

Abstract
Itraconazole (ITZ) is a well-known antifungal agent that also has anticancer activity. In this study, we identify ITZ as a broad-spectrum inhibitor of enteroviruses (e.g., poliovirus, coxsackievirus, enterovirus-71, rhinovirus). We demonstrate that ITZ inhibits viral RNA replication by targeting oxysterol-binding protein (OSBP) and OSBP-related protein 4 (ORP4). Consistently, OSW-1, a specific OSBP/ORP4 antagonist, also inhibits enterovirus replication. Knockdown of OSBP inhibits virus replication, whereas overexpression of OSBP or ORP4 counteracts the antiviral effects of ITZ and OSW-1. ITZ binds OSBP and inhibits its function, i.e., shuttling of cholesterol and phosphatidylinositol-4-phosphate between membranes, thereby likely perturbing the virus-induced membrane alterations essential for viral replication organelle formation. ITZ also inhibits hepatitis C virus replication, which also relies on OSBP. Together, these data implicate OSBP/ORP4 as molecular targets of ITZ and point to an essential role of OSBP/ORP4-mediated lipid exchange in virus replication that can be targeted by antiviral drugs.
Graphical Abstract
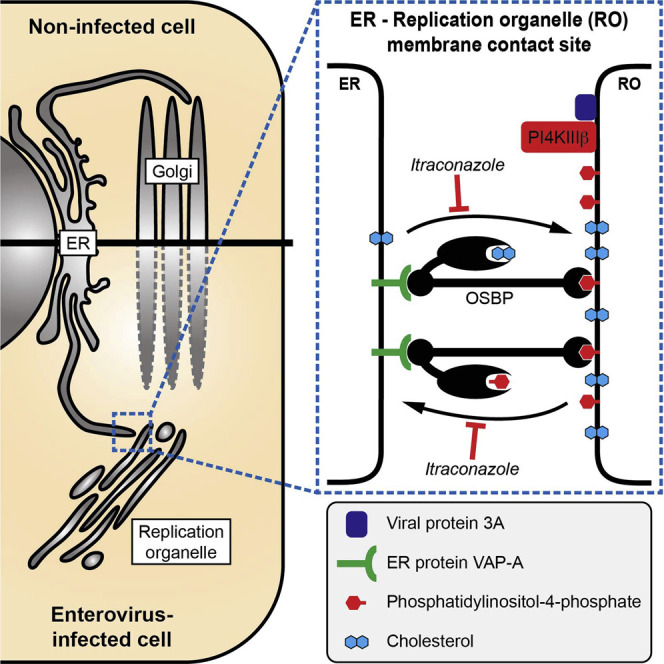
Strating et al. present the antifungal drug itraconazole as a novel inhibitor of a broad range of viruses, including poliovirus and hepatitis C virus. Itraconazole acted on a novel target, the oxysterol-binding protein (OSBP), a protein that has an essential role in lipid transfer.
Introduction
The family Picornaviridae contains many important human and animal pathogens. The genus Enterovirus includes poliovirus (PV), coxsackievirus (CV), echovirus, several numbered enteroviruses (e.g., enterovirus-71 [EV71]), and human rhinovirus (HRV). Except for PV, no vaccines are available to prevent infections with enteroviruses and no antiviral drugs are available for treatment. Other important human picornaviruses include hepatitis A virus and human parechovirus (HPeV). Well-known animal pathogens are foot-and-mouth disease virus and encephalomyocarditis virus (EMCV).
The genome of enteroviruses consists of a 7.5 kb single-stranded RNA molecule of positive polarity [(+)RNA]. It encodes a single polyprotein that is proteolytically processed by the viral proteases into the structural proteins (VP1–VP4) and the nonstructural proteins (2A–2C and 3A–3D). The viral genome is replicated by assemblies of viral and host proteins located on intracellular membranes termed replication organelles (ROs). The ROs are formed as a result of virus-induced remodeling of secretory pathway membranes, which most likely starts at the Golgi complex (Hsu et al., 2010), eventually resulting in a complex network of tubulovesicular membranes (Belov et al., 2012, Limpens et al., 2011). Viral modification of lipid homeostasis is thought to play a major role in RO formation. Viral proteins 2BC and 3A play a major role in the membrane rearrangements by recruiting essential host factor for enterovirus replication to ROs, such as phosphatidylinositol-phosphate-4-kinase III beta (PI4KIIIβ), a Golgi-localized lipid kinase that generates phosphatidylinositol-4 phosphate (PI4P) (Arita, 2014, Hsu et al., 2010). The functional importance of elevated PI4P levels at ROs remains to be established. The viral RNA-dependent RNA-polymerase, 3Dpol, binds PI4P in vitro, but it is unknown whether this is important for its recruitment and/or activation in infected cells (Hsu et al., 2010). Alternatively, the PI4P lipids may participate in RO formation by facilitating the recruitment of PI4P-binding host proteins with membrane-modifying properties.
Cholesterol is a critical membrane component that determines membrane fluidity and regulates the formation and function of membrane-bound complexes of lipids and proteins. Several (+)RNA viruses, such as hepatitis C virus (HCV), dengue, and West Nile virus, remodel the cellular cholesterol landscape to make intracellular host-cell membranes conducive for efficient genome replication (Rothwell et al., 2009, Wang et al., 2014). Enterovirus-induced rearrangements of secretory pathway membranes into the tubulovesicular RO structures may also depend on alterations in cholesterol homeostasis. Recent data suggest that enteroviruses stimulate clathrin-mediated endocytosis to transport cholesterol from the plasma membrane and extracellular medium to ROs (Ilnytska et al., 2013). However, other intracellular cholesterol trafficking pathways may also be subverted by enteroviruses to create their ROs.
Recently, oxysterol-binding protein (OSBP) was shown to play a key role in the transport of cholesterol and PI4P between the endoplasmic reticulum (ER) and Golgi (Mesmin et al., 2013). OSBP links ER and trans-Golgi membranes at so-called ER-Golgi membrane contact sites (MCSs) and shuttles sterol into the Golgi and PI4P back to the ER, where it is hydrolyzed by the PI4P-phosphatase Sac1. This cholesterol/PI4P exchange cycle drives the delivery of sterol in the trans-Golgi and self-regulates the localization of OSBP on the Golgi. OSBP and the OSBP-related proteins (ORPs) constitute a family of related proteins that, based on gene structure and sequence, can be subdivided into six subfamilies. OSBP and its closest relative, ORP4 (also called OSBP2), belong to subfamily I. All ORPs have a lipid-binding domain that was initially thought to be specific for sterols. However, recent structural analysis suggests that ORPs have the ability to bind PI4P and a second lipid that is either a sterol or a nonsterol ligand. Many ORPs, including OSBP, have an FFAT-motif that is recognized by ER-resident VAP receptors and an N-terminal pleckstrin homology (PH) domain for binding PI4P, through which they are linked to a variety of organelles. Although the functions of most ORPs are not very well understood, it has become clear that ORPs execute diverse functions in lipid sensing, lipid transport, and cell signaling (Raychaudhuri and Prinz, 2010, Weber-Boyvat et al., 2013).
We set out to identify novel inhibitors of enterovirus replication by screening the NIH Clinical Collection (NCC), a library of US Food and Drug Administration (FDA)-approved drugs that have a history of use in clinical trials for treatment of a wide variety of diseases. Similar collections of FDA-approved drugs have proven to be rich sources of undiscovered bioactivity and therapeutic potential. We identified itraconazole (ITZ) as a broad-spectrum inhibitor of enterovirus replication. ITZ is a well-known antifungal drug that inhibits CYP51, a cytochrome P450 required for sterol biosynthesis (Lestner and Hope, 2013). In addition, ITZ exerts anticancer activity by inhibiting angiogenesis—through disturbing mTOR signaling and vascular endothelial growth factor receptor 2 (VEGFR2) trafficking—and the Hedgehog (Hh) signaling pathway (Kim et al., 2010, Nacev et al., 2011, Xu et al., 2010). ITZ has been found to be efficacious in patients with several cancer types in multiple phase 2 clinical studies (Antonarakis et al., 2013, Kim et al., 2014, Rudin et al., 2013). We demonstrate that known targets of ITZ cannot explain the antiviral activity of ITZ. Instead, evidence is presented that OSBP and ORP4 are novel targets of ITZ and that direct binding of ITZ to OSBP, which localizes at ROs, disrupts its lipid-shuttling function, and accounts for the antiviral effect of ITZ.
