

Abstract
BH3 mimetic drugs may be useful to treat acute lymphoblastic leukemia (ALL) but the sensitivity of primary tumor cells has not been fully evaluated. Here B-lineage ALL cell cultures derived from a set of primary tumors were studied with respect to sensitivity to the BH3 mimetics ABT-263 and ABT-199 and to Bcl-2 dependence and function. These ALL cells each expressed high levels of Bcl-2 and exhibited great sensitivity to ABT-263 and ABT-199, which induced rapid apoptotic cell death. BH3 profiling indicated that the ALL cultures were Bcl-2 dependent. Co-immunoprecipitation studies revealed a multi-faceted role for Bcl-2 in binding pro-apoptotic partners including Bax, Bak, Bik and Bim. ABT-263 disrupted Bcl-2:Bim interaction in cells. Mcl-1 overexpression rendered ALL cells resistant to ABT-263 and ABT-199 with Mcl-1 assuming the role of Bcl-2 in binding Bim. Freshly isolated pediatric ALL blasts also expressed high levels of Bcl-2 and exhibited high sensitivity to Bcl-2 inhibition by the BH3 mimetic compounds. Overall our results showed that primary ALL cultures were both more sensitive to BH3 mimetics and more uniform in their response than established ALL cell lines which have been evaluated previously. Further, the primary cell model characterized here offers a powerful system for preclinical testing of novel drugs and drug combinations to treat ALL.
Keywords: BH3 mimetics, acute lymphoblastic leukemia, Bcl-2, Mcl-1, apoptosis
Introduction
Bcl-2 family proteins function as key regulators of intrinsic apoptosis (reviewed in (1-5)). They are divided into three main classes: multi-domain anti-apoptotic (or pro-survival) proteins which include Bcl-2, Bcl-xL, Mcl-1 and others; multi-domain pro-apoptotic proteins including Bax and Bak; and BH3-only pro-apoptotic proteins. The BH3-only proteins are divided further into activators (Bid and Bim), which can directly bind and activate Bax and Bak, and sensitizers (Bad, Bik, Hrk, Bmf, Noxa and Puma) which bind anti-apoptotic Bcl-2 proteins and prevent them from sequestering pro-apoptotic Bcl-2 proteins. Upon activation, Bax and Bak form pores in the mitochondrial membrane for the release of apoptogenic factors such as cytochrome c.
In order to survive in the face of abnormalities such as oncogene activation or genomic instability that would normally trigger cell death, cancer cells must acquire multiple anti-apoptotic mechanisms (6). Many of the selective blocks in apoptotic signaling in cancer cells involve Bcl-2 family proteins, including overexpression of anti-apoptotic members and/or defects in the activation or expression of pro-apoptotic members (7). Indeed, Bcl-2 itself was first recognized as a dysregulated and overexpressed gene that plays a key role in promoting survival of follicular lymphoma cells (8).
Importantly, dysregulation of death signaling not only promotes tumorigenesis but also confers resistance to anti-cancer drugs (9). It is well established that overexpression of pro-survival Bcl-2 proteins such as Bcl-2, Bcl-xL, or Mcl-1 blocks cell death induced by anti-cancer drugs resulting in chemoresistance (5, 7). Based on such observations, BH3 mimetics have been developed to target and disable pro-survival Bcl-2 proteins, as a means to overcome the mechanisms of apoptosis resistance and tilt the balance in favor of cell death. ABT-737, and the orally bioavailable derivative ABT-263, are mimetics based on the pro-apoptotic BH3-only protein Bad, which selectively target Bcl-2, Bcl-xL and Bcl-w, but do not inhibit Mcl-1 (10-16). ABT-263 has demonstrated in vitro activity in a wide range of cancer cell lines, primary leukemia cells, and xenograft models (10-17). Additionally, Phase I and II clinical trials conducted for several types of cancer have shown promising results (13, 14, 18-20). Because a limitation of ABT-263 is thrombocytopenia due to Bcl-xL inhibition in circulating platelets, the derivative ABT-199 was recently developed, which is selective for Bcl-2 and exhibits anti-tumor activity without significant thrombocytopenia (21).
Acute lymphoblastic leukemia (ALL) affects both adults and children (22, 23). Because cure rates have begun to plateau, new classes of therapeutic agents are needed, but these are difficult to evaluate systematically in patients especially in the context of polychemotherapy. Many continuously proliferating ALL cell lines have been established (24, 25), but after extensive propagation they have likely acquired properties which deviate from the originating primary cells. This emphasizes the need for preclinical cell models of ALL that more closely represent the disease. Recently, conditions were established for the expansion and long-term culture of primary adult ALL cells using a defined media that lacked serum and hematopoietic growth factors (26). This system provides a unique and powerful tool for the preclinical evaluation of novel therapies for ALL. In the present study, we examined ABT-263 and ABT-199 sensitivity, and Bcl-2 dependence and function, in several of these ALL cultures as well as in freshly isolated pediatric ALL blasts. These results demonstrate the utility of these expanded primary cultures for preclinical studies of ALL, provide mechanistic insight into the determinants of sensitivity and resistance to BH3 mimetics, and have important implications for the optimal use of these compounds in adult and pediatric ALL.
