

Abstract
Rhodopsin is a G protein-coupled receptor essential for vision and rod photoreceptor viability. Disease-associated rhodopsin mutations, such as P23H rhodopsin, cause rhodopsin protein misfolding and trigger endoplasmic reticulum (ER) stress, activating the Unfolded Protein Response (UPR). The pathophysiologic effects of ER stress and UPR activation on photoreceptors are unclear. Here, by examining a P23H rhodopsin knock-in mouse, we found that the UPR IRE1 signaling pathway is strongly activated in misfolded rhodopsin-expressing photoreceptors. IRE1 significantly upregulated ER-associated protein degradation (ERAD), triggering pronounced P23H rhodopsin degradation. Rhodopsin protein loss occurred as soon as photoreceptors developed, preceding photoreceptor cell death. By contrast, IRE1 activation did not affect JNK signaling or rhodopsin mRNA levels. Interestingly, pro-apoptotic signaling from the PERK UPR pathway was also not induced. Our findings reveal that an early and significant pathophysiologic effect of ER stress in photoreceptors is the highly efficient elimination of misfolded rhodopsin protein. We propose that early disruption of rhodopsin protein homeostasis in photoreceptors could contribute to retinal degeneration.
Keywords: Endoplasmic reticulum-associated degradation, rhodopsin, retinal degeneration, unfolded protein response, ER stress, rod photoreceptor
INTRODUCTION
The endoplasmic reticulum (ER) is essential for secretory and membrane protein folding and assembly. Impairment of ER protein folding creates ER stress, activating the Unfolded Protein Response (UPR) intracellular signal transduction network [1]. The UPR regulates transcriptional, translational, and post-translational intracellular programs such as ER-associated protein degradation (ERAD) whereby ER chaperones, protein folding enzymes, and lectins identify malformed proteins in the ER and retrotranslocate them into the cytosol for degradation by proteasomes [2]. The ERAD program therefore prevents misfolded protein accumulation, and disruption of ERAD components often leads to early embryonic lethality [3–7].
Inositol-Requiring Enzyme 1 (IRE1) is a key UPR regulator [1]. IRE1 encodes an ER-resident transmembrane protein with an ER-luminal domain coupled to cytosolic kinase and endoribonuclease (RNase) domains [8–10]. In response to ER stress, IRE1’s RNase removes an inhibitory intron from Xbp-1 mRNA [11,12] to generate the potent XBP1s transcription factor whose transcriptional targets include numerous ERAD genes [13,14]. By this mechanism, IRE1 signaling regulates the ERAD program. In addition to ERAD, IRE1 signaling can also activate the c-Jun Kinase signaling module and non-specifically degrade mRNAs through Rapid IRE1-mediated mRNA decay (RIDD) in response to strong ER stress [1]. PKR-like ER kinase (PERK) mediates a separate UPR signaling pathway [1]. In response to ER stress, PERK inhibits the ternary translation initiation complex formation on mRNAs [15] and therefore attenuates protein synthesis in the cell. PERK signal transduction also regulates a cell death program through its upregulation of proapoptotic transcriptional activators, ATF4 and CHOP [16,17].
Rod opsin mutations are common causes of heritable retinitis pigmentosa, a blinding disease arising from photoreceptor cell death [18]. Rod opsin is a G protein-coupled receptor that forms the visual photopigment, rhodopsin, when coupled with 11-cis-retinal [19]. Rhodopsin is essential for photoreceptor function and survival, and rod opsin knockout mice (Rho−/−) develop early retinal degeneration [20,21]. In cell culture, many rod opsin mutants misfold and aggregate to trigger ER stress and activate UPR [22–26]. However, the effects and role of UPR activation in photoreceptor cell death remain obscure.
Here, we analyzed UPR signaling in our recently developed P23H rod opsin knock-in mouse model of retinitis pigmentosa [27] that recapitulated patients’ gene dosage and spatiotemporal retinal degeneration [27]. By contrast, transgenic models of retinal degeneration produced retinal degeneration regardless of whether misfolded or wild-type rhodopsin was expressed [28]. Here, we found an unexpected bias in UPR signaling in photoreceptors, with strong IRE1 but little PERK signaling. The pathophysiologic consequence of IRE1 activation was robust ERAD elimination of almost all P23H rhodopsin from photoreceptors early after their birth to create a Rho−/−-like retinal degeneration phenotype.
MATERIALS AND METHODS
Animals
Homozygous breeding pairs of P23H rod opsin knock-in mice [27] were maintained to produce RhoP23H/P23H homozygotes or crossed with wild-type C57BL/6J (Rho+/+) mice to produce RhoP23H/+ heterozygotes. C57BL/6J mice were used as wild-type controls (Rho+/+). ERAI+/− mice were provided by Takao Iwawaki (RIKEN, Saitama, Japan) and Peter Walter (UCSF). Chop−/− mice were obtained from Jackson Laboratory. Mice were maintained in a barrier animal facility in a 12:12 light cycle at in-cage irradiance of less than 125 lux and provided standard mouse chow (UCSF and UCSD) or in a 12–h light (127–255 lux)/12–h dark (< 10 lux) cyclic environment (Case Western Reserve University). Retinal tissues were collected from the animals of either sex for at indicated ages. Mice were checked daily by the UCSF Animal Care staff. In addition, laboratory personnel checked births in the morning and evening during the light phase of the light cycle to determine exact birthdates because early eye developmental and degenerative events occurred rapidly once they commenced. To define phenotypes of each of the 3 genotypes from P4 to P15, we examined two litters of mice of either sex with exact birthdates from each line, examining one animal of either sex from each litter on successive dates, beginning on P4 (Table S1). All animal studies followed the guidelines of the institutional animal care committees at UCSF, UCSD, or Case Western Reserve University, and were conducted in accordance with the recommendations of the American Veterinary Medical Association Panel on Euthanasia and the Association of Research for Vision and Ophthalmology.