Results
ITZ Is an Inhibitor of Enterovirus and Cardiovirus Replication
We performed a screen of the NCC to identify novel inhibitors of CVB3 replication. Like all enteroviruses, CVB3 kills its host cell and thereby causes a “cytopathic effect” (CPE). We screened the NCC by microscopically observing which compounds prevented the development of CPE in a multicycle replication assay and identified ITZ (Figure S1) as one of the hits. To determine its spectrum of antiviral activity, we tested ITZ against a representative panel of picornaviruses in a multicycle CPE-reduction assay. ITZ exhibited antiviral effect against all enteroviruses examined (belonging to several species) with 50% effective concentration (EC50) values between 0.3 μM and 1.6 μM (Table S1). In addition, EMCV, a Cardiovirus genus member, was inhibited by ITZ. In contrast, equine rhinitis A virus (ERAV; Aphthovirus genus member) and HPeV-1 (Parechovirus genus member) were insensitive to ITZ. To exclude the possibility that the antiviral activity was due to toxic side effects, we determined the effect of ITZ on virus production during a single replication cycle. Similar to the multicycle CPE-reduction assay, ITZ was active against CVB3, EV71, HRV14, and EMCV, but not ERAV, in a single replication cycle (Figure 1 A) without apparent toxicity (Figure 1B). ITZ also inhibited Saffold virus (SAFV) replication, a human cardiovirus (Figure 1A). Thus, ITZ exerts broad antiviral activity against enteroviruses and cardioviruses.
Figure 1.

ITZ Inhibits Viruses at the Genome Replication Stage
(A) BGM (CVB3, EV71, EMCV, ERAV) or HeLa R19 cells (HRV14, SAFV) were infected with virus at multiplicity of infection (MOI) 1 and treated with ITZ. Virus titers at 8 hr postinfection (p.i.) (10 hr for SAFV) were determined by endpoint dilution.
(B) Cell viability with MTS assay after 8 hr incubation with ITZ.
(C) BGM cells were transfected with RNA of subgenomic replicons pRib-LUC-CB3/T7 or pRLuc-M16.1 (EMCV) and treated with DMSO, 25 μM ITZ, or as positive controls 2 mM GuHCl or 80 μM dipyridamole, and luciferase levels were determined at the indicated time points.
Experiments were performed in triplicate and mean values ± SEM are shown; asterisks indicate statistical significance compared to mock treated controls. See also Figures S1 and S2.
ITZ Inhibits Viral RNA Genome Replication
Next, we determined the effect of ITZ on translation and replication of transfected CVB3 and EMCV RNAs, namely a subgenomic replicon of CVB3, in which (part of) the capsid-coding region is replaced by a firefly luciferase gene, or a genomic RNA of EMCV, in which a Renilla luciferase gene is inserted upstream of the coding region. As positive controls, we used guanidine-HCl and dipyridamole, well-known and potent inhibitors of CVB3 and EMCV replication, respectively. Two hours after transfection of the RNAs, when no RNA replication has taken place yet (van Kuppeveld et al., 1995), luciferase levels were unaffected, indicating that ITZ does not inhibit viral genome translation (Figure 1C). However, at later time points, luciferase production by both replicons was decreased, demonstrating that ITZ affects RNA replication. Importantly, ITZ did not affect viral polyprotein synthesis and processing (Figure S2).
Inhibition of Virus Replication Is Independent of Known Targets of ITZ
ITZ is widely used as an antifungal drug that inhibits the fungal enzyme CYP51. ITZ has also been shown to have some inhibitory activity toward the human CYP51 (hCYP51) and the related cytochrome P450 CYP3A4. In addition to ITZ, other azole family antifungal drugs, including posaconazole, ketoconazole, fluconazole, and voriconazole (Figure S1), also inhibit hCYP51 and CYP3A4 with slightly lower or similar potency as ITZ (Warrilow et al., 2013, Zhang et al., 2012). We tested whether these drugs exert antiviral activity using recombinant viruses RLuc-CVB3 and RLuc-EMCV, which carry the Renilla luciferase gene upstream of the coding region. At 10 μM, only posaconazole inhibited replication of RLuc-CVB3 and RLuc-EMCV. The remaining azoles did not display any antiviral activity at concentrations up to 100 μM (Figures 2 A and S3A–S3C). Similar results were obtained in a multicycle CPE-reduction assay (not shown). These results ruled out the possibility that inhibition of hCYP51 or CYP3A4 underlies the antiviral activity of ITZ and its structurally related analog posaconazole.
Figure 2.

ITZ Does Not Inhibit Virus Replication through Known Targets or PI4KIIIβ, although CVB3 with Mutations in the Nonstructural Viral Protein 3A Are Cross-Resistant to ITZ and PI4KIIIβ Inhibitors
(A, B, and D) HeLa R19 (A) or BGM (B and D) cells were infected with RLuc-CVB3 at MOI 0.1 and treated with 10 μM ITZ, DMSO, or 10 μM antifungal azoles (A), Hedgehog pathway antagonists (100 nM Sant-1, Sant-2, or cyclopamine-KAAD) (B), or ERα (β-estradiol) (D), and Renilla luciferase levels were measured after 6 hr.
(C) HAP1 cells were treated with 10 μM antifungal azoles for 6 hr and fixed, and cholesterol was stained with filipin.
(E) BGM cells were infected, treated, and analyzed as in (A) with RLuc-CVB3 WT or the 3A[H57Y] mutant.
(F) BGM cells were infected with CVB3 WT or CVB3 3A[H57Y] at low MOI in the presence of ITZ, and cell viability was measured after 3 days.
(G and H) In vitro-transcribed RNA of subgenomic replicons pRib-LUC-CB3/T7 (WT and indicated 3A mutants) (G) or pPV-FLuc (WT and 3A[A70T]) (H) was transfected into RD cells. The cells were treated with DMSO, 25 μM ITZ, or 1.5 μM T-00127-HEV1 (PI4KIIIβ inhibitor), and firefly luciferase levels at 7 hr p.i. were determined.
(I) HeLa R19 cells were transfected with FAPP1-PH-GFP treated with DMSO, 25 μM ITZ, or 1 μM PIK93 for 1 hr and stained with an antibody against PI4KIIIβ and Hoechst.
Experiments were performed in triplicate and shown are mean values ± SEM; asterisks indicate statistical significance compared to mock-treated controls (A and B) or of mutant virus compared to WT. Scale bars correspond to 10 μm. See also Figures S3 and S4.
As ITZ also inhibits the Hedgehog (Hh) signaling pathway, most likely by interfering with the function of the G protein-coupled receptor-like protein Smoothened (Kim et al., 2010), we tested several Smoothened antagonists in the viral luciferase assays. The Smoothened antagonists KAAD-cyclopamine, Sant-1, and Sant-2 (Chen et al., 2002, Taipale et al., 2000) had no effect on the replication of RLuc-CVB3 or RLuc-EMCV (Figures 2B and S3D), indicating that the antiviral activity of ITZ is not mediated by its inhibition of the Hh pathway.
The antiangiogenic activity of ITZ has been attributed at least in part to its inhibition of the mTOR signaling pathway through disruption of the shuttling of cholesterol between plasma membrane and late endosomes/lysosomes, thereby inducing the accumulation of cholesterol in the endolysosomal system (Xu et al., 2010). We found that cholesterol, stained with filipin, was redistributed not only by ITZ and posaconazole but also by ketoconazole (which does not inhibit virus replication) in two human cell lines (HAP1 [Figure 2C] and HeLa R19 cells [Figure S3E]). Moreover, the mTOR inhibitor rapamycin had no effect on picornavirus replication (Beretta et al., 1996, Wong et al., 2008). Together, these results suggest that inhibition of virus replication by ITZ or posaconazole is not due to disruption of endosomal cholesterol shuttling or the cholesterol-related mTOR inhibition.