Materials and Methods
Materials, Cell extraction and immunoblotting, Caspase-3 assay, and Co-immunoprecipiation
Cell culture
KB3 cells (HeLa subline) were maintained in DMEM, and RS4;11 and NALM-6 cell lines were maintained in RPMI-1640 medium, supplemented with 10% bovine growth serum, 2 mM L-glutamine, 50 units/mL penicillin, and 50 μg/mL streptomycin. ALL cell cultures were maintained in suspension as described (26) in Iscove’s modified Dulbecco’s medium (IMDM) containing serum-free supplement (10 μg/mL cholesterol, 6 mg/mL human serum albumin, 0.5 μg/mL amphotericin, 1 μg/mL insulin, 200 μg/mL human apo-transferrin, 50 μM 2-mercaptoethanol, 2 mM glutamine and 50 units/mL penicillin). Mcl-1-dependent and Bcl-2-dependent leukemia cell lines were described previously (27). Cells were maintained at 37°C and 5% CO2. Authentication of the cell lines and ALL cultures was established via short tandem repeat (STR) profiling in September, 2014, by Genetica DNA Laboratories (LabCorp Speciality Testing Group, Burlington, NC). The STR profile of each cell line matched that of reference profiles available in the ATCC database. The primary ALL cell culture profiles did not match any repository cell lines, as expected, and each profile was unique with respect to the others.
Cell viability assay
Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as described (28). Cells (30,000 per well) were seeded in 96-well plates, and either ABT-263 or ABT-199 was added in a fixed final concentration of 0.1% DMSO. After 72h, MTT reagent (50 μg/10 μL/well) was added and incubated overnight at 37°C. The following day, 0.1 mL of 10% SDS in 0.01 M HCl was added, and after overnight incubation, absorbance readings were taken at 540 nm.
BH3 profiling
Whole cell (JC-1) BH3 profiling was performed as described previously (29, 30). Briefly, cells were harvested, washed, and resuspended in Newmeyer buffer (0.3 M trehalose, 10 mM HEPES-KOH pH 7.7, 80 mM KCl, 1 mM EGTA, 1 mM EDTA, 0.1% BSA, and 5 mM succinate). An equal volume of dye mastermix (4 μM JC-1, 0.2% digitonin, 40 μg/μL oligomycin, 20 mM β-mercaptoethanol in Newmeyer buffer) was then added and after 10 min in the dark, aliquots of 30 μL were added to the wells of a black, clear bottom 96-well plate containing 30 μL of 20 μM peptide in Newmeyer buffer. The peptides corresponded to the BH3 domain of different pro-apoptotic Bcl-2 proteins as follows: Bim – MRPEIWIAQELRRIGDEFNA; Bid – EDIIRNIARHLAQVGDSMDRY; Bad – LWAAQRYGRELRRMSDVEFEGSFKGL; Noxa – AELPPEFAAQLRKIGDKVYC; Puma – EQWAREIGAQLRRMADDLNA; Bmf – HQAEVQIARKLQLIADQFHRY; and Hrk – WSSAAQLRAARLKALGDELHQ. Fluorescence was measured at an excitation wavelength of 530 nm and an emission wavelength of 590 nm every 5 min for 2 h. Readings were normalized against that obtained with 10 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) as a positive control for mitochondrial membrane depolarization. Each condition was conducted in triplicate.
Retroviral transduction
Retroviral transduction to stably overexpress Mcl-1 in primary ALL cells was performed as described previously (31). Briefly, full-length human Mcl-1 in pcDNA3.1 vector was cloned into the bicistronic LZRS vector containing a multiple cloning site followed by an Internal Ribosome Entry Site coupled to a truncated form of the nerve growth factor receptor (ΔNGFR) as a marker gene. The constructs were transfected into the ϕ-NX-A retroviral packaging cell line using Fugene HD reagent, and positive cells selected in medium containing 2 μg/mL puromycin. After 24 h in puromycin-free medium, the supernatant was collected, centrifuged to remove cell debris, aliquoted, and quick frozen. For transduction, 0.5 mL of supernatant was added to the wells of a 24-well plate coated with human fibronectin fragments (CH-296, Retronectin, TaKaRa Biotec, Otsu, Japan). Plates were centrifuged at 3,000 × g for 20 min, supernatant was removed, and 1.5 × 105 ALL cells were added per well. After 24 h, cells were removed and expanded for four weeks in medium containing serum-free supplement. Transduced cells were purified by magnetic activated cell sorting using anti-hNGFR-APC clone ME20.4 and anti-APC MicroBeads using a LS-column in combination with a QuadroMACS™ separator (Miltenyi Biotec, San Diego, CA), and Mcl-1 overexpression confirmed by flow cytometry for anti-hNGFR immunoreactivity and by Mcl-1 immunoblots.
Isolation of pediatric ALL blasts
Samples of marrow (10 mL) were obtained from pediatric ALL patients under a protocol approved by the UAMS Institutional Review Board. 25 mL of IMDM containing 2% FBS, 1% Penicillin/Streptomycin, 2 mM L-glutamine, and 25 mM Hepes, pH 7.2, was added, and the mixture was pipetted onto 12 mL of Ficoll-Paque PLUS (1.078 g/mL) which was centrifuged at 1,000 × g for 22 min. The cloudy layer of mononuclear cells containing the blasts was collected, two volumes of IMDM media added, and the solution centrifuged at 500 × g for 10 min. The cell pellet was suspended in growth media (IMDM with serum-free supplement), aliquots were frozen and stored, and the remainder cultured for experiments the next day. This procedure yielded highly purified ALL blast preparations, comprising 98-100% of the cells visualized by Giemsa staining and light microscopy.
