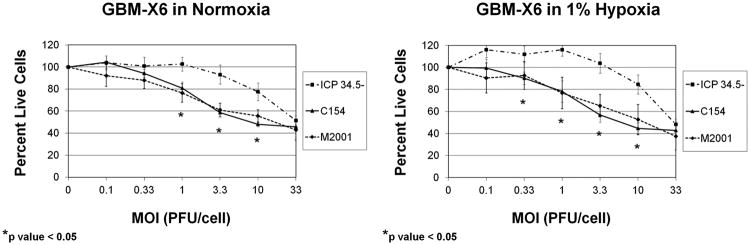
Sensitivity of GBM-X6 to ICP34.5- HSV-1, chimeric C154 and wild-type HSV-1 (M2001) in normoxia and 1% hypoxia as measured by the alamarBlue assay at a given MOI compared to a control (no virus) after a 3 day incubation period (mean + SD).
Consistent with the infectivity and cytotoxicity data, significantly more C154 was recovered 48 hours post-infection compared to the ICP34.5- virus in all three xenolines in both normoxia and hypoxia (Figure 4). The greatest increase was seen in GBM-X6 with over 5,500-fold (p<0.0001) and 6,300-fold (p=0.0001) more C154 virus recovered than ICP34.5- virus in normoxia and hypoxia respectively, followed by GBM-XD456 (>30-fold in normoxia, p<0.0001; >200-fold in hypoxia, p<0.0001) and GBM-X12 (>29-fold in normoxia, p<0.0001; >55-fold in hypoxia, p=0.0044). The C154 viral recovery quantitatively approached that of the wild-type virus although more wild-type virus was recovered than C154 except in GBM-XD456 in hypoxia. Taken together, these data demonstrate that the chimeric virus produces significantly more infectious viral particles and kills pediatric and adult GBM xenoline cells irrespective of oxygen tension more efficiently than the ICP34.5 virus, and the chimeric virus kills an ICP34.5- resistant xenoline similarly to wild-type HSV-1.
Figure 4.

Virus recovery (mean + SD) of wild-type HSV-1 (M2001), chimeric C154 and ICP34.5- HSV-1 48 hours post infection in normoxia and 1% hypoxia as measured by limiting plaque dilution on Vero cells.
To confirm the in vitro finding of improved infectivity of C154 compared to the 34.5-deleted virus, in vivo virus infectivity of GBM-XD456 growing in the right cerebral hemisphere of athymic nude mice was characterized by fluorescent immunohistochemistry (IHC) for GFP expression in hypoxic areas of the tumor, as measured by carbonic anhydrase 9 (CA9) expression, 24 hours post-inoculation. This time point was chosen to allow for ample time for GFP production, which occurs with immediate-early gene expression, but prior to cell lysis. More GFP expression was seen after C154 infection compared to ICP34.5- virus in areas with and without CA9 expression (Figure 5), suggesting superior C154 infectivity of GBM-XD456 cells in hypoxic and less-hypoxic areas of the tumor.
Figure 5.

In vivo infectivity, as measured by GFP expression (green), 24 hours post-inoculation of 1×107 PFU of ICP34.5- or C154 virus in GBM-XD456MG cells (DAPI; blue) grown in the right cerebral hemisphere of nude mice. Areas of hypoxia are marked by carbonic anhydrase 9 (CA9; red). Controls were injected with saline. Rabbit IgG was used as a negative control. Photos were taken microscopically with a 10× objective and representative of 10 sections.
We next sought to determine the cause of the decreased efficacy of the γ134.5-deleted virus in hypoxia by exploring the activation of p38 MAPK. Previously, it has been shown that the p38 pathway enhances expression of late viral genes and activation of p38 MAPK and Hsp27, a known substrate of p38 MAPK, improve wild-type viral yields.15,16 We hypothesized that a differential activation of p38 MAPK and Hsp27 post-infection in normoxia could explain the reduced efficacy of the γ134.5-deleted virus in hypoxia. Because GBM-X6 was resistant to the ICP34.5- virus, we focused our experiments on GBM-XD456 and GBM-X12, which were both significantly more sensitive to infection and killing by the ICP34.5- virus in normoxia. We examined the activation status of p38 MAPK in normoxia and hypoxia without and with infection at 10 MOI by the ICP34.5- virus and the chimeric virus by western blotting.