In addition to the aforementioned molecular and pathway targets of ITZ, ITZ has been reported to disturb N-glycosylation (Nacev et al., 2011). However, the N-glycosylation inhibitor tunicamycin did not affect poliovirus (Doedens et al., 1997) or CVB3 replication (our data not shown). ITZ has also been shown to antagonize the estrogen receptor α (ERα) (Cheng et al., 2012). But ERα agonist β-estradiol did not affect CVB3 (Figure 2D) or EMCV replication (Figure S3F). Finally, ITZ has been reported to target p-glycoprotein, UDP-glucuronosyltransferase, and ERβ, none of which are likely to mediate the antiviral activity of ITZ, because these are as potently inhibited by ketoconazole (Cheng et al., 2012, Walsky et al., 2012, Wang et al., 2002b), which did not affect virus replication.
Mutations in 3A that Confer Resistance to PI4KIIIβ Inhibitors Also Confer Resistance to ITZ, but ITZ Does Not Inhibit PI4KIIIβ Activity
As a first step to identifying the antiviral target of ITZ, we studied its effect on replication of CVB3 mutant viruses that we previously selected for resistance against other inhibitors. CVB3 carrying mutation 3A[H57Y] —which confers resistance to PI4KIIIβ inhibitors (e.g., PIK93, enviroxime, GW5074) (van der Schaar et al., 2012)—proved less sensitive to ITZ than wild-type (WT) CVB3 in both a single-cycle replication assay (Figure 2E) and a multicycle CPE-reduction assay (Figure 2F). Other mutations in 3A that were shown to protect against PI4KIIIβ inhibitors (i.e., V45A and I54F) (van der Schaar et al., 2012), also provided cross-resistance to ITZ (Figure 2G). Similarly, mutation A70T in PV 3A, which was also shown to protect against PI4KIIIβ inhibitors (Arita et al., 2009), protected PV against ITZ (Figure 2H). These results imply a link between 3A, PI4P lipids, and the mechanism of antiviral action of ITZ.
To determine whether ITZ inhibits PI4KIIIβ activity, we transiently transfected cells with a genetically encoded PI4P sensor, i.e., the GFP-tagged PH domain of FAPP1 (FAPP1-PH-GFP). Localization of this sensor specifically depends on activity of PI4KIIIβ (Balla et al., 2005, van der Schaar et al., 2012). In control cells, FAPP1-PH-GFP overlapped with the Golgi-localized PI4KIIIβ (Figure 2I). Upon treatment with a PI4KIIIβ inhibitor, PIK93, FAPP1-PH-GFP was redistributed to the cytosol. ITZ, however, did not decrease FAPP1-PH-GFP localization. In fact, ITZ caused a small increase in the amount of Golgi-localized FAPP1-PH-GFP, which was more apparent in a cell line stably expressing this PI4P sensor (which showed a more homogenous and moderate expression level) (Figure S4A). Also upon staining PI4P with a specific antibody, a PI4KIIIβ inhibitor, BF738735 (van der Schaar et al., 2013), decreased PI4P levels, whereas ITZ increased PI4P levels (Figure S4B).
CVB3 replication is not completely blocked by ITZ (see e.g., Figures 1A and 2E), thus permitting the monitoring of PI4P lipids in treated cells. Both in untreated and ITZ-treated cells infected with CVB3 (visualized using 3A antibody), PI4P levels (visualized using PI4P antibody) were clearly increased compared to uninfected cells (Figure S4C), indicating that ITZ also does not inhibit PI4KIIIβ activity in infected cells.
ITZ Inhibits Virus Replication by Targeting OSBP and ORP4
Having ruled out PI4KIIIβ as a target of ITZ, we next turned to signaling steps downstream of PI4P, i.e., proteins that bind to PI4P lipids. To assess whether any of the known PI4P-binding proteins could be a target of ITZ, we performed a target identification by small interfering RNA (siRNA) sensitization (TISS) assay (Arita et al., 2010). TISS encompasses siRNA knockdown of candidate target proteins to potentiate the biological effect of a low concentration of a compound. Among a number of PI4P-binding, Golgi-localized proteins, knockdown of OSBP, but not any of the other PH domain-containing proteins, enhanced the inhibitory effect of a low concentration (1.25 μM) of ITZ on PV replication (Figure 3 A), implying OSBP as a possible antiviral target of ITZ. We further assessed this possibility by several experiments. First, the OSBP antagonist OSW-1 (Burgett et al., 2011) potently inhibited CVB3 replication (Figure 3B), confirming that pharmacological targeting of OSBP can inhibit enterovirus replication. As for ITZ, the 3A[H57Y] mutation in CVB3 provided resistance against OSW-1 (Figure 3B). Akin to ITZ, OSW-1 inhibited all enteroviruses tested as well as EMCV, but not ERAV (data not shown). Importantly, OSW-1 did not affect endolysosomal cholesterol distribution (Figure 3C), supporting our previous conclusion that this effect unlikely explains the antiviral effect of ITZ. Second, similar as for PV (Wang et al., 2014), siRNA knockdown of OSBP inhibited replication of EV71 and HRV2 (Figure 3D). CVB3 replication was also inhibited by OSBP knockdown, but this difference was not statistically significant, in line with the lower sensitivity of CVB3 than EV71 to ITZ (Figure 1A). Third, overexpression of OSBP restored replication of CVB3 and EV71 in the presence of ITZ or OSW-1 (Figure 3E), confirming that inhibition of viral replication by ITZ and OSW-1 is mediated through OSBP. Overexpression of PI4KIIIβ failed to rescue replication, and OSBP overexpression did not provide rescue against PI4KIIIβ inhibitors (data not shown), indicating the specificity of the experimental setup.
Figure 3.

ITZ Inhibits Virus Replication by Targeting OSBP and ORP4
(A) HEK293 cells were transfected with siRNAs targeting PI4P-binding proteins, infected with PV, and incubated in the presence of 1.25 μM ITZ. Normalized PV infection represents the level of firefly luciferase activity at 7 hr p.i. for siRNA-transfected and compound-treated cells divided by the firefly luciferase activity measured in siRNA-transfected and untreated cells.
(B) HeLa R19 cells were infected with RLuc-CVB3 WT or the 3A[H57Y] mutant at MOI 0.1 and treated with OSW-1, and Renilla luciferase levels were determined after 7 hr. Cell viability was determined in parallel.
(C) HAP1 cells were treated for 6 hr with 10 nM OSW-1 or 10 μM ITZ, fixed, and stained with filipin.
(D) HeLa R19 cells were transfected with constructs encoding OSBP or EGFP (negative control) for 24 hr, infected with RLuc-CVB3 at MOI 0.25 or EV71 at MOI 1, and treated with 10 μM (CVB3) or 3 μM (EV71) ITZ, 3 nM OSW-1, or DMSO. Renilla luciferase levels were determined at 7 hr p.i. (CVB3) or virus titers at 10 hr p.i. were determined by endpoint titration (EV71).
(E) HeLa R19 cells were transfected with siRNAs against OSBP, PI4KIIIβ (positive control), or a scrambled siRNA for 2 days and infected with CVB3, EV71, or HRV2 at MOI 1. Virus titers at 10 hr p.i. were determined by endpoint titration. Knockdown efficiency was determined by quantitative PCR and immunofluorescence (Figure S5), and an MTS assay was used to test for effects on cell viability.
(F) HEK293 cells were transfected with siRNAs targeting ORP family members (roman numbering indicates ORP subfamilies), infected, treated with ITZ, and analyzed as in (A).
(G) HeLa R19 cells were transfected with constructs encoding OSBP, ORP4, or enhanced GFP, infected and treated with 3 nM OSW-1, and data were analyzed as in (C).