Results
Primary ALL cells express high levels of Bcl-2
We initially selected seven primary adult ALL cell cultures from a total of twelve available samples for which long-term proliferation had been established (26). These were derived from patients 19, 2, 5, 14, 3, 12, and 6 (26). KB3, a HeLa subline, and RS4;11, a human B-cell ALL cell line, were used as comparative controls. The relative expression level of the three major pro-survival Bcl-2 proteins, Bcl-2, Bcl-xL and Mcl-1, was first examined by immunoblotting (Fig. 1). RS4;11 cells and all of the primary ALL cell cultures expressed high levels of Bcl-2 relative to KB3 cells, with quantitation of band intensity indicating levels 150- to 200-fold greater, with the exception of ALL-2 which was about 30-fold greater. In contrast and despite some variation, the ALL cells expressed levels of Bcl-xL more similar to, and levels of Mcl-1 lower than, KB3 cells.
Fig. 1. Primary ALL cells express high levels of Bcl-2.
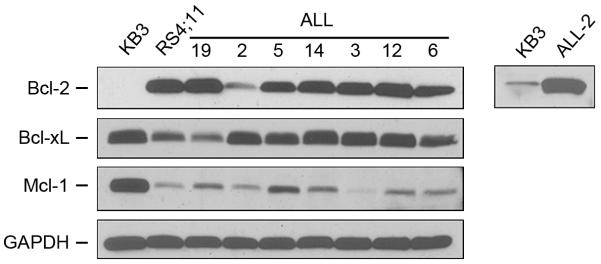
Extracts were made from KB3 or RS4;11 cell lines or from the indicated primary ALL cell cultures and subjected to immunoblotting for Bcl-2, Bcl-xL or Mcl-1. GAPDH was used as a loading control. The right panel shows an overexposure (20 × duration) of the indicated lanes.
Primary ALL cells are acutely sensitive to the Bcl-2 inhibitors ABT-263 and ABT-199
Cell viability assays were conducted after 3 day exposure of cells to the Bcl-2/Bcl-xL inhibitor ABT-263 (10) (Supplementary Fig. S1). KB3 cells were refractory to the effects of ABT-263, consistent with their strict dependence on Mcl-1 for survival (28) (Fig. S1 and Table 1). RS4;11 cells exhibited an IC50 value of 223 nM while an additional B-ALL cell line, NALM-6, were relatively more resistant with an IC50 value of 839 nM (Fig. S1 and Table 1). Strikingly, the primary ALL cell cultures were uniformly highly sensitive to ABT-263, with IC50 values ranging from 2 to 6 nM (Fig. S1 and Table 1). We next tested ABT-199, which is more selective for inhibition of Bcl-2 than Bcl-xL (21). KB3 cells were unaffected, RS4;11 cells were sensitive, and NALM-6 were relatively resistant (Supplementary Fig. S2 and Table 1). As with ABT-263, the primary ALL cell cultures were highly sensitive to ABT-199, with IC50 values ranging from 3-22 nM (Fig. S2 and Table 1).
Table 1.
Sensitivity of RS4;11 and NALM-6 cell lines and primary ALL cells to ABT-263 and ABT-199. Values shown are mean ± S.D., and are derived from data in Figs. S1 and S2.
| IC50 ABT-263 (nM) |
IC50 ABT-199 (nM) |
|
|---|---|---|
| RS4;11 | 223.5 ± 19.7 | 40.2 ± 9.8 |
| NALM-6 | 839 ± 11 | > 3,000 |
| ALL-19 | 1.92 ± 0.12 | 4.02 ± 0.44 |
| ALL-2 | 3.24 ± 0.15 | 9.69 ± 1.45 |
| ALL-5 | 4.25 ± 0.20 | 2.87 ± 0.38 |
| ALL-14 | 5.33 ± 0.71 | 6.76 ± 0.51 |
| ALL-3 | 5.94 ± 0.32 | 22.1 ± 4.7 |
| ALL-12 | 5.95 ± 0.38 | 6.15 ± 1.07 |
| ALL-6 | 3.27 ± 0.23 | 3.11 ± 0.23 |
Bcl-2 inhibition leads to rapid apoptotic death of ALL cells
In order to examine the kinetics of cell death, ALL cells were treated with lethal concentrations of ABT-263 or ABT-199 based on the cell survival curves (30 nM for primary ALL and 1 μM for RS4;11 cells) for periods up to 24 h, and cell extracts subjected to immunoblotting for PARP, which is cleaved into characteristic products upon apoptotic cell death. ABT-263 failed to induce PARP cleavage in KB3 cells as expected, induced complete PARP cleavage within 8 h in RS4;11 cells, and also induced rapid PARP cleavage in the primary ALL cultures (Supplementary Fig. S3A). Similarly, lethal concentrations of ABT-199 induced rapid PARP cleavage in RS4;11 cells and in many of the primary ALL cultures (Fig. S3B). For ALL-2, ABT-199 induced loss of expression of the 116 kDa form of intact PARP without evidence of the 85 kDa fragment being present, and for ALL-5, PARP cleavage was largely incomplete. Similar results for ALL-2 and ALL-5 were obtained at a higher ABT-199 concentration of 300 nM (data not shown). To complement these results and confirm apoptotic death in response to the BH3 mimetics, especially in the ALL cell cultures that displayed anomalies in PARP cleavage, caspase-3 assays were performed (Fig. S3C). Both ABT-263 and ABT-199 caused rapid caspase-3 activation in the RS4;11 cell line and in all of the primary ALL cultures, but failed to activate caspase-3 in KB3 cells.