Six hours post-infection, total p38 MAPK protein levels, detected at 38 kDa, were similar in both GBM-XD456 and GBM-X12 with and without infection in normoxia and hypoxia (Figure 6). While baseline levels of activated p38 MAPK (as measured by phosphorylated Thr 180/Tyr 182 of p38 MAPK detected at 43kDa) were similar in normoxia and hypoxia in uninfected cells, a stronger signal was seen in normoxia compared to hypoxia after infection with either γ134.5-deleted virus or C154. This was more pronounced in GBM-XD456 (1.9-fold greater signal intensity in C154 infected cells in normoxia compared to hypoxia and 1.5-fold greater signal intensity in cells infected by the ICP34.5- virus, as measured by densitometry). Furthermore, the signal was most intense in C154 infected cells. Activated Hsp27 (phosphorylation on Ser82, detected at 27 kDa) followed a similar pattern to activated p38 MAPK with higher signal apparent in GBM-XD456 after infection in normoxia and with highest levels seen in C154 infected cells. Signal intensity was 3.4-fold greater in normoxia compared to hypoxia for ICP34.5- infected cells and 3.1-fold greater for C154 infected cells.
Figure 6.

Total P38 MAPK (T-p38 MAPK), activated P38 MAPK (p-P38 MAPK), and activated Hsp27 (p-Hsp27) in normoxia or hypoxia at baseline (control) or 24 hours after infection at 10 MOI with ICP34.5-deleted virus (34.5-) or chimeric C154 virus in GBM-D456MG and GBM-X12 cells. 35 μg of whole cell lysate was used for western blot analysis.
Western blot experiments were repeated at 24 hours post-infection with similar results (Figure 6). Total p38 MAPK was comparable in uninfected and infected cells. In GBM-XD456, activated p38 MAPK remained high in infected cells in normoxia but were nearly undetectable by 24 hours in hypoxia for both ICP34.5- and C154 infected cells. Similarly, much more intense p-p38 MAPK signals were seen in infected GBM-X12 cells by 24 hours whereas faint bands were seen in hypoxia (14-fold greater signal intensity in normoxia compared to hypoxia for ICP34.5- infected cells and 9-fold greater for C154 infected cells). Similar to p-p38 MAPK levels, increased p-Hsp27 was seen in GBM-X12 infected cells in normoxia compared to hypoxia (2.1-fold greater in ICP34.5- infected cells and 1.2-fold greater in C154 cells). These data indicate that the relative activation of p38 MAPK, which is known to enhance expression of late viral genes, is diminished in hypoxia after infection, and the chimeric virus results in greater activation compared to the ICP34.5- virus.
Since p38 MAPK signaling was greater in normoxia, we next sought to determine if inhibiting p38 MAPK activation in normoxia could decrease virus yields. GBM-XD456 cells were treated with a single 10μM dose of p38 MAPK inhibitor (SB 203580). At 2, 4, 6 or 24 hours after the inhibitor was added, cells were infected with ICP34.5- virus for 24 hours and p-p38 MAPK and p-Hsp27 were assessed by western blotting. Adding inhibitor 2, 4 or 6 hours prior to infection resulted in decreased p-p38 MAPK signal with the greatest decrease at 6 hours (61% of the control (total p38 MAPK); Figure 7a). By 24 hours the signal intensity of p-p38 MAPK was back to 81% of the control. Hsp27 was inhibited at all time points with greatest inhibition at 24 hours (52% of the control). The results demonstrate that activation of p38 MAPK and Hsp27 was inhibited by a single dose of SB 203580.
Figure 7.