All figures are representative examples of experiments that were performed in triplicate. Shown are mean values ± SEM. Scale bars correspond to 10 μm. See also Figures S5 and S6.
Besides OSBP, OSW-1 also targets ORP4 (Burgett et al., 2011). Knockdown of ORP4, but none of the other ORPs, also sensitized PV to ITZ (Figure 3F), and overexpression of ORP4 counteracted the inhibitory effect of OSW-1 on CVB3 and EV71 replication (Figure 3G). We also attempted to test the effect of ORP4 depletion. Although in the TISS assay, ORP4 knockdown potentiated the effect of ITZ, we were not able to achieve robust knockdown (>75% at mRNA level), and therefore we cannot conclude unambiguously whether ORP4 is important for virus replication. Problems with ORP4 knockdown were also observed by others and are likely due to an essential role of ORP4 in cell proliferation and survival (Charman et al., 2014). Collectively, these results indicate that both OSBP and ORP4 are novel targets of ITZ and are involved in its mechanism of antiviral action.
ITZ Inhibits In Vitro HCV Replication
Replication of HCV also requires OSBP and is inhibited by OSW-1 (Wang et al., 2014). In line with our findings for enteroviruses, we found that ITZ and posaconazole, but not the other selected azoles, inhibited HCV replication in cell culture (Figure S6). EC50 values for inhibition of HCV replication by ITZ were comparable to those obtained for the enteroviruses (Table S1). Together, our data clearly demonstrate that ITZ inhibits OSBP function and that viruses from different families that depend on OSBP function can be inhibited by ITZ. Importantly, not all (+)RNA viruses are sensitive to inhibition of OSBP. Dengue virus replication was recently observed to be insensitive to OSW-1 (Wang et al., 2014), and we also showed that replication of mouse hepatitis virus (a coronavirus) is insensitive to OSW-1 and ITZ (data not shown).
Treatment with ITZ Results in Relocalization of OSBP to the Golgi Complex
After having established that OSBP and, possibly, ORP4 are novel targets of ITZ, we wanted to study how ITZ targets these proteins. Because of available tools, we focused on OSBP for the remainder of this study. In line with published data (Burgett et al., 2011), OSW-1 caused a massive recruitment of overexpressed GFP-OSBP to the Golgi apparatus (as marked by staining endogenous PI4KIIIβ) (Figure 4 A). A similar relocalization was observed for endogenous OSBP (Figure 4B). ITZ and posaconazole, but not the azoles that lacked antiviral activity, redistributed OSBP in a manner that is similar to OSW-1. Live-cell imaging was performed to study the dynamics of GFP-OSBP relocalization by the compounds. Before addition of the compounds, GFP-OSBP primarily localized in the cytosol with a Golgi pattern faintly visible. A few minutes after the addition of the compounds, GFP-OSBP fluorescence at the Golgi was clearly increased in cells treated with either ITZ or OSW-1 and continued to increase at the expense of the cytoplasmic signal (Figures 4C and 4D; Movie S1). OSW-1 was previously reported to disrupt the structure of the Golgi apparatus (Burgett et al., 2011), which we also observed from ∼30 to 60 min onward as GFP-positive punctae that became more numerous over time (Figure 4C). In ITZ-treated cells, the Golgi pattern became affected only hours later and appeared less dispersed than that in OSW-1 treated cells.
Figure 4.

Azoles that Inhibit Virus Replication Rapidly Accumulate OSBP at the Golgi
(A) HeLa R19 cells were transfected with OSBP-GFP; treated with DMSO, 10 μM of ITZ or antifungal azoles, or 10 nM OSW-1 for 1 hr; and fixed and counterstained with an antibody against PI4KIIIβ and DAPI.
(B) HeLa R19 cells were treated as in (A), fixed and immunostained for endogenous OSBP.
(C) HeLa R19 cells were transfected with GFP-OSBP and treated with DMSO, 10 μM of ITZ, or 10 nM OSW-1, and the relocalization of OSBP was imaged by live-cell confocal laser scanning microscopy. Cells were imaged overnight. During the first 30 min, images were taken as fast as possible (∼1.5 min intervals), then intervals were stepwise increased to 30 min from 3.5 hr onward. Representative groups of cells are shown. The images are frames from Movie S1.
(D) Quantification of the relative GFP-OSBP signal at the Golgi apparatus in five cells for each condition from (C). Error bars indicate SEM. Scale bar corresponds to 10 μm.
See also Movie S1.
ITZ Directly Inhibits Lipid Shuttling by OSBP
To investigate whether ITZ can block the lipid transfer activity of OSBP, we used a set of in vitro liposomal assays (Mesmin et al., 2013) (Supplemental Experimental Procedures) to measure the transport of dehydroergosterol (DHE) (Figure 5 A) and PI4P (Figure 5D) between ER-like and Golgi-like liposomes. ITZ inhibited the sterol-transfer activity of purified OSBP in a dose-dependent manner with a 50% inhibitory concentration (IC50) of ∼200 nM (Figure 5B). At 1 μM, ITZ and posaconazole, but not the other selected azoles, strongly inhibited DHE transfer transport in this liposomal assay, although they were less potent than the known OSBP ligand 25OH (Figure 5C). We also observed a dose-dependent inhibition of PI4P transfer by ITZ (IC50 = ∼4 μM) (Figure 5E). Posaconazole slightly inhibited PI4P, whereas the other azoles showed no activity (Figure 5F). For unknown reasons, a stimulatory effect of 25OH on PI4P transfer was observed, which depended on the 2% cholesterol content of the ER-like liposomes. The IC50 values suggest that ITZ is more potent toward sterol than PI4P transfer. Importantly, for technical reasons, the sterol and PI4P-shuttling assays are performed under different conditions and therefore cannot be directly compared. Further investigations would be needed to establish whether ITZ indeed more potently inhibits sterol than PI4P shuttling.
Figure 5.

ITZ Binds OSBP and Inhibits Sterol and PI4P Transfer by OSBP
(A–F) The effect of ITZ, antifungal azoles, or positive (25OH) or solvent controls (DMSO) on in vitro OSBP-mediated transfer of the fluorescent cholesterol analog DHE (A–C) or PI4P (D–F) was tested using liposomal assays depicted in (A) and (D). In both cases, initial exchange rates were determined in the presence of increasing concentrations of ITZ (B and E) or in the presence of 1 μM of the indicated drugs and then plotted in bar diagram (C and F).
(G–J) The effect of ITZ on binding of an N-terminal OSBP fragment (amino acids 76–408; PH-FFAT) to ER-like (G and H) and Golgi-like (I and J) liposomes was examined by liposomal float-up assays as outlined in (G) and (I). Liposomal fractions were analyzed for binding of proteins by SDS-PAGE (H and J).
(K and L) The effect of ITZ and control compounds on DHE (K) or PI4P (L) transfer by trypsinized OSBP was studied as with full-length OSBP (A–F).
(M) The interaction of ITZ with GFP-tagged OSBP was investigated using MST. Data from three separate measurements were normalized and plotted, and a sigmoidal dose-response curve was fitted.
Shown are mean values ± SEM. Statistical significance for the drug-treated conditions was calculated compared to the “no drug” control. See also Figure S7.
ITZ may inhibit the lipid transfer functions of OSBP directly by inhibiting the function of the ORD, which transfers the lipids, or indirectly by disrupting the binding of OSBP to the liposomes. To investigate whether ITZ inhibits binding of OSBP to the liposomes, we studied whether it interferes with the interactions between (1) the FFAT-motif and VAP-A on the ER-like liposomes and (2) the PH-domain and PI4P on the Golgi-like liposomes. To this end, we performed liposomal float-up experiments using a recombinant fragment of OSBP containing the PH domain and FFAT motif (amino acids 76–408; PH-FFAT) (Figures 5G and 5I). In the presence of VAP-A, PH-FFAT bound to the ER-like liposomes, and this interaction was not disrupted by 1 μM ITZ (Figure 5H). The interaction of PH-FFAT with PI4P-containing Golgi-like liposomes was not disrupted by 10 μM ITZ either (Figure 5J). Likewise, VAP-A interaction with PH-FFAT recruited to Golgi-like liposomes was also insensitive to 10 μM ITZ (Figure 5J). Together, the liposomal float-up assays show that ITZ does not interfere with the binding of OSBP to the liposomes via VAP-A and PI4P.