Primary ALL cells are dependent on Bcl-2
To determine whether the primary ALL were dependent on Bcl-2, BH3 profiling was employed (29, 30). Peptides derived from a panel of BH3-only proteins are added to permeabilized cells and the pattern of peptides that causes loss of mitochondrial outer membrane potential identifies which anti-apoptotic protein is responsible for maintaining survival. Loss of mitochondrial membrane potential is measured by the decay of fluorescence of JC-1, with FCCP, a protonophore which depolarizes mitochondrial membranes, as a positive control. Two leukemia cell lines derived from transgenic mice, dependent on either Mcl-1 or Bcl-2 (27), were used as controls. With the Mcl-1 dependent cell line, loss of mitochondrial membrane potential occurred with peptides corresponding to the BH3 domains of Bim, Bid, Puma, Bmf, and Noxa, whereas peptides from Bad and Hrk were less effective (Fig. 2). This corresponds to the known binding profile of Mcl-1, which has poor affinity for Bad and Hrk, and to previously published BH3 profiling data for this cell line (27, 29, 30). With the Bcl-2 dependent cell line, loss of mitochondrial membrane potential occurred with peptides from the BH3 domains of Bim, Bid, Puma, Bmf, and Bad, whereas those from Noxa and Hrk were ineffective (Fig. 2). This corresponds to the known binding profile of Bcl-2, which has poor affinity for Noxa and Hrk, and to previously published BH3 profiling data for this cell line (27, 29, 30). RS4;11 cells and all of the ALL cell cultures clearly showed the Bcl-2 dependent profile (Fig. 2), indicating their Bcl-2 dependence and consistent with their high level of sensitivity to ABT-199.
Fig. 2. Primary ALL cells are Bcl-2-dependent.

Mcl-1-dependent or Bcl-2-dependent leukemia cell lines, RS4;11 cells, or primary ALL cultures, as indicated, were subjected to whole cell BH3 profiling with JC-1 dye, as described in Materials and Methods. The traces shown in the top of each panel represent an average of triplicates, and are color coded according to the key on the right of each set. The bar graphs in the lower portion of each panel represent mean ± s.d. (n = 3), normalized to that of FCCP (100% loss of mitochondrial membrane potential) and are derived from readings obtained at 120 min. Note that Noxa and Hrk peptides sometimes gave fluorescence readings (top panels) that were slightly greater than DMSO control, and these appear as small negative values in the bar graph representation in the lower panels.
Bcl-2 sequesters several pro-apoptotic Bcl-2 family members
We next sought to identify by co-immunoprecipitation which pro-apoptotic Bcl-2 family proteins were bound to Bcl-2. RS4;11 cells and the primary ALL cultures expressed, with varying relative levels, several pro-apoptotic Bcl-2 family members, including Bak, Bax, Bid, Bim and Bik (see lane 1 in Figs. 3A and 4A). Bcl-2 was first immunoprecipitated from RS4;11 cells (Fig. 3A) and efficient immunoprecipitation was demonstrated as Bcl-2 appeared mainly in the pellet (lane 5) and was depleted from the supernatant (lane 3), whereas in a mock immunoprecipitation performed in the absence of antibody, the protein remained in the supernatant (lane 2) and was absent from the pellet (lane 4). Immunoblotting for pro-apoptotic Bcl-2 proteins in the precipitated material showed bands which appeared to correspond to Bak, Bax, several forms of Bim (Bim-EL, Bim-L, and Bim-S), and Bik, although Bid, despite being present in the extract, was not detected (Fig. 3A). Identification of Bak, however, was complicated by the presence of two bands, one at 23 kDa and an additional band, presumably immunoglobulin light chain (IgG-LC) at around 25 kDa, migrating with a slower mobility (Fig. 3A, lane 5). We reasoned that the binding of Bak to Bcl-2 in the immunoprecipitate might be disrupted under mild detergent conditions such as 1% Triton X-100, whereas IgG-LC and Bcl-2 may remain more firmly bound to the Protein A/G beads under such conditions. To test this, Bcl-2 was immunoprecipitated from RS4;11 cells, and the washed beads incubated in buffer containing 1% Triton X-100, and the detergent-solubilized eluant subjected to immunoblotting (Fig. 3A, lane 7). Immunoblotting for Bak detected only the band at 23 kDa, indicating that the detergent elution step is useful in positive identification of proteins specifically bound to Bcl-2. Consistent with the ability of Triton X-100 to remove more loosely bound proteins from the beads, Bcl-2 itself was absent from the eluted material (Fig. 3A, lane 7). Bax was not detected in the detergent eluant, confirming that the band present in the immunoprecipitate (lane 5, asterisk), which ran slower than authentic Bax, was likely due to IgG-LC cross-reactivity. Strong bands corresponding to the three forms of Bim as well as Bik were clearly identified in the detergent eluant, confirming them as authentic Bcl-2 partners. Reciprocal immunoprecipitations were performed for further validation, and Bcl-2 was identified in immunoprecipitates of Bim (Fig. 3B) as well as Bik (Fig. 3C) but was not detected in an immunoprecipitate of Bax (data not shown). Thus in RS4;11 cells, Bcl-2 binds several pro-apoptotic partners including Bak, Bim and Bik, but does not appear to bind either Bax or Bid. To further verify the specificity of these interactions, immunoprecipitation with β-actin antibody as a negative control was performed. As shown in Supplementary Fig. S4, when β-actin was immunoprecipitated from an extract of RS4;11 cells, β-actin was detected whereas Bak, Bax, Bid and Bim were not detected. Interestingly, Bcl-2 was present in the β-actin immunoprecipitate, consistent with several reports suggesting these proteins interact (32, 33). Thus Bcl-2 exists in at least two complexes, with pro-apoptotic Bcl-2 proteins and with β-actin.
Fig. 3. Bcl-2 interacts with multiple pro-apoptotic Bcl-2 family proteins in RS4;11 cells.