Tissue Harvesting and Eye Histology
Immediately after euthanizing mice, an enucleated eye of each mouse was immersed in a fixative of mixed aldehydes for histology [29], and the other eye was removed, and the retina was dissected by the Winkling procedure [30] and rapidly frozen in dry ice for biochemical and molecular analyses. For histological analysis, eyes were bisected, post-fixed with osmium tetroxide, and embedded in an epoxy resin as previously described [29]. One-μm thick sections were cut through the optic nerve head along the vertical meridian and stained with toluidine blue. Beginning at P8, the outer nuclear layer (ONL) thickness, a surrogate for photoreceptor number [31], was quantified by measuring 3 positions in nine 250-μm adjacent microscopic fields on each side of the optic nerve head that produced 54 measurements of each retina [32]. Means of these measurements allowed quantitative comparisons of multiple animals at different ages and of different genotypes in the two hemispheres of the eye, and the resulting values could then be plotted in a “retinal spidergram” to illustrate hemispheric differences across the retina [33]. All quantitative measurements were from a single retina from 5–7 different mice of each experimental cohort.
The young retinas, particularly at P4–P8, changed so rapidly in formation of the ONL from the outer neuroblastic layer in a strong central-to-peripheral gradient (which all features follow in the developing eye) that they were compared based on the following 4 phenotypic features: 1) length of rod inner + outer segments (if present) measured with an eyepiece micrometer in several regions in the central retina; 2) development of the outer plexiform layer (OPL) based on the percentage of the length in which it was present in each hemisphere (measured from the posterior pole); 3) incidence of ectopic photoreceptor nuclei still in the inner nuclear layer [34] measured as a percentage of the length in each hemisphere in which they still comprised a continuous row (measured from the ora serrata); and 4) incidence of pyknotic nuclei in the ONL, as seen in most rodent retinal degenerations [35,36], per 430-μm microscopic field.
Immunofluorescence and Confocal Microscopy
Eyes were enucleated and fixed by immersion in 4% paraformaldehyde in PBS buffer for 1 h at room temperature. After overnight incubation with 30% sucrose at 4°C, eyes were frozen in O.C.T. (Tissue-Tek). Eight-micrometer thick sections were cut through the optic nerve head and stained with indicated antibodies. Primary antibodies used included 1D4 anti-rhodopsin 1:500 dilution (Santa Cruz Biotechnologies, CA); anti-calnexin 1:250 (GeneTex, Irvine, CA); and anti-GFP 1:250 (Invitrogen, Carlsbad, CA). Secondary antibodies included Alexa 546 goat anti-mouse (Molecular Probes, Invitrogen) and Alexa 488 goat anti-rabbit (Molecular Probes, Invitrogen) used at 1:500. Images were collected with an Olympus FluoView-1000 confocal microscope and processed using Olympus FluoView Ver.2.0a Viewer software.
Transmission Electron Microscopy
Four mice from each genotype (Rho+/+, Rho P23H/+ and Rho P23H/P23H) were analyzed by transmission electron microscopy at P14. Fixation, sectioning and staining were performed according to Sakami et al., 2013. Low magnification (x 2000) TEM images of photoreceptor cell inner segments were captured by a JEOL 1200 EX EM at 80 kV (JEOL, Musashino, Tokyo, Japan). 8 images from each genotype (two images from each mouse) were analyzed.
Molecular Biology
Retinas were lysed and total RNA was collected with a RNeasy mini kit (Qiagen, Germany). mRNA was reverse-transcribed with the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). For quantitative PCR analyses, primers included: mouse rhodopsin mRNA, 5′-TTCACCACCACCCTCTACACATCAC-3′ and 5′-CGGAAGTTGCTCATCGGCTTG-3′; mouse GNAT1 mRNA, 5′-GAGGATGCTGAGAAGGATGC-3′ and 5′-TGAATGTTGAGCGTGGTCAT-3′; mouse Xbp-1s mRNA, 5′-GAGTCCGCAGCAGGTG-3′ and 5′-GTGTCAGAGTCCATGGGA-3′; mouse Derl1 mRNA, 5′-CGCGATTTAAGGCCTGTTAC-3′ and 5′-GGTAGCCAGCGGTACAAAAA-3′; mouse VCP mRNA, 5′-AAGTCCCCAGTTGCCAAGGATG-3′ and 5′-AGCCGATGGATTTGTCTGCCTC-3′; mouse ERdj4, 5′-TAAAAGCCCTGATGCTGAAGC -3′ and 5′-TCCGACTATTGGCATCCGA -3′; mouse Grp78/BiP: 5′-CCTGCGTCGGTGTGTTCAAG-3′ and 5′-AAGGGTCATTCCAAGTGCG-3′; mouse Rpl19: 5′-ATGCCAACTCCCGTCAGCAG- 3′ and 5′-TCATCCTTCTCATCCAGGTCACC-3′; and mouse Chop: 5′-ACGGAAACAGAGTGGTCAGTGC-3′ and 5′-CAGGAGGTGATGCCCACTGTTC-3′. For rhodopsin mRNA levels, GNAT1 mRNA levels, a transcript only expressed in rod cells, served as the normalization control. For all other qPCR analysis, RPL19 mRNA levels, a transcript with levels unaltered by ER stress, served as internal normalization standards. qPCR conditions were 95°C for 5 min; 95°C for 10 sec; 60°C for 10 sec; 72°C for 10 sec, with 50 cycles of amplification.