To establish whether ITZ inhibits the lipid transfer activity of the ORD, we made use of a previously established assay (Mesmin et al., 2013). Limited tryptic proteolysis of OSBP cleaves OSBP into three major fragments; a ∼43 kDa fragment containing the PH-domain and FFAT-motif, and two fragments of ∼35 kDa and ∼20 kDa that are derived from the ORD. Previously, it was shown that the ORD-derived fragments retain lipid transfer activity, also in the absence of the inactive ∼43 kDa fragment (Mesmin et al., 2013). We found that ITZ still inhibited both DHE (Figure 5K) and PI4P (Figure 5L) transfer by OSBP that had been subjected to tryptic proteolysis (Figure S7A). These results suggest that ITZ inhibits both the sterol- and PI4P-transfer activities of OSBP by targeting the ORD.
ITZ Binds Directly to OSBP
The inhibitory effect of ITZ on OSBP function in a minimal in vitro system implied that ITZ directly inhibits OSBP. To biochemically define the binding in more detail, we measured binding of ITZ to GFP-OSBP using microscale thermophoresis (MST). Each molecule or complex distributes differently in a temperature field, depending on size, charge, and the hydration shell. Binding of ITZ to OSBP will affect the hydration shell and thereby its thermophoretic behavior. ITZ altered the thermophoretic profiles of purified GFP-OSBP (Figures S7B and S7C) in a dose-dependent manner, indicating direct binding. Normalization and fitting of data from three independent measurements demonstrated that ITZ binds to OSBP with a KD of ∼430 nM (Figure 5M). The monophasic shape of the binding curve indicates that there is likely only a single binding site for ITZ on OSBP, although our data cannot rule out that there are two sites with nearly identical KD’s.
OSBP Localizes to ROs in a PI4P-Dependent Manner
To test whether OSBP plays a role in formation and/or maintenance of the ROs, we examined its localization in infected cells. In uninfected cells, OSBP is mainly distributed throughout the cytosol with some OSBP localized to the Golgi apparatus (Figure 6 A), where it colocalized with PI4KIIIβ and the trans-Golgi network marker TGN46 (data not shown). In infected cells, OSBP localization was markedly changed, i.e., the Golgi pattern was lost and OSBP appeared in dispersed structures throughout the cytoplasm where it colocalized with viral protein 3A as a marker for ROs (Manders’ coefficient for overlap of 3A with OSBP: 0.36) (Figures 6A and 6B). To examine whether OSBP localized to ROs in a PI4P-dependent manner, cells were infected with CVB3 and replication was allowed to progress uninhibited for 4.5 hr before the PI4KIIIβ inhibitor BF738735 was added for 30 min, after which cells were processed for microscopy. Inhibition of PI4KIIIβ decreased colocalization of OSBP with 3A (Manders’ coefficient: 0.11), whereas treatment with ITZ increased colocalization of OSBP with the RO-marker 3A (Manders’ coefficient: 0.62) (Figures 6A and 6B). The enhanced recruitment of OSBP to ROs upon ITZ treatment is reminiscent of the enhanced Golgi-localization of OSBP upon treatment of ITZ or other OSBP inhibitors, and may therefore be caused by an inhibition of PI4P removal from ROs.
Figure 6.

ITZ Affects OSBP Localization in Infected Cells
(A) BGM cells were infected with CVB3 at MOI 10. At 4.5 hr p.i., cells were treated for 30 min with DMSO as vehicle control, 1 μM BF738753 (BF; a PI4KIIIβ inhibitor), or 10 μM ITZ. At 5 hr p.i., cells were fixed, processed for immunofluorescence with antibodies against OSBP and viral protein 3A, and imaged using confocal laser scanning microscopy.
(B) Manders’ coefficients for overlap of 3A with OSBP were calculated for DMSO (12 cells), BF (7 cells) and ITZ (10 cells). Shown are means ± SEM. Asterisks indicate statistical significance compared to DMSO-treated controls. Scale bars correspond to 10 μm.
ITZ Inhibits PI4P and Cholesterol Shuttling at ROs
To test directly whether ITZ inhibits the PI4P shuttling function of OSBP at ROs, cells were infected with CVB3 and replication was allowed to progress uninhibited for 3 hr. Then ITZ or BF738735 were added for 1 hr, cells were processed for microscopy, and PI4P intensity at ROs was quantified. ITZ treatment caused a strong increase in PI4P signal at the ROs (∼50% increase), whereas BF738735 treatment reduced it by ∼50% (Figures 7A and 7B), in line with the effects of these drugs on OSBP recruitment (Figure 6A). No such effects on PI4P were observed upon treatment with guanidine, an inhibitor of the viral 2C protein, which was included to rule out that the observed effects were merely due to an inhibition of replication. Thus, these results demonstrate that in infected cells, ITZ prevents the removal of PI4P from ROs, which is comparable to our observations in uninfected cells (Figures 2I, S4A, and S4B).
Figure 7.

ITZ Inhibits PI4P and Cholesterol Shuttling in Infected Cells
(A) BGM cells were infected with CVB3 at MOI 10. At 3 hr p.i., cells were treated for 1 hr with DMSO, 2 mM guanidine HCl (Gua), 1 μM BF738735 (BF), or 10 μM ITZ. At 4 hr p.i., cells were fixed, processed for immunofluorescence with antibodies against 3A and PI4P, imaged by wide-field microscopy, and deconvoluted.
(B) PI4P intensity at 3A-positive structures was calculated for DMSO (11 cells), Gua (13 cells), BF (12 cells), and ITZ (11 cells).
(C) HeLa R19 cells were infected with CVB3 at MOI 10. At 3 hr p.i., cells were treated for 1 hr with DMSO, 2 mM Gua, 1 μM BF, or 10 μM ITZ. At 4 hr p.i., cells were fixed, processed for immunofluorescence with an antibody against 3A and filipin to stain cholesterol, imaged by wide-field microscopy, and deconvoluted.
(D) Pearson’s correlation coefficients for overlap of filipin and 3A were calculated for DMSO (17 cells), Gua (15 cells), BF (18 cells), and ITZ (19 cells).
Shown are means ± SEM. Scale bars correspond to 10 μm.
To test whether ITZ also inhibits cholesterol shuttling to ROs, cells were infected and treated similar as described above, cholesterol was visualized by filipin staining, and colocalization of filipin with 3A was quantified using a Pearson’s correlation coefficient. In DMSO-treated cells, filipin partially overlapped with 3A (Pearson’s 0.53). ITZ significantly reduced the colocalization of filipin with 3A (Pearson’s 0.38), indicating that ITZ inhibited the redistribution of cholesterol to the ROs (Figures 7C and 7D). Similarly, BF738735, which reduces the localization of OSBP to ROs (Figure 6A), also inhibited cholesterol shuttling to ROs (Pearson’s 0.35), whereas guanidine did not decrease the overlap between filipin and 3A. Thus, we demonstrate that OSBP is recruited to ROs through the action of PI4KIIIβ and that ITZ inhibits both the PI4P and the cholesterol-transfer functions of OSBP in infected cells.
Discussion
Enteroviruses alter cellular lipid homeostasis and remodel host-cell membranes into replication organelles by usurping a number of host proteins, such as PI4KIIIβ (Arita et al., 2011, Hsu et al., 2010). However, as yet little is known about the underlying mechanisms and the identity of other host factors involved. Elucidation of the mechanism of action of inhibitors of virus replication has proven instrumental in obtaining novel insights into the mechanisms of viral replication. In this study we identified ITZ, a widely used antifungal drug that is currently also being explored as an anticancer agent, as a novel, broad-spectrum inhibitor of enteroviruses, cardioviruses, and HCV. We show that none of the well-established targets of ITZ (i.e., hCYP51, mTOR, VEGFR2, Hh) explains its antiviral activity. Instead, we identified the PI4P-binding proteins OSBP and ORP4 as novel targets of ITZ through which the antiviral effect is mediated.