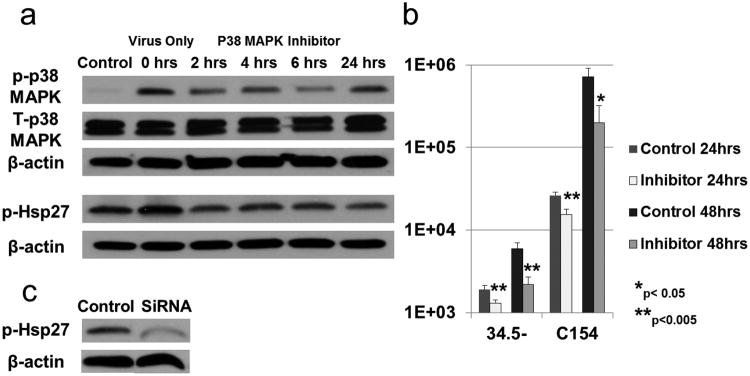
(a) Activated p38 MAPK (p-p38 MAPK) and Hsp27 (p-Hsp27) in GBM-XD456 cells (control) infected for 24 hours with ICP34.5- virus at 10 MOI. At time point 0 hours (virus infection only), a single dose of p38 MAPK inhibitor was added at 10μM. At 2, 4, 6 and 24hrs after the inhibitor was added, cells were infected with ICP34.5- virus for 24 hrs. (b) Virus recovery (mean + SD) in GBM-XD456 cells 24 and 48 hours post-infection with ICP34.5- or chimeric C154 virus in normoxia with (Inhibitor) and without (Control) a single 10μM dose of p38 MAPK inhibitor 24 hours prior to infection. (c) Activated Hsp27 (p-Hsp27) in GBM-XD456 cells 48 hour post-transfection with Hsp27 SiRNA or scrambled siRNA (control).
Next, we determined virus recovery in GBM-XD456 cells 24 and 48 hours post infection with ICP34.5- or C154 virus in normoxia with and without a single 10μM dose of p38 MAPK inhibitor 24 hours prior to infection. The single dose of the inhibitor significantly reduced virus yields in both the ICP34.5- and C154 virus at both 24 hours and 48 hours (Figure 7b). To confirm that inhibition of Hsp27 results in decreased virus recovery, GBM-XD456 cells transfected with small interfering RNA (siRNA) specific for Hsp27 (Figure 7c) or non-specific, control siRNA were infected for 24 hours with C101 and virus recovery was determined. Knockdown of Hsp27 resulted in a 38.7% (p=0.002) and 40.9% (p=0.0021) decrease in virus yield at 24 hours and 48 hours respectively. Together, these data show that inhibition of p38 signaling and knock-down of its substrate Hsp27 reduces virus yields, and based on the increased activation of p38 MAPK in normoxia compared to hypoxia, these data indicate that the p38 pathway may play a role in the reduced efficacy of the γ134.5-deleted virus in hypoxia.
To establish if p38 MAPK inhibition reduced virus recovery by affecting virus entry, the expression of CD111, an adhesion molecule that is the most efficient mediator of HSV-1 entry, was compared in GBM-XD456 cells treated or untreated with a single 10μM dose of p38 MAPK inhibitor for 6, 12 or 24 hours.17 At each time point, CD111 expression was not significantly different (p=0.9) in untreated and treated cells. Next, cells treated or untreated with a 10μM dose of p38 MAPK inhibitor for 24 hours were infected with ICP34.5- virus at 10 MOI for 6 hours, and the percentage of cells infected was determined by florescent activated cell sorting (FACS) for GFP expression. A similar percentage of cells that received the p38 MAPK inhibitor were infected as compared to the control (25.4 ± 6.9% versus 25.5 ± 3.4% respectively; p=1.0). Since GFP is produced with immediate-early gene expression kinetics, the infectivity data combined with the entry receptor expression data suggest that effects of the p38 MAPK inhibitor on virus recovery occur post-virus entry.