Cell Culture
Cells were maintained at 37°C, 5% CO2 in Dulbecco’s modified Eagle medium (Mediatech, Manassas, VA) supplemented with 10% fetal calf serum (Invitrogen), and 1% penicillin/streptomycin (Invitrogen). Tetracycline-inducible P23H rhodopsin HEK293 cell lines was previously described [25]. To induce the expression of P23H rhodopsin, 1μg/ml of doxycycline is added to the cell culture media for 20 hours.
Immunoprecipitation
Rhodopsin proteins were immunoprecipitated for mass spectrometry (MS) analysis or immunoblotting analysis. Retinas from 3 to 5 mice per experiment were collected from Rho+/+, RhoP23H/+ and RhoP23H/P23H mice. As controls, one 10-cm dish of MEF cells was collected. To lyse the retinas or MEF cells, 1 ml of PBSD (PBS containing 1.0% n-dodecyl-b-D-maltoside and protease and phosphatase inhibitor mixture) was added to MEF cells or retinas. After sonication, the suspension was rotated for 1 h at 4°C. The supernatants were collected, and the protein concentrations were determined with a BCA assay kit (Pierce). Equal amounts of total protein were added to Dynabeads Protein G (Invitrogen) preincubated with 1D4 anti-rhodopsin antibody (Santa Cruz Biotechnologies) and incubated overnight at 4°C. To elute rhodopsin protein for immunoblotting analysis, beads were heated with SDS sample buffer at 80°C. For MS analysis, rhodopsin was eluted twice from the beads by heating the beads with elution buffer (PBS containing 8 M urea and 0.5% n-dodecyl-b-D-maltoside) for 10 minutes at 80°C. Total proteins from the two eluates were combined and subjected to methanol chloroform extraction to remove detergent.
Liquid Chromatography-Tandem Mass Spectrometry
Protein pellets were resuspended in 8 M urea. The samples then were reduced by a 20 min incubation with 5 mM TCEP (tris(2-carboxyethyl)phosphine) at room temperature and alkylated in the dark by treatment with 10mM iodoacetamide for 20 additional min. Proteins were digested over-night at 37°C with Sequencing Grade Modified Trypsin (Promega, Madison, WI, USA) and the reaction was stopped by acidification. A 100-μm i.d. capillary with a 5-μm pulled tip was packed with 10 cm of 5-μm Aqua C18 material (Phenomenex). This desalting column then was equilibrated for 30 min with Buffer A (5% acetonitrile/0.1% formic acid), and the protein digest was loaded onto it under pressure. The column was placed in line with an Agilent 1200 quaternary HPLC and analyzed after elution and separation. As peptides eluted from the micro-capillary column, they were electrosprayed directly into an LTQ -Orbitrap mass spectrometer from Thermo Finnigan (Waltman, MA) with the application of a distal 2.4-kV spray voltage.
Protein identification and quantification and analyses were done with Integrated Proteomics Pipeline - IP2 (Integrated Proteomics Applications, Inc., www.integratedproteomics.com/) using ProLuCID and DTASelect2. Spectrum raw files were extracted into ms1 and ms2 files from raw files with RawExtract 1.9.9 (http://fields.scripps.edu/downloads.php) [37], and the tandem mass spectra were searched against European Bioinformatic Institute protein databases To estimate peptide probabilities and FDRs accurately, we used a target/decoy database containing the reversed sequences of all the proteins appended to the target database [38]. Tandem mass spectra were matched to sequences by using the ProLuCID [39] algorithm with 50 ppm peptide mass tolerance for precursor ions and 400 ppm for fragment ions.
Immunoblotting Analysis
Mouse retinas were lysed in 300 μl lysis buffer (PBS, 0.5 g/ml n-dodecyl-b-D-maltoside (Calbiochem EMD Bioscience), protease inhibitors (Sigma-Aldrich), and phosphatase inhibitor (Thermo Scientific, Rockford, IL)). MEF cells and HEK293 cells with indicated drug treatments were lysed in SDS lysis buffer (2% SDS, 62.5 mM Tris-HCl pH 6.8, containing protease inhibitors and phosphatase inhibitor). The following antibodies and dilutions were used: anti-Gαt1 and 1D4 anti-rod opsin 1:1000 dilution (Santa Cruz Biotechnologies); B630N anti-rhodopsin 1:1000 (gift of W.C. Smith, Gainesville, FL); anti-calnexin 1:1000, anti-VCP at 1:1000, anti-HSP90 at 1:5000, and anti-β-tubulin at 1:5000 (GeneTex); anti-P-JNK at 1:1000 and anti-JNK at 1:1000 (Cell Signaling); anti-flag at 1:5000 (Sigma-Aldrich); and anti-ubiquitin at 1:1000 (Dako). After overnight incubation with primary antibody, membranes were washed followed by incubation with a horseradish peroxidase-coupled secondary antibody (Cell signaling). Immunoreactivity was detected with the SuperSignal West chemiluminescent substrate (Pierce).