OSBP is a master regulator of lipid homeostasis at MCSs between the ER and the trans-Golgi apparatus. It exchanges cholesterol and PI4P between these membranes and has been proposed to control MCS stability (Mesmin et al., 2013). OSBP is the prototype member of the family of ORPs, a group of proteins whose cellular functions have remained poorly understood. We identified OSBP and ORP4 as targets of ITZ. Pharmacologic inhibition, siRNA knockdown, and rescue of replication by overexpression demonstrate the importance of these proteins for virus replication. Furthermore, OSBP localized to ROs in a PI4KIIIβ- and PI4P-dependent manner. ITZ directly bound purified OSBP and inhibited both the cholesterol and PI4P-transport activities of OSBP in vitro (in liposomal assays). Also in living (uninfected) cells, ITZ inhibited the transport function of OSBP (i.e., transport of cholesterol from ER to Golgi and transport of PI4P from Golgi to ER), leading to an increase in PI4P levels at the Golgi, thereby causing the accumulation of OSBP. Likewise, in infected cells, ITZ increased PI4P levels on ROs, again leading to an enhanced recruitment of OSBP, and inhibited the accumulation of cholesterol on ROs. Thus, we demonstrate that ITZ inhibits the lipid-shuttling functions of OSBP not only in vitro but also in both infected and uninfected cells.
The enteroviral proteins 2BC and 3A play a critical role in RO formation by recruiting PI4KIIIβ, which leads to the accumulation of PI4P lipids on ROs (Arita, 2014, Arita et al., 2011, Hsu et al., 2010). We here show that OSBP is subsequently recruited to ROs via PI4P. Our data indicate that at ER-RO MCSs, OSBP exchanges PI4P for cholesterol, either newly synthesized in the ER or originating from a lipid droplet storage pool and being mobilized through the ER, leading to an accumulation of cholesterol at the ROs (Arita, 2014). Our findings are in agreement with those of a recent paper that suggested that OSBP shuttles cholesterol to HRV ROs based on the inhibitory effects on HRV replication of OSBP knockdown and 25OH treatment (Roulin et al., 2014). The finding that the levels of cholesterol are elevated at the expense of cholesterylesters (i.e., the form in which cholesterol is stored in lipid droplets) in enterovirus-infected cells (Ilnytska et al., 2013, Roulin et al., 2014) suggests that stored cholesterol is mobilized for transport to ROs. In addition, uptake of cholesterol by endocytosis has been suggested to contribute to the accumulation of cholesterol at ROs (Ilnytska et al., 2013). The role of cholesterol accumulation at ROs is far from established. Cholesterol is of profound importance for membranes properties such as membrane fluidity and formation of lipid microdomains, and it is thereby likely important for the membrane rearrangements and deformations underlying RO formation. In addition, cholesterol alterations have been suggested to affect viral polyprotein processing efficiency (Ilnytska et al., 2013).
The activity of OSBP is also important for the homeostasis of other lipids. At ER-Golgi MCSs, it acts in concert with the PI transfer protein Nir2, which supplies PI for PI4P synthesis at Golgi membranes, and CERT, which transfers ceramide to Golgi for sphingomyelin synthesis, thereby generating diacylglycerols (DAGs) (Peretti et al., 2008). Importantly OSBP ligands, e.g., 25OH and OSW-1, change the localization of CERT and modify sphingomyelin synthesis (Burgett et al., 2011, Perry and Ridgway, 2006). As an inhibitor of OSBP-mediated lipid shuttling, ITZ may thus not only affect the accumulation of cholesterol but also perturb the homeostasis of other lipids, such as sphingomyelin and DAGs. Whether and how this contributes to the inhibition of RO formation and/or function remains to be established.
Our study and the work by Arita et al. (2013) implicate a role for ORP4 in addition to OSBP in enterovirus replication. Unfortunately, little is known about the biological function of ORP4. Roles for ORP4 are proposed in organization of the cytoskeletal vimentin network, cell proliferation and survival, and sterol transfer (Charman et al., 2014, Wang et al., 2002a). However, unlike OSBP, ORP4 does not localize to the Golgi under normal conditions or in response to a ligand such as 25OH (Charman et al., 2014, Wang et al., 2002a). It therefore seems unlikely that ORP4 transports cholesterol between the ER and Golgi in a similar manner as OSBP. How ORP4 overexpression can counteract the inhibitory effect of ITZ on virus replication thus remains to be established. It is possible that OSBP-ORP4 heteromultimers (Wyles et al., 2007) are important for virus replication, but this requires further investigation. Besides OSBP and ORP4, other ORPs did not appear to be targeted by ITZ, though they may still be important for virus replication.
ITZ has been shown to inhibit angiogenesis (via mTOR and VEGFR2) and growth of Hh-dependent cancer cells, but the exact molecular mechanisms of the antitumor activities of ITZ await elucidation. It remains to be established whether OSBP inhibition contributes to the anticancer activities of ITZ via these pathways. OSBP overexpression, which we showed to counter the antiviral activity of ITZ, failed to prevent the inhibitory effects of ITZ on mTOR and Hh signaling (not shown). These observations suggest that ITZ does not inhibit these antitumor pathways through OSBP, but we cannot exclude that the overexpression approach can only neutralize the antiviral effect of ITZ. Therefore, more work is needed to establish whether or not ITZ exerts its antitumor activities via OSBP and/or ORP4. OSW-1 and several other natural products were recently reported to inhibit the growth of cultured human cancer cell lines through OSBP and ORP4 and therefore collectively termed ORPphilins (Burgett et al., 2011). Our data that ITZ targets OSBP and ORP4 justify classifying ITZ as a novel ORPphilin. It is plausible that ITZ inhibits OSBP/ORP4-dependent cancer cell growth and survival in a manner independent of, and in addition to, mTOR, VEGFR2, and Hh. Recently, two inhibitors of PV replication were shown to target OSBP and ORP4 (Arita et al., 2013), although binding to OSBP has yet to be shown, and may therefore also classify as ORPphilins.
In conclusion, we identified ITZ as a broad-spectrum inhibitor of enterovirus, cardiovirus, and HCV replication that exerts its antiviral activity through the novel targets OSBP and ORP4, presumably by inhibiting the lipid-shuttling functions of OSBP. Together, our study provides insight into enterovirus replication and presents ITZ, OSW-1, and other ORPphilins as potential novel inhibitors to treat enterovirus infections.
Experimental Procedures
Details about published and standard methods (cell culture, plasmids, virus infections, replicon transfections, the TISS assay, rescue experiments, analysis of viral polyprotein processing, siRNA experiments, immunofluorescence microscopy, and liposomal assays) are provided in Supplemental Experimental Procedures.
Reagents
The following compounds were purchased: itraconazole (Santa Cruz Biotechnology); posaconazole (Merck); ketoconazole (Enzo Life Sciences); fluconazole and voriconazole (Pfizer); T-00127-HEV1 (Pharmeks); dipyridamole, guanidine hydrochloride (GuHCl), and β-estradiol (Sigma Aldrich); Sant-1, Sant-2 (Tocris Bioscience); and cyclopamine-KAAD (Calbiochem). PIK93 was a kind gift from Dr. K. Shokat (Universeity of California, Berkeley), BF738735 (MacLeod et al., 2013) was provided by Galapagos NV, and OSW-1 was isolated from nature (Burgett et al., 2011). β-Estradiol was dissolved according to the manufacturer’s instructions. GuHCl was dissolved in water and all other compounds in DMSO.
Compound Library Screen
The NIH Clinical Collection was purchased from the NIH. The 446 highly drug-like compounds were screened for inhibitors of CVB3 using reduction of CPE as readout. Subconfluent monolayers of Buffalo green monkey kidney (BGM) cells in 96-well plates were infected with 10 CCID50 of CVB3 per well, compounds were added to a final concentration of 10 μM, and the level of CPE was visually assessed after 2 days of incubation at 37°C when full CPE had developed in the infected, untreated control wells.