Discussion
Increasing evidence has implicated hypoxia in mediating phenotypic cellular changes that result in tumorigenesis; increased invasiveness, metastatic potential, induction of angiogenesis, resistance to chemotherapy and radiation; and decreased patient survival.12 Many of these resultant changes appear to be due to hypoxia's regulation of GSCs; hypoxia increases the CD133+ GSC fraction and promotes the self-renewal, proliferation and survival capability of GSCs.13,14 Thus, for an oHSV to have maximum therapeutic effect, it must be able to infect, replicate in, and kill GSCs in hypoxia. While previous phase I adult recurrent GBM trials utilizing γ134.5-deleted viruses demonstrated safety and some evidence of efficacy, the responses were more modest compared to the preclinical studies, and one explanation may be the differential sensitivity of GBM cells under normoxia, the oxygen tension traditionally used in in vitro experiments, compared to hypoxia.7,8
We previously showed that CD133+ GSCs express the primary HSV-1 entry molecule CD111/nectin-1 in similar amounts to CD133- tumor cells and that the GSCs that expressed CD111 were sensitive to γ134.5-deleted viruses in normoxic conditions.18 We subsequently showed that CD111 increased significantly in hypoxia; however, just having more HSV entry molecules per cell did not improve efficacy.9 Our data showed that the first-generation ICP34.5-virus had decreased infectivity, replication, and cytotoxicity in GBM xenolines under hypoxia. In this study, we demonstrate that the chimeric HCMV/HSV-1 oncolytic virus outperformed the γ134.5-deleted virus in infecting, replicating in and killing pediatric and adult GBM xenoline cells including CD133+ GSCs in hypoxia and normoxia. Significantly, the chimeric virus infected and killed a GBM xenoline, which is resistant to ICP34.5- virus oncolysis, in a fashion similar to that of wild-type HSV-1. These compelling data suggest that the chimeric virus, which has been produced in clinical grade, will have improved therapeutic effect and, combined with earlier data showing safety and efficacy of the chimeric virus in vivo, strongly support the use of the chimeric virus for upcoming brain tumor trials.6
To gain a better understanding of the differential sensitivity of GBM xenolines to the ICP34.5- virus in normoxia compared to hypoxia, we explored the p38 MAPK signaling pathway and its known substrate, Hsp27, which to our knowledge has not been previously examined in tumor cells infected with attenuated oncolytic HSVs. The p38 MAPK pathway is one of three signaling pathways driven by members of the mitogen activated protein kinase superfamily, which includes the extracellular signal-regulated protein kinases (ERKs) and the c-Jun N-terminal kinases (JNKs). p38 is known to be phosphorylated and activated in normal tissue by oxidative and environment stresses such as hypoxia and has an important role in apoptosis regulation.19,20 In an animal model, hypoxia upregulated p38 MAPK phosphorylation in cortical and hippocampal neurons.21 While there are limited studies of the effects of hypoxia on p38 MAPK in gliomas and none in GBM xenografts or xenolines, activation of p38 MAPK in established glioma cell lines under normoxic conditions resulted in increased levels of VEGF secretion.22 In the GBM xenolines, we found that total p38 MAPK levels were similar in hypoxia and normoxia without infection, which is consistent with the findings by Levin et al. in established glioma cell lines, and post-infection.23 A unique finding is that hypoxia did not increase activation of p38 MAPK in the glioma xenoline cells without infection. This may be due to the fact that the cells were allowed to equilibrate in the hypoxic environment for 4-7 days prior to any experiments being performed.
Importantly, while p38 MAPK activation increased after infection with either the ICP34.5- virus or the chimeric virus, the relative increases were less in hypoxia and p38 activation had returned almost to baseline 24 hours post-infection whereas activation of p38 MAPK remained elevated at 24 hours after infection in normoxia. Under hypoxic conditions, the stresses on the cell to maintain levels of ATP needed for regulatory states of multiple kinase-driven pathways likely govern the continued activation processes and protective mechanisms are likely evoked by these stresses to limit the expenditure of energy resources. Evanescent activation of mechanisms utilized by the virus for its replication likely limited the amount of virus produced under hypoxia. Furthermore, we showed that inhibiting p38 MAPK or Hsp27 knockdown significantly decreased viral recovery in the ICP34.5- virus and the chimeric virus. The effect of p38 MAPK inhibition on virus replication is likely post-entry as inhibition did not affect expression of the entry molecule CD111 or alter early infectivity.
The immediate-early HSV-1 protein ICP27 induces activation of p38 MAPK.24 Zachos et al.25,26 demonstrated that wild-type HSV-1 infection stimulates activation of the p38 pathway to enhance transcription of specific viral gene promoters and to improve viral replication; however a more recent study found that inhibition of p38 did not affect the transcriptional programs of wild-type HSV-1.27 Mezhir et al. showed that in pancreatic tumors treated with oHSV, ionizing radiation activates late HSV-1 promoters through activation of the p38 MAPK pathway, although the mechanism by which p38 MAPK acts to enhance expression of late viral genes has yet to be elucidated.28 Mathew et al. found that Hsp27 is rapidly phosphorylated in a p38-dependent manner and in HeLa cells depleted of all forms of Hsp27, virus replication was significantly reduced suggesting that the chaperone protein is necessary for productive viral replication.16 In this study, we show that similar to p38 activation, Hsp27 activation was increased in normoxia post-infection. Thus, the increased signaling of p38 MAPK, as measured by phosphorylated p38 MAPK and Hsp27, in normoxia in contrast to hypoxia coupled with the reduced viral yields after p38 inhibition and Hsp27 knockdown suggest that the p38 pathway plays an important role in the reduced efficacy of the γ134.5-deleted virus in hypoxia.