Statistical Analyses
All results are presented as means ± standard deviations from at least 3 mice per experimental condition as indicated. Student two-tailed t-tests (for paired samples) were performed to determine p-values. A p value of ≤0.05 was considered statistically significant.
RESULTS
Disruption of photoreceptor compartment morphogenesis in P23H rhodopsin knock-in mice during early retinal degeneration
In wild-type mice (Rho+/+), photoreceptor development and histogenesis follow well-described, stereotyped, temporal programs [40–42]. Photoreceptor neurogenesis starts in the embryo and lasts through approximately the first week of post-natal life (Figure 1A). Post-mitotic photoreceptor cell bodies accumulate in the outermost neural lamina of the retina, termed the outer nuclear layer (ONL) (Figure 1A). Newly formed rod photoreceptors cells develop morphologically and functionally distinct cellular compartments including the rod inner segment (RIS) and the more distal rod outer segment (ROS). Biosynthetic organelles, such as the ER, Golgi, and mitochondria, are found in the RIS (Figure 3A), and the RIS cellular compartment can be readily distinguished in Rho+/+ retinas by post-natal day (P) 8 (Figure 1A). The ROS is a specialized cilium that house hundreds of membranous discs, embedded with rhodopsin protein for phototransduction [43]. In Rho+/+ mice, nascent ROS were clearly visible at P10 (Figure 1D) and scattered ROS disc membranes were also seen as early as P7–P8 by electron microscopy [40]. The ROS elongated to about 2/3 of their adult length by P15 (Figure 2A), and reached their maximum adult length shortly afterwards as shown at P30 and P60 (Figures 2D, 2G).
Figure 1.

Light micrographs of wild-type and P23H mouse retinas at postnatal day (P) 8, P10, and P12. Degeneration of photoreceptor morphology in RhoP23H/P23H mice begins by postnatal day 10. (A–C) At P8, the ROS have not formed, and the thicknesses of the rod inner segment (IS) layer and outer nuclear layer (ONL) are identical between Rho+/+, RhoP23H/+ and RhoP23H/P23H mice. (D–F) Also in P10, nascent ROS can be identified in Rho+/+ and RhoP23H/+ mice but are absent in RhoP23H/P23H mice. By P10, the rod IS layer is thinner in RhoP23H/P23H retinas compared to those in Rho+/+ and RhoP23H/+ mice. By P10, pyknotic nuclei (white arrows) are identified in RhoP23H/P23H mice at a significantly greater incidence than in Rho+/+ or RhoP23H/+ mice. (G–I) By P12, ROS are present in Rho+/+ and RhoP23H/+ mice but are absent in RhoP23H/P23H mice. The rod IS layer and ONL are appreciably thinner in RhoP23H/P23H mice compared to Rho+/+ and RhoP23H/+ mice, and many pyknotic nuclei (white arrows) are identified in the inner aspect of the ONL in RhoP23H/P23H mice. Scale bar: 20 μM
Figure 3.

Transmission electron microscopy images of photoreceptor cell inner segments at postnatal day 14. (AC) Mitochondria (Mi), rough endoplasmic reticulum (rER), Golgi apparatus (G) and ribosomes (r) were abundant in the inner segment. Golgi apparatus was observed at the proximal part of most inner segments. (A) Image of longitudinal-cut inner segments from Rho+/+ mouse retinas. Column shaped inner segments were observed between photoreceptor cell outer segments (OS) and outer limiting membranes (OLM). In rare cases, the entire cone photoreceptor outer segment (COS) and inner segment (CIS) of a single cell was observed. (B) Image of tilted-cut inner segments from RhoP23H/+ mouse retinas. 2–4 rows of oval or column shaped inner segments were observed between OS and OLM. The structure of the rER and Golgi apparatus in the inner segments of RhoP23H/+ mice was almost identical to Rho+/+ mouse retinas especially compared to tilted-cut inner segment images from Rho+/+ mouse retinas. (C) Image of slightly tilted-cut inner segments from RhoP23H/P23H mouse retinas. 1–3 rows of oval or column shaped inner segments were observed between retinal pigment epithelium microvilli (RM) and OLM. Ciliary protrusions (CP) and cone outer segments (COS) were observed in the space between RM and the distal ends of inner segments. Not all, but some inner segments showed multiple vacuolization (white arrow). Such vacuolization was observed in all four analyzed RhoP23H/P23H mice, but not in TEM images of RhoP23H/+ and Rho+/+ mice (four mice were analyzed for each genotype). Hypothetical diagram of photoreceptor cells are included, indicating the cutting planes with dashed lines. Scale bar, 2 μm.