Live-Cell Imaging
For live-cell imaging experiments, HeLa R19 cells were transfected with pEGFP-hOSBP; treated with ITZ, OSW-1, or solvent control (DMSO); and imaged using a Nikon A1R confocal laser scanning microscope. Images were processed and quantified using the Nikon NIS-Elements software. For additional details, see Supplemental Experimental Procedures.
Microscale Thermophoresis
The interaction between ITZ and recombinant GFP-hOSBP-SII (human OSBP with an N-terminal GFP and a C-terminal Strep-tagII) was investigated by MST using a NanoTemper Monolith NT.115 instrument and the NTAnalysis software (NanoTemper Technologies).
Statistical Analyses
Data are presented as means ± SEM. Replication data were analyzed by pairwise comparisons of conditions using one-tailed Student’s t test. Statistics significance is indicated as ∗p < 0.05, ∗∗p < 0.01, or ∗∗∗p < 0.001.
Author Contributions
J.R.P.M.S., L.v.d.L., L.A., J.B., M.A., L.D., P.L., H.M.v.d.S., K.H.W.L., H.J.T., R.U., G.D., N. Schlinck, R.W., N. Sever, S.H., J.O.L., P.A.B., J.N., and F.J.M.v.K. designed, performed, and analyzed experiments. M.A.D.M., M.D.S., and V.M.O. contributed essential reagents. J.R.P.M.S., L.v.d.L., L.A., J.B., M.A., L.D., P.L., H.M.v.d.S., H.J.T., G.D., N. Schlinck, R.W., N. Sever, S.H., J.O.L., P.A.B., M.A.D.M., M.D.S., V.M.O., J.N., and F.J.M.v.K. participated in critical discussions regarding data and the manuscript. J.R.P.M.S., L.v.d.L., L.A., and F.J.M.v.K. wrote the paper.
Acknowledgments
We thank Stijn Delmotte, Katrien Geerts, Caroline Collard, Gerrit Koen, and Katja Wolthers for assistance in acquisition of part of the antiviral data; Patrick Celie and Alex Fisch (Netherlands Cancer Institute, Amsterdam) for help with the MST measurements; and the Center for Cell Imaging (Faculty of Veterinary Medicine, Utrecht University) for support with microscopy experiments. This work was supported by grants from the “Convenant K.U. Leuven-Radboud University Nijmegen” framework (L.v.d.L., J.N., and F.J.M.v.K.), the European Union FP7 Marie Curie Initial Training Network “EUVIRNA” (grant agreement number 264286) (F.J.M.v.K.) and FP7 Large Scale Collaborative Project “SILVER” (grant agreement number 260644) (F.J.M.v.K.), the KU Leuven geconcerteerde onderzoeksactie (GOA/10/014) (J.N.), the Belgian Science Policy Office (BELSPO, Belvir consortium, IAP, phase VII) (J.N.), the Fund for Scientific Research of Flanders (FWO) (L.D.), CNRS and ANR (2010-1503-01) (J.B. and G.D.), the Sigrid Juselius Foundation (V.M.O.), the Finnish Foundation for Cardiovascular Research (V.M.O.), the Magnus Ehrnrooth Foundation (V.M.O.), and the Netherlands Organisation for Scientific Research (N.W.O.): ECHO-700.57.001, ALW-820.02.018 and VICI-91812628 (F.J.M.v.K.), VENI-722.012.066 (J.R.P.M.S.), and VENI-863.12.005 (H.M.v.d.S.). M.A. was supported in part by Grants-in-Aid for the Promotion of Polio Eradication and Research on Emerging and Re-emerging Infectious Diseases from the Ministry of Health, Labor and Welfare, Japan; a grant from the World Health Organization for a collaborative research project of the Global Polio Eradication Initiative; and by JSPS KAKENHI grant 25460579. N. Sever and P.A.B. are supported by the NIH. P.A.B. is an investigator of the Howard Hughes Medical Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. N. Schlinck is an employee of NanoTemper Technologies GmbH.
Published: January 29, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, one table, and one movie and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2014.12.054.
Supplemental Information
HeLa R19 cells transfected with GFP-OSBP and treated with DMSO, 10 μM of ITZ, or 10 nM OSW-1 were imaged overnight by live-cell confocal laser scanning microscopy. During the first 30 min, images were taken as fast as possible (∼1.5 min intervals), then intervals were stepwise increased to 30 min from 3.5 hr onward. Representative groups of cells are shown. The time after addition of the compounds for each image is indicated as hr: min: s. The size of the scale bar corresponds to 10 μm.
References
- Antonarakis E.S., Heath E.I., Smith D.C., Rathkopf D., Blackford A.L., Danila D.C., King S., Frost A., Ajiboye A.S., Zhao M., et al. Repurposing itraconazole as a treatment for advanced prostate cancer: a noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncologist. 2013;18:163–173. doi: 10.1634/theoncologist.2012-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 2014;58:239–256. doi: 10.1111/1348-0421.12144. [DOI] [PubMed] [Google Scholar]
- Arita M., Wakita T., Shimizu H. Cellular kinase inhibitors that suppress enterovirus replication have a conserved target in viral protein 3A similar to that of enviroxime. J. Gen. Virol. 2009;90:1869–1879. doi: 10.1099/vir.0.012096-0. [DOI] [PubMed] [Google Scholar]
- Arita M., Takebe Y., Wakita T., Shimizu H. A bifunctional anti-enterovirus compound that inhibits replication and the early stage of enterovirus 71 infection. J. Gen. Virol. 2010;91:2734–2744. doi: 10.1099/vir.0.023374-0. [DOI] [PubMed] [Google Scholar]
- Arita M., Kojima H., Nagano T., Okabe T., Wakita T., Shimizu H. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J. Virol. 2011;85:2364–2372. doi: 10.1128/JVI.02249-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita M., Kojima H., Nagano T., Okabe T., Wakita T., Shimizu H. Oxysterol-binding protein family I is the target of minor enviroxime-like compounds. J. Virol. 2013;87:4252–4260. doi: 10.1128/JVI.03546-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A., Tuymetova G., Tsiomenko A., Várnai P., Balla T. A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol. Biol. Cell. 2005;16:1282–1295. doi: 10.1091/mbc.E04-07-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., Nair V., Hansen B.T., Hoyt F.H., Fischer E.R., Ehrenfeld E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012;86:302–312. doi: 10.1128/JVI.05937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beretta L., Svitkin Y.V., Sonenberg N. Rapamycin stimulates viral protein synthesis and augments the shutoff of host protein synthesis upon picornavirus infection. J. Virol. 1996;70:8993–8996. doi: 10.1128/jvi.70.12.8993-8996.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgett A.W., Poulsen T.B., Wangkanont K., Anderson D.R., Kikuchi C., Shimada K., Okubo S., Fortner K.C., Mimaki Y., Kuroda M., et al. Natural products reveal cancer cell dependence on oxysterol-binding proteins. Nat. Chem. Biol. 2011;7:639–647. doi: 10.1038/nchembio.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charman M., Colbourne T.R., Pietrangelo A., Kreplak L., Ridgway N.D. Oxysterol-binding protein (OSBP)-related protein 4 (ORP4) is essential for cell proliferation and survival. J. Biol. Chem. 2014;289:15705–15717. doi: 10.1074/jbc.M114.571216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.K., Taipale J., Young K.E., Maiti T., Beachy P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F., Liu C., Jiang J., Lu W., Li W., Liu G., Zhou W., Huang J., Tang Y. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 2012;8:e1002503. doi: 10.1371/journal.pcbi.1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens J.R., Giddings T.H., Jr., Kirkegaard K. Inhibition of endoplasmic reticulum-to-Golgi traffic by poliovirus protein 3A: genetic and ultrastructural analysis. J. Virol. 1997;71:9054–9064. doi: 10.1128/jvi.71.12.9054-9064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu N.-Y., Ilnytska O., Belov G., Santiana M., Chen Y.-H., Takvorian P.M., Pau C., van der Schaar H., Kaushik-Basu N., Balla T., et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilnytska O., Santiana M., Hsu N.Y., Du W.L., Chen Y.H., Viktorova E.G., Belov G., Brinker A., Storch J., Moore C., et al. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe. 2013;14:281–293. doi: 10.1016/j.chom.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Tang J.Y., Gong R., Kim J., Lee J.J., Clemons K.V., Chong C.R., Chang K.S., Fereshteh M., Gardner D., et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17:388–399. doi: 10.1016/j.ccr.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.J., Kim J., Spaunhurst K., Montoya J., Khodosh R., Chandra K., Fu T., Gilliam A., Molgo M., Beachy P.A., Tang J.Y. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 2014;32:745–751. doi: 10.1200/JCO.2013.49.9525. [DOI] [PubMed] [Google Scholar]