Interestingly, the chimeric virus was not limited to the same extent as the γ134.5-deleted virus in hypoxia and was able to infect CD133+ GSCs equally in normoxia and hypoxia. Unlike ICP34.5- mutants, the chimeric virus expresses the CMV PKR-evasion gene IRS1 and has improved late viral protein synthesis which likely provides the virus with a replication advantage over the ICP34.5- virus in hypoxia despite decreased p38 MAPK activation.5 It is not known if other genetic modifications to improve replication of γ134.5-deleted HSV would be as effective in hypoxia. For example, talimogene laherparepvec (T-VEC) has a deletion of the α47 gene that juxtaposes its promoter to the Us11 coding sequences causing Us11 to be expressed with alpha-gene kinetics rather than in its native late expression. Us11, expressed early, is able to block shut-off of host protein synthesis by a mechanism different from that of γ134.5. 29 This improves virus replication but whether it overcomes the inhibitory effects of hypoxia or has improved infectivity of GSCs is unknown.29 The greater activation of p38 MAPK seen after infection with the chimeric virus relative to the γ134.5-deleted virus may at least partially contribute to the improved efficacy in hypoxia. What role the IRS1 gene plays in the chimeric virus’ ability to replicate in hypoxia requires further investigation. In conclusion, the chimeric HCMV/HSV-1 oncolytic virus was superior to the ICP34.5- virus at infecting and killing pediatric and adult GBM xenolines in hypoxia and our data suggest that the chimeric virus is better equipped to target resistant cells in the hypoxic microenvironment.
Materials and Methods
Human Glioblastoma Xenolines
All xenograft lines or “xenolines” were established as tumor cell lines by implanting freshly resected human GBM tissues directly into the flanks of athymic nude (nu/nu) mice. The xenolines were then maintained by serial transplantation in athymic nude mice. GBM-X12 and GBM-X6 were established from adult patients and were provided by C. David James, Ph.D. and Jann Sarkaria, M.D., Mayo Clinic. GBM-XD456 was from a pediatric patient and was provided by Darell D. Bigner, M.D., Ph.D., Duke University Medical Center. The University of Alabama at Birmingham Institutional Animal Care and Use Committee approved the uses of all animal subjects (APN130908973; APN090108642; APN 130509395).
Genetically Engineered Herpes Simplex Viruses
C101 is a γ134.5-deleted (ICP34.5-) recombinant virus that has been previously described.5 C154 was constructed from the C134 chimeric HSV, which has been previously described.5 C154 was derived from the Δγ134.5 recombinant C101 and contains the HCMV IRS1 gene and the gene encoding enhanced green florescent protein (eGFP) under control of the HCMV immediate early (IE) promoter in the UL3-UL4 domain. M2001 has previously been described as an HSV-1 wild type strain F virus with the gene encoding eGFP under control of the CMV IE promoter.9 GFP expression from the viruses was used to determine the percentage of infected and uninfected cells after a specified time period by FACS analysis.
Tumor disaggregation and tissue culture
Xenoline tumors were aseptically harvested from the flank of mice and disaggregated as previously described9,17 or via a gentleMacs Disscociator® (Miltenyi Biotec, Auburn, CA) per standard protocol. Collected cells were washed twice with serumless DMEM/F12 (200×G, 8 minutes, room temp) and added to NeuroBasal (NB) medium (Invitrogen, Grand Island, NY) prepared with fibroblast growth factor-β (Invitrogen) and epidermal growth factor (Invitrogen) at 10ng/ml, 2% B-27 supplement without vitamin A (Invitrogen), 2mM L-glutamine, amphotericin B (250 μg/ml) and gentamicin (50 μg/ml). This medium was used to promote growth and slow differentiation of GSCs. Medium was exchanged every 3-7 days as needed, and cells grew non-adherently as neurospheres. Cells were maintained in either normoxia (20.8% O2) or hypoxia (1% O2) in a hypoxia chamber (BioSpherix, New York, NY) for 4-7 days prior to performing studies. Before exposure to the cells, all media and solutions were equilibrated to the 1% oxygen tension. All procedures were conducted within the hypoxia chamber through fitted glove ports. Experiments were performed in this hypoxic environment to recapitulate the severe hypoxia experienced by high-grade glioma cells which enhances stem-like properties and promotes aggressiveness and chemo- and radio-resistance.10-14