Figure 2.

Light micrographs of wild-type and P23H mouse retinas at P15, P30, and P60. Degeneration of photoreceptor morphology in RhoP23H/+ mice begins by P15. (A–C) At P15, the rod OS are shorter and more disorganized in RhoP23H/+ mice (B) compared to Rho+/+ mice (A), and only short fragments of ROS disc membranes are present in RhoP23H/P23H mice (C). At P15, the rod inner segments (IS) are shorter in RhoP23H/+ mice compared to Rho+/+ and significantly shortened in RhoP23H/P23H mice. Pyknotic nuclei (white arrows) are evident in RhoP23H/+ mice. The ONL is significantly thinner in RhoP23H/P23H mice (C). (D–F) By P30, the OS, IS, and ONL are significantly thinner in RhoP23H/+ mice (E) compared to Rho+/+ mice (D). Extensive photoreceptor cell loss is present in RhoP23H/P23H mice, with less than 1 full row of nuclei present in the ONL (F). (G–I) By P60, the OS, IS, and ONL have progressively thinned in RhoP23H/+ mice (H) compared to Rho+/+ mice (G), and even further loss of photoreceptors is evident in the ONL of RhoP23H/P23H mice (I). Scale bar: 20 μM
In heterozygous RhoP23H/+ mice, the thickness of the ONL was indistinguishable from wild-type Rho+/+ mice from P4 through P12 (Figures 1B, 1E, 1H, Table S1). By P15, scattered pyknotic nuclei could be identified (Figure 2B, Table S1), accompanied by a slight reduction in the mean ONL layer thickness (P<0.01), (Figure 4A), indicating the earliest onset of photoreceptor cell loss and retinal degeneration. The lengths of the photoreceptor RIS and ROS compartments in RhoP23H/+ mice were also indistinguishable from those of wild-type mice until P15 (Figures 1B, 1E, 1H, Table S1), when both RIS and ROS compartments were clearly shorter compared to their wild-type photoreceptor counterparts (compare Figure 2A and 2B). Later, at P30 (Figure 2E) and P60 (Figure 2H), the retinas from heterozygous in RhoP23H/+ mice showed progressive shortening of ROS, RIS, and loss of photoreceptor nuclei in the ONL (Figure 2E, 2H) and as previously described [27].
Figure 4.
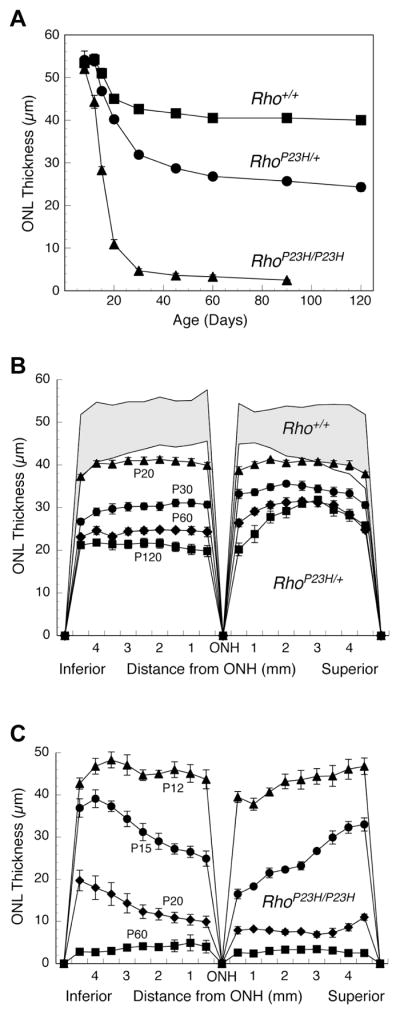
Anatomic distribution of retinal degeneration in Rho+/+, RhoP23H/+ and RhoP23H/P23H mice. (A) Mean ONL thickness of Rho+/+, RhoP23H/+ and RhoP23H/P23H retinas. Each value is the mean ± SEM of 5–7 retinas, one from each mouse; the mean for each retina was determined from 54 measurements around the eye from inferior to superior poles. If error bars are not shown, they fall within the symbol. (B) Retinal “spidergram” showing ONL thickness from the optic nerve head (ONH) to the anterior optic margin (ora serrata) in the inferior and superior hemispheres of Rho+/+ retinas and RhoP23H/+ retinas at P12, P20, P30, P60, and P120. The top border of the shaded area represents the Rho+/+ ONL thickness at P12, and the bottom border shows the Rho+/+ ONL thickness at P120. Thinning of the retina ONL in Rho+/+ mice between P12 and P120 is due to growth of the eye, not retinal degeneration. The ONL thickness progressively diminishes in RhoP23H/+ from P20 to P120 with more pronounced retinal degeneration in the inferior hemisphere. (C) Retinal spidergram showing a rapid decrease in ONL thickness in RhoP23H/P23H retinas from P12 to P60. In RhoP23H/P23H retinas, the superior hemisphere shows greater loss of the ONL, the opposite of that seen in the RhoP23H/+ retinas.