A. Bcl-2 was immunoprecipitated from RS4;11 cells, and subjected to immunoblotting for Bcl-2 and the indicated pro-apoptotic Bcl-2 family members. WCE, whole cell extract; Sup, supernatant; IP, immunoprecipitate; eluant, detergent eluant of immunoprecipitate. Samples were obtained from immunoprecipitations performed in the absence (−) or presence (+) of Bcl-2 antibody. Asterisks indicate non-specific bands. B. Bim was immunoprecipitated from RS4;11 cells, and subjected to immunoblotting for Bim and Bcl-2. C. Bik was immunoprecipitated from RS4;11 cells, and subjected to immunoblotting for Bik and Bcl-2.
Fig. 4. Bcl-2 interacts with multiple pro-apoptotic Bcl-2 family proteins in primary ALL cells.

A. Bcl-2 was immunoprecipitated from ALL-19 cells and subjected to immunoblotting for Bcl-2 and the indicated pro-apoptotic Bcl-2 family members. WCE, whole cell extract; Sup, supernatant; IP, immunoprecipitate; eluant, detergent eluant of immunoprecipitate. Samples were obtained from immunoprecipitations performed in the absence (−) or presence (+) of Bcl-2 antibody. Asterisks indicate non-specific bands. B, C, and D. Bax (panel B), Bim (panel C) or Bik (panel D) were immunoprecipitated from ALL-19 cells, and subjected to immunoblotting for the proteins indicated.
One of the ALL primary cultures, ALL-19, was chosen for similar examination of Bcl-2 complexes, and with one exception, similar results were found. Thus Bak, Bim and Bik, but not Bid, were each positively identified in Bcl-2 immunocomplexes (Fig. 4A). A band appearing to correspond to Bax was also present in the Bcl-2 immunoprecipitate (Fig. 4A, lane 5), and its identification was strengthened as the same band was detected in the detergent eluant (Fig. 4A, lane 7). Reciprocal immunoprecipitations confirmed that Bax, Bim and Bik complexes with Bcl-2 in ALL-19 cells (Figs. 4B, C, D, respectively). Thus with the exception of Bax which complexes with Bcl-2 in the primary ALL cells but not in the RS4;11 cell line, the binding partners of Bcl-2 are similar.
ABT-263 displaces Bim from Bcl-2
The sensitivity of ALL cells to ABT-263 is likely based on the ability of the mimetic to bind directly to Bcl-2 and displace bound pro-apoptotic Bcl-2 proteins. We initially focused on Bim because it is a key activator BH3-only protein and found associated with Bcl-2 in ALL cells. To test the effect of ABT-263 on Bim:Bcl-2 interaction, RS4;11 cells were treated with vehicle or 1 μM ABT-263 for 4 or 6 h, and whole cell extracts were prepared, Bcl-2 immunoprecipitated, and subjected to immunoblot analysis for both Bcl-2 and Bim. As shown in Supplementary Fig. 5, lanes 9-11, while the levels of Bcl-2 in the immunoprecipitate remained unchanged, the levels of Bim, and most notably Bim-EL, were reduced following ABT-263 treatment. These results show that ABT-263 reduces the amount of Bim associated with Bcl-2 in RS4;11 cells, consistent with the mimetic acting to directly displace Bim from Bim:Bcl-2 complexes.
Mcl-1 overexpression renders ALL cells ABT-263/199 resistant
To further establish that the primary ALL cells are sensitive to the BH3 mimetics because of their strict dependence on Bcl-2 for survival, we next determined whether Mcl-1 overexpression would reverse sensitivity. Thus, human Mcl-1 was expressed via retroviral transduction in several of the ALL primary cultures, namely ALL-2, ALL-14 and ALL-19, as described in Materials and Methods. After FACS sorting for selection of cells with relatively high Mcl-1 expression, MTT viability assays were conducted. Results obtained for ALL-2 are presented in Fig. 5. Immunoblotting confirmed overexpression of Mcl-1 in the transduced cells while the expression level of Bcl-2 was decreased and that of Bcl-xL was relatively unchanged (Fig. 5A). Cell viability assays showed that even at the highest concentrations tested of 1 μM, ABT-263 reduced viability by only 40% (Fig. 5B) and ABT-199 was essentially ineffective (Fig. 5C). Thus in the context of Mcl-1 overexpression, IC50 values in ALL-2 cells were > 1 μM for each compound, compared to low nanomolar values in the absence of Mcl-1 overexpression. Essentially identical results were obtained with ALL-14 and ALL-19 cells which also became highly resistant to the Bcl-2 antagonists upon Mcl-1 overexpression (data not shown). Thus in Bcl-2 dependent ALL cells, Mcl-1 can compensate and maintain cell survival under conditions where the normal guardian, Bcl-2, is inhibited.
Fig. 5. Mcl-1 overexpression confers resistance to ABT-263 and ABT-199.

ALL-2 cells overexpressing Mcl-1 (ALL-2/Mcl-1) were prepared by retroviral transduction, as described in Materials and Methods. A. Extracts were made from ALL-2 and ALL-2/Mcl-1 cells and subjected to immunoblotting for Bcl-2, Bcl-xL or Mcl-1. GAPDH was used as a loading control. B. ALL-2 or ALL-2/Mcl-1 cells were treated with vehicle (100% viability) or increasing concentrations of ABT-263 for 72 h and cell viability was assessed by MTT assay as described in Materials and Methods. Results are given as mean ± s.d. (n = 6). C. ALL-2 or ALL-2/Mcl-1 cells were treated with vehicle (100% viability) or increasing concentrations of ABT-199 for 72 h and cell viability was assessed by MTT assay as described in Materials and Methods. Results are given as mean ± s.d. (n = 6). D. Overexpressed Mcl-1 sequesters Bim. Extracts from ALL-2 (lane 2) or ALL-2/Mcl-1 (lane 3) cells were immunoprecipitated with either Mcl-1 antibody (left panel) or Bcl-2 antibody (right panel) as indicated and subjected to immunoblotting for Mcl-1 and Bim (left) or Bcl-2 and Bim (right). Mock immunoprecipitations in the absence of primary antibody were also performed, using extracts from ALL-2 cells (lane 1 in each panel). The asterisk indicates a non-specific band.