In Vitro Infectivity Assay
To determine the ability of a C101, C154 and M2001 to infect glioma tumor cells and GSC, we dissociated neurospheres grown in normoxia or in hypoxia (1% O2) into a single cell suspension using Accutase (Innovative Cell Technologies, San Diego, CA) supplemented with 2.5μg/ml of DNAse I (Worthington Biochemical Co) as we have previously described.9,17 Single cells were replated at 5×105 cells/0.8ml of NB medium in a 12 well flat bottom plate (Corning Incorporated, Corning, NY). After overnight culture, virus was added at various MOI: 0, 1, 3.3, and 10 (PFU/cell in 200μl of medium. At 30 hours post-infection, cells and neurospheres were collected from each well, dissociated with Accutase-DNAse I and prepared for FACS analysis as previously described using a fluorochrome-conjugated monoclonal allophycocyanin (APC)-CD133 antibody (Miltenyi Biotec).9,17 To determine the effect of p38 MAPK inhibition on cell entry, CD111 (PRR1) (Immunotech, Marseille, France) expression was determined by FACS in D456MG treated or untreated with a single 10μM dose of SB 203580 (Sigma-Aldrich, St. Louis, MO) for 6, 12 or 24 hours. Cells were infected 24 hours later with C101 at 10 MOI for 6 hours and GFP expression was determined by FACS. Data were analyzed with FlowJo v10.0.6 software (Tree Star Inc., Ashland, OR), and results were expressed as a percentage of gated cells based on antibody binding and GFP. Mean values from multiple determinations were calculated and paired student t-tests were used to determine significance.
In Vitro Cytotoxicity Assay
Cultured cells grown in normoxia and hypoxia were dissociated as described above and 104 cells/well were plated in 96 well plates overnight. Graded doses (0 to 100 PFU/cell) of C101, C154 or M2001 were added to each row. 72 hours post-infection, 25μl of alamarBlue® (Life Technologies, Grand Island, NY) was added to each well and incubated for 6-8 hours to detect metabolic activity as indicated by reduction of the dye from dark blue to pink. These color changes were quantified with a BioTek microplate spectrophotometer (Winooski, VT), and OD595-562nm values were used to calculate an LD50. Each experiment always included mock-treated controls, and the graded doses of virus were internally compared to the controls, which represented 100% tumor cell survival. These controls normalized each experimental oxygen condition.
Viral Recovery Assay
Xenoline cells (3 × 105) grown in normoxia or hypoxia were infected in parallel with C101, C154, or M2001 virus at an MOI of 0.1. After 2h, the virus inocula were removed and replaced with medium. At 48 hours post-infection, the cultures were frozen (-80°C) and virus recovery was measured by limiting plaque dilution on Vero cells as previously described.5 For p38 MAPK inhibitor studies, 2.5 × 105 cells were grown in normoxia with and without a single 10μM dose of SB 203580 (Sigma-Aldrich) and infected with C101 or C154 for 24 or 48 hours and then viral recovery was determined as above.
Immunoblotting
Cells were seeded at 1×106 cells/well in 1600ul of medium in 6-well plates in normoxia or hypoxia. The next day, cells were infected at 10 PFU/cell with C101 or C154 for 24 hours. Uninfected cells served as controls. An equal amount of protein (35ug) from each lysate sample was loaded in a well of 4-15% SDS-PAGE mini gels (Bio-Rad, Hercules, CA), electrophoretically separated, transferred to PVG membranes, and immunoblotting was performed as previously described.5 Antibodies used were p38 total (H147; Santa Cruz, Dallas, TX); p-p38 (Thr180/Tyr182; Cell Signaling Technology, Danvers, MA); p-Hsp27 (ser82: Cell Signaling Technology); and β-actin, (Sigma-Aldrich) which was used to confirm equal protein loading.