In homozygous RhoP23H/P23H retinas, thicknesses of the ONL and RIS were indistinguishable from wild-type mice from birth up to P10 (Figures 1A, 1C, Table S1). By P10, rare pyknotic photoreceptor nuclei were evident in the ONL (Figure 1F, Table S1), and photoreceptor RIS lengths were shorter in homozygous RhoP23H/P23H retinas compared to wild-type (Figures 1D, 1F, Table S1). Little ROS was seen in homozygous RhoP23H/P23H retinas compared to wild-type at this age (Figures 1D, 1F, Table S1). By P12, the prevalence of pyknotic photoreceptor nuclei increased significantly (Figure 1I, Table S1), accompanied by a measurable reduction in ONL thickness (P<0.001), (Figure 4A). Also, by P12, photoreceptor morphology was further compromised with markedly stunted ROS (Figure 1I) bearing abnormally formed discs visualized by electron microscopy in ciliary protrusions [44]. By P14–15, vacuolization of IS become pronounced (Figure 3C). The RIS remained short, and few visible ciliary protrusions were present (Figure 2C, Figure 3C). 40–50% of the ONL thickness was lost (Figure 2C, Table S1), and the nuclei of many of the remaining photoreceptors were clearly pyknotic (Figure 2C). By P30 (Figure 2F) and P60 (Figure 2I), less than a full row of photoreceptor nuclei remained in the ONL, and some attenuation of the retinal pigment epithelium is evident at P60 (Figure 2I).
When the mean ONL values across the entire retina are plotted, sharp differences in the kinetics of photoreceptor loss and amount of retinal degeneration was evident between the heterozygous and homozygous P23H genotypes, with an early and rapid loss of ONL thickness in the RhoP23H/P23H retinas significantly preceding the onset of loss seen in the heterozygous RhoP23H/+ retinas (Figure 4A). The ONL thicknesses at P30 and later in the homozygous RhoP23H/P23H and heterozygous RhoP23H/+ retinas were similar to our prior findings [27], with the loss of most photoreceptors in the RhoP23H/P23H mice by P30. By contrast, the ONL thickness stabilized and retinal degeneration plateaued in RhoP23H/+ retinas by P30 (Figure 4A). Previously, we reported greater loss of photoreceptors in the inferior hemisphere of the eye in heterozygous RhoP23H/+ retinas than in the superior hemisphere at P30 [27]. Our retinal “spidergram” measurements of ONL thickness at different ages in the heterozygote RhoP23H/+ retinas (Figure 4B) revealed this hemispheric asymmetrical degeneration was not present at P20, but present at all older ages examined (Figure 4B, P45 and P90 not shown). In the homozygous RhoP23H/P23H retinas, however, the hemispheric asymmetry in retinal degeneration was reversed, with the superior hemisphere more severely affected in all mice from P12 to P60 (Figure 4C; P30 and P45 not shown), but not at ages older than P60. The difference in the least marked asymmetry at P12 was significant, however (P<0.005).
Our analyses of early retinal development revealed an unexpected early onset of photoreceptor pathology in P23H mice. Although photoreceptor neurogenesis and retinal patterning appear histologically normal in both heterozygous and homozygous P23H mice, severe defects in photoreceptor compartment morphogenesis, especially the proper generation and elongation of ROS and to a lesser extent RIS, appear in RhoP23H/P23H mice as soon as P10, coinciding with the onset and progressive death of nearly all photoreceptors by P30. Defects in photoreceptor ROS and RIS morphogenesis also appear in heterozygous RhoP23H/+ mice beginning at a slightly later date, ~P15, and these also coincide with the onset of retinal degeneration. However, by contrast to homozygous mice, far fewer photoreceptors are lost in heterozygous RhoP23H/+ retinas such that the ONL thickness stabilizes by ~P30 (Figure 4A). These studies revealed a critical period within the first postnatal month of life when photoreceptors in both heterozygous and homozygous P23H mice undergo degeneration. We next examined UPR activity during this early period of photoreceptor cell death in P23H rhodopsin mice.
The IRE1 signaling pathway of the UPR is strongly induced in photoreceptors expressing P23H rhodopsin in vivo
To investigate the role of the IRE1 signaling UPR pathway in native photoreceptors, we crossed RhoP23H/P23H mice with ERAI reporter mice [45]. ERAI mice carry a modified FLAG-tagged truncated Xbp-1 that lacks the DNA-binding and transcriptional activator domains but retains the native Xbp-1 intron fused upstream of Venus, a green fluorescent protein (GFP) variant. Activated IRE1 splices out the Xbp-1 intron, leading to the production of FLAG-tagged XBP1-Venus fusion protein [45]. Thus, the ERAI reporter mouse provides a highly specific monitor of the status of IRE1 signaling at the cellular, tissue, and organ level, and in the eye, the ERAI reporter mouse offers a useful way to distinguish IRE1 signaling status between the different laminar layers of the retina [46,47].