- Lestner J., Hope W.W. Itraconazole: an update on pharmacology and clinical use for treatment of invasive and allergic fungal infections. Expert Opin. Drug Metab. Toxicol. 2013;9:911–926. doi: 10.1517/17425255.2013.794785. [DOI] [PubMed] [Google Scholar]
- Limpens R.W.A.L., Van der Schaar H.M., Kumar D., Koster A.J., Snijder E.J., Van Kuppeveld F.J.M., Bárcena M. The transformation of enterovirus replication structures: a three-dimensional study of single- and double-membrane compartments. MBio. 2011;2 doi: 10.1128/mBio.00166-11. e00166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod A.M., Mitchell D.R., Palmer N.J., Van de Poël H., Conrath K., Andrews M., Leyssen P., Neyts J. Identification of a series of compounds with potent antiviral activity for the treatment of enterovirus infections. ACS Med. Chem. Lett. 2013;4:585–589. doi: 10.1021/ml400095m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B., Bigay J., Moser von Filseck J., Lacas-Gervais S., Drin G., Antonny B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013;155:830–843. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- Nacev B.A., Grassi P., Dell A., Haslam S.M., Liu J.O. The antifungal drug itraconazole inhibits vascular endothelial growth factor receptor 2 (VEGFR2) glycosylation, trafficking, and signaling in endothelial cells. J. Biol. Chem. 2011;286:44045–44056. doi: 10.1074/jbc.M111.278754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretti D., Dahan N., Shimoni E., Hirschberg K., Lev S. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol. Biol. Cell. 2008;19:3871–3884. doi: 10.1091/mbc.E08-05-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry R.J., Ridgway N.D. Oxysterol-binding protein and vesicle-associated membrane protein-associated protein are required for sterol-dependent activation of the ceramide transport protein. Mol. Biol. Cell. 2006;17:2604–2616. doi: 10.1091/mbc.E06-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S., Prinz W.A. The diverse functions of oxysterol-binding proteins. Annu. Rev. Cell Dev. Biol. 2010;26:157–177. doi: 10.1146/annurev.cellbio.042308.113334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell C., Lebreton A., Young Ng C., Lim J.Y., Liu W., Vasudevan S., Labow M., Gu F., Gaither L.A. Cholesterol biosynthesis modulation regulates dengue viral replication. Virology. 2009;389:8–19. doi: 10.1016/j.virol.2009.03.025. [DOI] [PubMed] [Google Scholar]
- Roulin P.S., Lötzerich M., Torta F., Tanner L.B., van Kuppeveld F.J., Wenk M.R., Greber U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe. 2014;16:677–690. doi: 10.1016/j.chom.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Rudin C.M., Brahmer J.R., Juergens R.A., Hann C.L., Ettinger D.S., Sebree R., Smith R., Aftab B.T., Huang P., Liu J.O. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J. Thorac. Oncol. 2013;8:619–623. doi: 10.1097/JTO.0b013e31828c3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J., Chen J.K., Cooper M.K., Wang B., Mann R.K., Milenkovic L., Scott M.P., Beachy P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- van der Schaar H.M., van der Linden L., Lanke K.H., Strating J.R., Pürstinger G., de Vries E., de Haan C.A., Neyts J., van Kuppeveld F.J. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res. 2012;22:1576–1592. doi: 10.1038/cr.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaar H.M., Leyssen P., Thibaut H.J., de Palma A., van der Linden L., Lanke K.H., Lacroix C., Verbeken E., Conrath K., Macleod A.M., et al. A novel, broad-spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4-kinase IIIβ. Antimicrob. Agents Chemother. 2013;57:4971–4981. doi: 10.1128/AAC.01175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kuppeveld F.J., Galama J.M., Zoll J., Melchers W.J. Genetic analysis of a hydrophobic domain of coxsackie B3 virus protein 2B: a moderate degree of hydrophobicity is required for a cis-acting function in viral RNA synthesis. J. Virol. 1995;69:7782–7790. doi: 10.1128/jvi.69.12.7782-7790.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsky R.L., Bauman J.N., Bourcier K., Giddens G., Lapham K., Negahban A., Ryder T.F., Obach R.S., Hyland R., Goosen T.C. Optimized assays for human UDP-glucuronosyltransferase (UGT) activities: altered alamethicin concentration and utility to screen for UGT inhibitors. Drug Metab. Dispos. 2012;40:1051–1065. doi: 10.1124/dmd.111.043117. [DOI] [PubMed] [Google Scholar]
- Wang C., JeBailey L., Ridgway N.D. Oxysterol-binding-protein (OSBP)-related protein 4 binds 25-hydroxycholesterol and interacts with vimentin intermediate filaments. Biochem. J. 2002;361:461–472. doi: 10.1042/0264-6021:3610461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.J., Lew K., Casciano C.N., Clement R.P., Johnson W.W. Interaction of common azole antifungals with P glycoprotein. Antimicrob. Agents Chemother. 2002;46:160–165. doi: 10.1128/AAC.46.1.160-165.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Perry J.W., Lauring A.S., Neddermann P., De Francesco R., Tai A.W. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology. 2014;146:1373–1385, e1–e11. doi: 10.1053/j.gastro.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrilow A.G., Parker J.E., Kelly D.E., Kelly S.L. Azole affinity of sterol 14α-demethylase (CYP51) enzymes from Candida albicans and Homo sapiens. Antimicrob. Agents Chemother. 2013;57:1352–1360. doi: 10.1128/AAC.02067-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber-Boyvat M., Zhong W., Yan D., Olkkonen V.M. Oxysterol-binding proteins: functions in cell regulation beyond lipid metabolism. Biochem. Pharmacol. 2013;86:89–95. doi: 10.1016/j.bcp.2013.02.016. [DOI] [PubMed] [Google Scholar]
- Wong J., Zhang J., Si X., Gao G., Mao I., McManus B.M., Luo H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008;82:9143–9153. doi: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyles J.P., Perry R.J., Ridgway N.D. Characterization of the sterol-binding domain of oxysterol-binding protein (OSBP)-related protein 4 reveals a novel role in vimentin organization. Exp. Cell Res. 2007;313:1426–1437. doi: 10.1016/j.yexcr.2007.01.018. [DOI] [PubMed] [Google Scholar]
- Xu J., Dang Y., Ren Y.R., Liu J.O. Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc. Natl. Acad. Sci. USA. 2010;107:4764–4769. doi: 10.1073/pnas.0910872107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Pillai V.C., Mada S.R., Strom S., Venkataramanan R. Effect of voriconazole and other azole antifungal agents on CYP3A activity and metabolism of tacrolimus in human liver microsomes. Xenobiotica. 2012;42:409–416. doi: 10.3109/00498254.2011.631224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HeLa R19 cells transfected with GFP-OSBP and treated with DMSO, 10 μM of ITZ, or 10 nM OSW-1 were imaged overnight by live-cell confocal laser scanning microscopy. During the first 30 min, images were taken as fast as possible (∼1.5 min intervals), then intervals were stepwise increased to 30 min from 3.5 hr onward. Representative groups of cells are shown. The time after addition of the compounds for each image is indicated as hr: min: s. The size of the scale bar corresponds to 10 μm.