In vivo infectivity
GBM-XD456 cells (3 × 105) were injected in the right cerebral hemisphere of athymic nude mice as previously described.30 The tumor bed was injected 10 days later with 1 × 107 PFU of C101, C154 or saline. Mice were euthanized 24 hours post-injection and the tumors harvested, fixed in formalin, and paraffin embedded for IHC and fluorescence microscopy. Tumor sections were deparaffinized, treated in a Citrate buffer (Dako, Carpinteria, CA) to retrieve antigens, incubated in Power Block (BioGenex, Fremont, CA) to block non-specific binding. Sections were then exposed to CA9 (abcam, Cambridge, MA) primary antibody at 1:500 or normal rabbit IgG as a negative control. Cy3 conjugated AffinityPure F(ab)2 fragment donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Suffolk, UK) was used for a secondary antibody. DNA dye DAPI (Invitrogen) was used to counterstain sections. Slides were mounted with Fluoromount-G (Southern Biotech, Birmingham, AL). Photos were taken microscopically with Olympus IX70 connected to a DP71 digital camera (Olympus, Center Valley, PA) and analyzed with software from the manufacturer.
Transfection with siRNA
GBM-XD456 cells were transfected with siRNA designed to interfere with hsp-27 (5′-GUCUCAUCGGAUUUUGCAGC-3′; Dharmacon, Lafayette, CO) or scrambled siRNA (5′-CAGCGCUGACAACAGUUUCAU-3′; D-001206; Dharmacon) as a control at a concentration of 200 nM per well using Oligofectamine (Invitrogen) transfection reagent. Cells were infected 48 hours post-transfection with C101 and virus recovery was determined after 24 and 48 hours.
Statistical analyses
Student's t-test analyses for significance of mean differences were performed using Microsoft Excel (Microsoft Corp, Redmond, WA). All experiments were performed at a minimum in triplicate. A p value of ≤ 0.5 was considered significant.
Acknowledgments
Funding was provided by the St. Baldrick's Foundation, The Rally Foundation, Hyundai Hope on Wheels, and Kaul Pediatric Research Institute to GKF, and the National Institutes of Health (NIH) (CA071933, CA097247, CA151129). We thank Enid Keyser and the Analytic and Preparative Core Facility (supported by P30 AR048311 and P30 AI027767) for assistance with FACS analysis and Drs. Darell Bigner, Jann Sarkaria and C. David James for sharing the human xenograft lines with us for these studies.
Footnotes
Conflict of Interest: Dr. Markert and Dr. Gillespie are founders of and own stock and stock options (<7% interest) in Catherex, Inc., and in Aettis, Inc., biotechnology companies that are developing other oncolytic HSV that are not the subject of this current investigation. They serve as consultants for Catherex, Inc. as well. Dr. Gillespie currently serves as one of 5 unpaid members of the Board of Directors for Catherex, Inc. The virus employed in this study is not licensed by either company or the subject of any company plans for clinical use. Dr. Gillespie has served as a paid advisor to the Program Project at the Ohio State University that seeks to find improved methods for application of oncolytic HSV to treat localized and metastatic cancers. This is generally, but not specifically, related to the subject matter of this investigation.
References
- 1.Ries L, Melbert D, Krapcho M, Mariotto A, Miller B, Feuer E, et al. SEER Cancer Statistics Review, 1975–2004. Bethesda, MD: National Cancer Institute; 2006. [Google Scholar]
- 2.Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol. 2011;13:317–323. doi: 10.1093/neuonc/noq191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 5.Cassady KA. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J Virol. 2000;9:8707–8715. doi: 10.1128/JVI.79.14.8707-8715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah AC, Parker JN, Gillespie GY, Lakeman FD, Meleth S, Markert JM, et al. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther. 2007;14:1045–54. doi: 10.1038/sj.gt.3302942. [DOI] [PubMed] [Google Scholar]
- 7.Markert JM, Medlock MD, Rabkin S, Gillespie G, Todo T, Hunter W, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 8.Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–866. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 9.Friedman GK, Haas MC, Kelly VM, Markert JM, Gillespie GY, Cassady KA. Hypoxia moderates γ(1)34.5-deleted herpes simplex virus oncolytic activity in human glioma Xenoline primary culture. Transl Oncol. 2012;5:200–207. doi: 10.1593/tlo.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans SM, Judy KD, Dunphy I, Jenkins WT, Nelson PT, Collins R, et al. Comparative measurements of hypoxia in human brain tumors using needle electrodes and EF5 binding. Cancer Res. 2004;64:1886–1892. doi: 10.1158/0008-5472.can-03-2424. [DOI] [PubMed] [Google Scholar]