In RhoP23H/+ERAI+/− mice, we detected strong expression of XBP1-Venus protein in retinas compared to Rho+/+ERAI+/− littermates at P30 (Figure 5A–5H). Strong XBP1-Venus expression was localized to rod photoreceptor cells in the outer nuclear layer (Figure 5D) with strong staining seen in the RIS and perinuclear region of all rod photoreceptors (Figure 5F, 5H). XBP1-Venus expression was excluded from photoreceptor nuclei consistent with the deletion of the DNA-binding domain of XBP1 in the reporter construct (Figure 5F, 5H and [45]. By contrast, in wild-type retinas, only rare XBP1-Venus staining was observed in photoreceptors in the ONL (Figure 5C, 5E, 5G). At P120, we also observed strong XBP1-Venus staining in the RhoP23H/+ERAI+/− mice, but not the Rho+/+ERAI+/− littermates (Figure 5I, 5J, 5K, 5L). In other retinal cells of RhoP23H/+ERAI+/− mice, XBP1-Venus staining was also rarely seen in cone photoreceptors (indicated by a white arrow in Figure 5L), the inner nuclear layer, ganglion cell layer, and retinal pigment epithelium (data not shown). Whole retinal lysates also showed significantly increased FLAG-tagged XBP1-Venus protein expression in RhoP23H/+ERAI+/− mice compared to controls (Figure 5M), consistent with the increased XBP1-Venus protein by imaging. These findings demonstrate that the ERAI reporter is efficiently and specifically activated in rod photoreceptors expressing P23H rhodopsin.
Figure 5.

The IRE1 signaling pathway of the UPR is strongly activated in photoreceptors of RhoP23H/+ERAI+/− mice. (A–H) Rhodopsin (red) and XBP1-Venus (by anti-GFP, green) were visualized in eyes from RhoP23H/+ERAI+/− mice and Rho+/+ERAI+/− mice at P30. DAPI (blue) highlights photoreceptor nuclei. Strong XBP1-Venus staining is visualized in the IS and perinuclear region of ONL photoreceptors expressing P23H rhodopsin. (I–L) Peanut agglutinin (PNA) staining of cones (red) and XBP1-Venus (by anti-GFP, green) were visualized in eyes from RhoP23H/+ERAI+/− mice and Rho+/+ERAI+/− mice at P120. DAPI (blue) highlights photoreceptor nuclei. White arrow in (L) indicated the cone cell overlapping with XBP1-Venus signal. (M) XBP1-Venus (by anti-flag) protein levels were detected in retinal lysates from RhoP23H/+ERAI+/− mice and littermate control Rho+/+ERAI+/− mice at P30. HSP90 served as a protein loading control. (N) XBP1-Venus protein levels were detected by anti-flag or anti-GFP in retinal lysates from RhoP23H/+ERAI+/− mice at indicated ages.
To confirm the activation status of the endogenous IRE1 signal transduction pathway in retina, we examined the splicing status of native Xbp-1 mRNA in RhoP23H/+ERAI+/− mice. We found increased levels of spliced Xbp-1 mRNA in animals expressing P23H rhodopsin (Figure 6A). Spliced Xbp-1 produces a potent transcription factor, XBP1s, with many transcriptional targets that are shared between diverse mammalian cells [14,13,48,49]. In both RhoP23H/+ and RhoP23H/P23H mice, we found significantly elevated mRNA levels of Erdj4, VCP, Derl1, and Grp78 (Figure 6B, 6C, 6D, 6E) at P30. Furthermore, we found a P23H rhodopsin dose-dependent increase in the upregulation of Xbp-1 splicing and XBP1s target genes, with significantly higher levels of spliced Xbp-1, Erdj4, VCP, Derl1, and Grp78 in retinas from RhoP23H/P23H mice expressing two copies of P23H rod opsin gene compared to the RhoP23H/+ mice expressing only one copy (Figure 6A–E). From these findings, we conclude that P23H rhodopsin induces ER stress and strongly activates the IRE1 pathway in photoreceptors in vivo.
Figure 6.

The IRE1 signaling pathway of the UPR is strongly activated in photoreceptors expressing P23H rhodopsin. (A–E) Xbp-1s mRNA level (A) and XBP1s transcriptional target genes, Erdj4 (B), VCP (C), Derl1 (D), and Grp78/BiP (E), were measured in Rho+/+, RhoP23H/+, and RhoP23H/P23H mice at P30 by quantitative PCR. (F–G) Upregulation of Erdj4 was measured in Rho+/+, RhoP23H/+, and RhoP23H/P23H mice at P60 (F) or P90 (G) by quantitative PCR. (H) Retinas from three Rho+/+, RhoP23H/+ or RhoP23H/P23H mice were collected at P15, pooled, and lysed. 40 μg of total retinal lysates were loaded onto the SDS-PAGE. Phosphorylated JNK protein levels were detected by immunoblotting. Total JNK protein levels served as a loading control.
Next, we examined the onset and duration of IRE1 signaling in RhoP23H/+ERAI+/− mice. We detected no expression of FLAG-tagged XBP1-Venus protein by anti-Flag or anti-GFP antibodies at P5 (Figure 5N). By P14, we saw faint expression of FLAG-tagged XBP1-Venus protein by anti-GFP immunoblotting, indicating that the IRE1 pathway was becoming activated in these retinas. The XBP1-Venus protein levels detected by both anti-GFP and anti-Flag progressively increased by P30, and these levels along with mRNA levels of downstream transcriptional targets of XBP1s, such as ERdj4, remained strongly elevated at older ages (P60 and P90) (Figure 6F, 6G). These findings demonstrated that photoreceptors expressing P23H rhodopsin activated the IRE1 signal transduction pathway by ~P14, and IRE1 signaling remained strongly activated thereafter. Interestingly, the onset of IRE1 signaling closely mirrored the onset of photoreceptor degeneration seen in RhoP23H/+ mice (Figure 2B). Furthermore, IRE1 signaling remained strongly activated in photoreceptors even after retinal degeneration had plateaued in heterozygous RhoP23H/+ mice (Figure 4A). IRE1 signaling can also activate the JNK pathway independent of its induction of XBP1s in vitro [50]. However, we saw no increases in activated phosphorylated JNK levels in P23H animals compared to control (Figure 6H). We subsequently focused our studies on the consequences of IRE1’s induction of XBP1s in the retina.