Mcl-1 may provide anti-apoptotic function in the context of Bcl-2 inhibition due to its capacity to sequester pro-apoptotic Bcl-2 proteins normally bound to Bcl-2. To test this, Mcl-1 or Bcl-2 was immunoprecipitated from parental and Mcl-1-overexpressing ALL-2 cells, and immunoprecipitates probed for Bim. When Mcl-1 was immunoprecipitated from parental cells (Fig. 5D, left, lane 2), bands corresponding to Bim-L and Bim-S were not detected, whereas when Mcl-1 was immunoprecipitated from Mcl-1-overexpressing cells, Bim-L and Bim-S were clearly detected (lane 3) (note that the presence of Bim-EL was obscured by non-specific bands). Conversely, when Bcl-2 was immunoprecipitated, the amount of Bim associated with Bcl-2 was diminished in cells overexpressing Mcl-1 (Fig. 5D, right, compare lane 2 with lane 3). These results indicate that when overexpressed, Mcl-1 shares or attains Bcl-2’s responsibility in sequestering Bim.
Freshly isolated pediatric ALL blasts are highly sensitive to ABT-263
To further establish that primary ALL cells are highly sentive to Bcl-2 inhibtion, and to extend the study to include samples derived from pediatric patients, blasts were freshly isolated from two pediatric patients diagnosed with ALL. Immunoblotting indicated that the pediatric ALL cell cultures displayed anti-apoptotic Bcl-2 protein expression profiles similar to that observed for the adult ALL cultures, with high levels of Bcl-2, similar levels of Bcl-xL, and lower levels of Mcl-1, versus KB3 cells (Fig. 6A). Cell viability assays showed that both samples of the pediatric ALL blasts were highly sensitive to ABT-263, with IC50 values of 6.2 ± 0.3 nM and 6.7 ± 0.4 nM (Fig. 6B, 6C).
Fig. 6. Leukemic blasts from pediatric ALL patients express high levels of Bcl-2 and are highly sensitive to ABT-263.

Blasts from two patients, termed P1-ALL and P2-ALL, were isolated and cultured, as described in Materials and Methods. A. Extracts were made from KB3 or RS4;11 cell lines or from P1-ALL and P2-ALL cultures as indicated and subjected to immunoblotting for Bcl-2, Bcl-xL or Mcl-1. GAPDH was used as a loading control. B, C. Cell viability was assessed by MTT assay as described in Materials and Methods. P1-ALL or P2-ALL cells as indicated were treated with vehicle (100% viability) or increasing concentrations of ABT-263 for 72 h. Results are given as mean ± s.d. (n = 6).
Discussion
In this study, a series of adult ALL cell cultures, derived and expanded from primary cells and representing a unique model of ALL, were tested for sensitivity to ABT-263 and ABT-199, and further characterized with respect to Bcl-2 dependence and function. The results show that the ALL cell cultures are highly sensitive to both BH3 mimetics, with IC50 values in the low nanomolar range. Our results are consistent with other reports that primary B-ALL and T-ALL cells are sensitive to BH3 mimetics (16, 17). In addition to this high degree of sensitivity, we observed a very narrow range of IC50 values for each of the two compounds, reflecting a striking consistency across all seven cultures examined. This contrasts with established ALL cell lines which vary over a much broader range and which are typically far more resistant. For example, Jayanthan et al. (34) found sensitivity to ABT-737 varied over 1000-fold, from sub-nanomolar to sub-micromolar, for five ALL cell lines with MLL rearrangement. A 20-fold difference in IC50 value for ABT-263 for two B-cell ALL cell lines was reported in one study (35), and in another study four ALL cell lines varied over a 100-fold range in ABT-263 sensitivity (17). In addition and aside from ALL, a study of twenty five multiple myeloma cell lines reported IC50 values for ABT-263 ranged from 7 nM to 150 nM (36). In the current work, the B-cell ALL cell lines RS4;11 and NALM-6 were much more resistant to ABT-263 versus the most sensitive ALL culture (Table 1). Interestingly, RS4;11 cells were much more sensitive to ABT-199 than NALM-6 cells (Table 1), suggesting that NALM-6 may be more dependent on Bcl-xL than Bcl-2. Importantly, freshly isolated pediatric ALL blasts exhibited IC50 values for ABT-263 similar to those of the adult ALL primary cultures. The uniformity of response observed with the ALL cultures versus the large variation seen with established cell lines has two important implications. Firstly, it supports the prevailing notion that, over time, cell lines acquire characteristics that may not reflect those of the primary cells from which they were derived. Secondly, this disparity indicates that primary ALL cultures are much more sensitive to BH3 mimetics, and more uniform in their response, than suggested by data generated from the study of cell lines. In turn these findings highlight the utility of such cultures for preclinical studies of ALL, and add to the growing body of evidence validating the use of Bcl-2 inhibitors as a therapeutic strategy for this disease. Preliminary results using peripheral blood mononuclear cells from healthy donors have indicated that monocytes, T-cells and natural killer cells have IC50 values for ABT-263 of > 1 μM, and B-cells have an IC50 value of 80 nM, suggesting that the compound exhibits selective cytotoxicity toward ALL cells and thus a favorable therapeutic index.