- 11.Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang WT, Nelson PT, et al. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res. 2004;10:8177–1884. doi: 10.1158/1078-0432.CCR-04-1081. [DOI] [PubMed] [Google Scholar]
- 12.Jensen RL. Brain tumor hypoxia: tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a therapeutic target. J Neurooncol. 2009;92:317–335. doi: 10.1007/s11060-009-9827-2. [DOI] [PubMed] [Google Scholar]
- 13.McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, Tofilon PJ. Physiologic oxygen concentration enhances the stem-like properties of CD133+ human glioblastoma cells in vitro. Mol Cancer Res. 2009;7:489–497. doi: 10.1158/1541-7786.MCR-08-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009;8:3274–3284. doi: 10.4161/cc.8.20.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zachos G, Koffa M, Preston CM, Clements JB, Conner J. Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication. J Virol. 2001;75:2710–2712. doi: 10.1128/JVI.75.6.2710-2728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Mathew SS, Della Selva MP, Burch AD. Modification and reorganization of the cytoprotective cellular chaperone Hsp27 during herpes simplex virus type 1 infection. J Virol. 2009;83:9304–9312. doi: 10.1128/JVI.01826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krummenacher C, Baribaud F, Ponce de Leon M, Baribaud I, Whitbeck JC, Xu R, et al. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology. 2004;322:286–299. doi: 10.1016/j.virol.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Friedman GK, Langford C, Coleman J, Cassady KA, Parker JN, Markert JM, et al. Engineered herpes simplex virus efficiently infects and kills CD133+ glioma cells that express CD111. J Neurooncol. 2009;95:199–209. doi: 10.1007/s11060-009-9926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cowan KJ, Storey KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol. 2003;206:1107–1115. doi: 10.1242/jeb.00220. [DOI] [PubMed] [Google Scholar]
- 20.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 21.Haddad JJ, Hanbali LB. Hypoxia upregulates MAPK(p38)/MAPK(ERK) phosphorylation in vitro: neuroimmunological differential time-dependent expression of MAPKs. Protein Pept Lett. 2014;21:444–451. doi: 10.2174/092986652105140218112521. [DOI] [PubMed] [Google Scholar]
- 22.Yoshino Y, Aoyagi M, Tamaki M, Duan L, Morimoto T, Ohno K. Activation of p38 MAPK and/or JNK contributes to increased levels of VEGF secretion in human malignant glioma cells. Int J Oncol. 2006;29:981–987. [PubMed] [Google Scholar]
- 23.Levin VA, Panchabhai S, Shen L, Baggerly KA. Protein and phosphoprotein levels in glioma and adenocarcinoma cell lines grown in normoxia and hypoxia in monolayer and three-dimensional cultures. Proteome Sci. 2012;10:5. doi: 10.1186/1477-5956-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillis PA, Okagaki LH, Rice SA. Herpes simplex virus type 1 ICP27 induces p38 mitogen-activated protein kinase signaling and apoptosis in HeLa cells. J Virol. 2009;83:1767–1777. doi: 10.1128/JVI.01944-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zachos G, Clements B, Conner J. Herpes simplex virus type 1 infection stimulates p38/c-Jun N-terminal mitogen-activated protein kinase pathways and activates transcription factor AP-1. J Biol Chem. 1999;274:5097–5103. doi: 10.1074/jbc.274.8.5097. [DOI] [PubMed] [Google Scholar]
- 26.Zachos G, Koffa M, Preston CM, Clements JB, Conner J. Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication. J Virol. 2001;75:2710–2728. doi: 10.1128/JVI.75.6.2710-2728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Karaca G, Hargett D, McLean TI, Aguilar JS, Ghazal P, Wagner EK, et al. Inhibition of the stress-activated kinase, p38, does not affect the virus transcriptional program of herpes simplex virus type 1. Virology. 2004;329:142–156. doi: 10.1016/j.virol.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 28.Mezhir JJ, Advani SJ, Smith KD, Darga TE, Poon AP, Schmidt H, et al. Ionizing radiation activates late herpes simplex virus 1 promoters via the p38 pathway in tumors treated with oncolytic viruses. Cancer Res. 2005;65:9479–9484. doi: 10.1158/0008-5472.CAN-05-1927. [DOI] [PubMed] [Google Scholar]
- 29.Mohr I, Sternberg D, Ward S, Leib D, Mulvey M, Gluzman Y. A herpes simplex virus type 1 gamma34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J Virol. 2001;75:5189–5196. doi: 10.1128/JVI.75.11.5189-5196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A. 2000;97:2208–2213. doi: 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]