While anti-apoptotic Bcl-2 proteins exhibit overlapping functions (1-5), different cell types may be dependent on one dominant member. For example, HeLa cells are strictly dependent on Mcl-1 for survival, whereas HT29 colon carcinoma cells are not dominantly dependent on one particular anti-apoptotic Bcl-2 member (28). The recently developed technique of BH3 profiling has been immensely useful in defining which anti-apoptotic proteins play the primary role in survival (29). Applying this technique (Fig. 2), we found that RS4;11 cells and the primary ALL cell cultures exhibited a BH3 profiling signature characteristic of cells dependent on Bcl-2. Interestingly though, the primary cultures were much more sensitive to ABT-263, up to 100-fold, than RS4;11 cells. Furthermore, ALL-2, which had the lowest relative expression of Bcl-2 (Fig. 1), was among the most sensitive to ABT-263 (Table 1). Taken together, these findings indicate that factors other than Bcl-2 expression and Bcl-2 dependency play a role in ABT-263 response. One of the established factors in ABT-263 resistance is Mcl-1 expression (36-39), and we formally demonstrated that overexpressed Mcl-1 conferred ABT-263/199 resistance (Fig. 5). However, endogenous levels of Mcl-1 were similar for RS4;11 cells and the primary ALL cultures, so this parameter alone cannot explain the differential response. A recent study has shed light on determinants of ABT-263 sensitivity (40). It was found that cells sensitive to ABT-263 expressed high levels of both Bcl-2 and Bim, and furthermore that a critical function of Bim was to be available to bind and inhibit Mcl-1. These findings are consistent with others which have shown a key role for Bim:Bcl-2 complexes in ABT-263 responsiveness (37, 41, 42), and furthermore suggest that in addition to its established role as an activator, the Bim released from Bim:Bcl-2 complexes by ABT-263 has additional responsibilities as a Mcl-1 neutralizer. These and other observations indicate that the relative occupancy and availability of not only the targeted anti-apoptotic protein, Bcl-2 or Bcl-xL, but also the non-targeted anti-apoptotic protein, Mcl-1, is critical in dictating the degree of ABT-263 sensitivity. Overall, the data available to date highlight the complex underpinnings of the ABT-263 response.
It is well established that a key role for anti-apoptotic Bcl-2 proteins is sequestration and inactivation of pro-apoptotic Bcl-2 family members. While Bcl-2 potentially can bind many pro-apoptotic family members, the number and nature of binding partners in specific contexts have not been fully explored. Co-immunoprecipitation was used to identify the spectrum of pro-apoptotic Bcl-2 proteins bound to Bcl-2, and the results demonstrated the multifaceted role of Bcl-2 in binding several pro-apoptotic Bcl-2 proteins (Fig. 3 and 4). Bcl-2 in primary ALL cells was found in complex with Bak, Bax, Bim and Bik. Thus Bcl-2 sequesters members of all three pro-apoptotic subgroups, namely effector (Bak, Bax), activator (Bim) and sensitizer (Bik). We showed that high levels of Mcl-1 can maintain survival of ABT-263/199-treated cells and does so, at least in part, by binding Bim. This further underscores the key roles of Bim:Bcl-2 complexes in ABT-263 sensitivity and Mcl-1 in ABT-263 resistance.
In addition to their use and success as single agents (12-16), BH3 mimetics hold much promise as chemosensitizers for new and conventional cancer drugs. Recent work has shown additive or synergistic effects of ABT-263 when combined with other anti-cancer agents. For example, ABT-263 enhanced the actions of etoposide, vincristine, bortezomib, cyclophosphamide, as well as several drug combination regimens, in vitro and in vivo in several hematologic tumor models (43). In ALL cell lines, ABT-263 was found to synergize with several different anti-neoplastic agents including daunorubicin, bortezomib, apicidin, and the multi-tyrosine inhibitor sunitinib (34). With the development and availability of an increasing number of novel targeted agents for cancer, including a broader array of compounds targeting anti-apoptotic Bcl-2 proteins (12-14), there are now many more agents and combinations than can be systematically studied clinically. In addition, the drug responsiveness of established cell lines may differ from the primary cells from which they were derived. Thus there is an urgent need for appropriate cancer cell culture models, and advances in preclinical drug development will increasingly depend on these. The results presented here indicate that the ALL cell cultures represent a valuable model system for preclinical testing of novel agents and drug combinations for ALL.
Supplementary Material
Acknowledgements
We thank Anthony Letai for providing the Mcl-1- and Bcl-2-dependent leukemia cell lines and for assistance with BH3 profiling, Jason Farrar for providing the NALM-6 cell line, Masahiro Higuchi for use of the fluorescence microplate reader, and Peter Emanuel for support and interest.
Grant support: This work was supported by National Institutes of Health Grant CA-109821 from the National Cancer Institute (to TCC) and in part by pilot funds from Translational Research Institute Grant UL1TR000039 (to TCC).
Abbreviations
- ALL
acute lymphoblastic leukemia
- DMSO
dimethyl sulfoxide
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- IMDM
Iscove’s modified Dulbecco’s medium
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IgG-LC
immunoglobulin light chain
- NGFR
nerve growth factor receptor
- PARP
poly(ADP-ribose) polymerase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Footnotes
Conflicts of interest: No potential conflicts of interest were disclosed.
References
- 1.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature reviews Molecular cell biology. 2008;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 2.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. Journal of cell science. 2009;122(Pt 4):437–41. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Molecular cell. 2010;37(3):299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harbor perspectives in biology. 2013;5(4):a008714. doi: 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nature reviews Molecular cell biology. 2014;15(1):49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26(9):1324–37. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335(6189):440–2. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 9.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108(2):153–64. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 10.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 11.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer research. 2008;68(9):3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 12.Azmi AS, Wang Z, Philip PA, Mohammad RM, Sarkar FH. Emerging Bcl-2 inhibitors for the treatment of cancer. Expert opinion on emerging drugs. 2011;16(1):59–70. doi: 10.1517/14728214.2010.515210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khaw SL, Huang DC, Roberts AW. Overcoming blocks in apoptosis with BH3-mimetic therapy in haematological malignancies. Pathology. 2011;43(6):525–35. doi: 10.1097/PAT.0b013e32834b1b34. [DOI] [PubMed] [Google Scholar]
- 14.Billard C. BH3 mimetics: status of the field and new developments. Molecular cancer therapeutics. 2013;12(9):1691–700. doi: 10.1158/1535-7163.MCT-13-0058. [DOI] [PubMed] [Google Scholar]
- 15.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nature reviews Drug discovery. 2008;7(12):989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 16.Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell death and differentiation. 2009;16(3):360–7. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 17.Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111(4):2300–9. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30(5):488–96. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29(7):909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. The lancet oncology. 2010;11(12):1149–59. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine. 2013;19(2):202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 22.Smith MA. Update on developmental therapeutics for acute lymphoblastic leukemia. Current hematologic malignancy reports. 2009;4(3):175–82. doi: 10.1007/s11899-009-0024-3. [DOI] [PubMed] [Google Scholar]
- 23.Nathan PC, Wasilewski-Masker K, Janzen LA. Long-term outcomes in survivors of childhood acute lymphoblastic leukemia. Hematology/oncology clinics of North America. 2009;23(5):1065–82. vi–vii. doi: 10.1016/j.hoc.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Drexler HG, Minowada J. History and classification of human leukemia-lymphoma cell lines. Leukemia & lymphoma. 1998;31(3-4):305–16. doi: 10.3109/10428199809059223. [DOI] [PubMed] [Google Scholar]
- 25.Drexler HG, MacLeod RA, Uphoff CC. Leukemia cell lines: in vitro models for the study of Philadelphia chromosome-positive leukemia. Leukemia research. 1999;23(3):207–15. doi: 10.1016/s0145-2126(98)00171-4. [DOI] [PubMed] [Google Scholar]
- 26.Nijmeijer BA, Szuhai K, Goselink HM, van Schie ML, van der Burg M, de Jong D, et al. Long-term culture of primary human lymphoblastic leukemia cells in the absence of serum or hematopoietic growth factors. Experimental hematology. 2009;37(3):376–85. doi: 10.1016/j.exphem.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. The Journal of cell biology. 2009;187(3):429–42. doi: 10.1083/jcb.200904049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eichhorn JM, Alford SE, Sakurikar N, Chambers TC. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Experimental cell research. 2014;322(2):415–24. doi: 10.1016/j.yexcr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer cell. 2006;9(5):351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 30.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(29):12895–900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heemskerk MH, Hoogeboom M, de Paus RA, Kester MG, van der Hoorn MA, Goulmy E, et al. Redirection of antileukemic reactivity of peripheral T lymphocytes using gene transfer of minor histocompatibility antigen HA-2-specific T-cell receptor complexes expressing a conserved alpha joining region. Blood. 2003;102(10):3530–40. doi: 10.1182/blood-2003-05-1524. [DOI] [PubMed] [Google Scholar]
- 32.Ke H, Zhang JY, Akiyama SK, French JE. BCL2 interaction with actin in vitro may inhibit cell motility by enhancing actin polymerization. Cell adhesion & migration. 2011;5(1):6–10. doi: 10.4161/cam.5.1.13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ke H, Parron VI, Reece J, Zhang JY, Akiyama SK, French JE. BCL2 inhibits cell adhesion, spreading, and motility by enhancing actin polymerization. Cell research. 2010;20(4):458–69. doi: 10.1038/cr.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jayanthan A, Incoronato A, Singh A, Blackmore C, Bernoux D, Lewis V, et al. Cytotoxicity, drug combinability, and biological correlates of ABT-737 against acute lymphoblastic leukemia cells with MLL rearrangement. Pediatric blood & cancer. 2011;56(3):353–60. doi: 10.1002/pbc.22760. [DOI] [PubMed] [Google Scholar]
- 35.Lock R, Carol H, Houghton PJ, Morton CL, Kolb EA, Gorlick R, et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatric blood & cancer. 2008;50(6):1181–9. doi: 10.1002/pbc.21433. [DOI] [PubMed] [Google Scholar]
- 36.Bodet L, Gomez-Bougie P, Touzeau C, Dousset C, Descamps G, Maiga S, et al. ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood. 2011;118(14):3901–10. doi: 10.1182/blood-2010-11-317438. [DOI] [PubMed] [Google Scholar]
- 37.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer cell. 2006;10(5):375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, Vernetti LA, et al. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 2007;26(27):3972–9. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 39.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer cell. 2006;10(5):389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood. 2012;119(24):5807–16. doi: 10.1182/blood-2011-12-400929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. The Journal of clinical investigation. 2007;117(1):112–21. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer cell. 2007;12(2):171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 43.Ackler S, Mitten MJ, Foster K, Oleksijew A, Refici M, Tahir SK, et al. The Bcl-2 inhibitor ABT-263 enhances the response of multiple chemotherapeutic regimens in hematologic tumors in vivo. Cancer chemotherapy and pharmacology. 2010;66(5):869–80. doi: 10.1007/s00280-009-1232-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.