ERAD induction leads to ubiquitination and rapid degradation of P23H rhodopsin in vivo
Previously, we found that artificial activation of IRE1 signaling to selectively induce XBP1s targeted P23H rhodopsin for protein degradation in vitro [25]. The genes we found transcriptionally upregulated in retinas expressing P23H rhodopsin all play well-defined roles in ERAD (p97/VCP and Derlin1) and/or ER protein folding (ERdj4 and Grp78/BiP) (Figure 6B, 6C, 6D, 6E, and [51]). To determine if ERAD components and ER chaperones targeted P23H rhodopsin in photoreceptors, we performed mass spectrometry analysis on pure P23H rhodopsin immunoprecipitated from retinas of homozygous RhoP23H/P23H mice and wild-type rhodopsin immunoprecipitated from Rho+/+ mice. We consistently detected increased interaction of P23H rhodopsin with ER chaperones and ERAD components in triplicate experimental mass spectrometry analyses from retinal lysates compared to analogous studies with wild-type rhodopsin (Figure 7A). We also confirmed some of the P23H rhodopsin interactions identified by mass spectrometry in retinal co-immunoprecipitation studies where we found increased levels of p97/VCP and calnexin after rhodopsin pull-down in photoreceptors from RhoP23H/P23H mice compared to wild-type mice (Figure 7B). The transcriptional induction of ERAD genes and increased association between ERAD proteins and P23H rhodopsin protein suggested that photoreceptors of RhoP23H/P23H mice target misfolded rhodopsin for ERAD. Consistent with this notion, we found a substantial increase in the amount of ubiquitination on the purified P23H rhodopsin (Figure 7C). In heterozygous RhoP23H/+ mice that express both WT and mutant P23H rhodopsin protein, we found significant increase in the amount of ubiquitinated rhodopsin protein after immunoprecipitation with an antibody that recognizes both WT and P23H rhodopsin proteins (Figure 7C, lower panel). In homozygous RhoP23H/P23H that express only mutant P23H rhodopsin protein, we found a massive amount of ubiquitination on the immunoprecipitated P23H rhodopsin protein (Figure 7C, lower panel). These data demonstrate that induction of ERAD by IRE1 signaling leads to profound ubiquitination of P23H rhodopsin in photoreceptors, indicating P23H rhodopsin is targeted in vivo for degradation. Indeed, when we examined steady-state levels of rhodopsin from wild-type, heterozygous RhoP23H/+, and homozygous RhoP23H/P23H retinas, we found substantial loss of total rhodopsin protein in P23H rhodopsin-expressing mice with almost complete loss of rhodopsin protein from photoreceptors in homozygous RhoP23H/P23H mice (Figure 8A), despite equivalent levels of rod opsin mRNA gene expression in wild-type and P23H rhodopsin expressing mice at P14 (Figure 8B). Immunofluorescence analysis confirmed the pronounced loss of rhodopsin protein in homozygous RhoP23H/P23H photoreceptor cells (Figure 8C). In stark contrast to the pronounced retention of P23H rhodopsin in the ER when P23H rhodopsin is expressed in heterologous cell types, we detected no ER-retained P23H rhodopsin in native photoreceptors under non-saturating imaging conditions (Figure 8C, note the absence of rhodopsin staining of the IS and perinuclear regions of the ONL as exhibited by calnexin expression). However, under these imaging conditions, P23H rhodopsin expression in RhoP23H/P23H photoreceptors was found outside of the ER at the base of the stunted OS (Figure 8C), corresponding to ciliary protrusion and abnormal discs where P23H predominantly localized as we reported previously by immuno-electron microscopy [44].
Figure 7.

P23H rhodopsin protein co-immunoprecipitates with ERAD components and is strongly ubiquitinated in photoreceptor cells. (A) ERAD-related proteins that co-immunoprecipitated with P23H rhodopsin were identified by mass spectrometry. Total spectra counts of the proteins co-immunoprecipitated with WT or P23H rhodopsin from three experimental mass spectrometry analyses are shown. Immunoprecipitation of MEF cell samples served as experimental control. (B) Rhodopsin was immunoprecipitated from retinal lysates of Rho+/+ and RhoP23H/P23H mice at P15. The elutions were concentrated by chloroform/methanol precipitation. Rhodopsin, VCP, and calnexin proteins were detected by immunoblotting. (C) Rhodopsin was immunoprecipitated from retinas of Rho+/+, RhoP23H/+ and RhoP23H/P23H mice at P15, and rhodopsin (top panel) and ubiquitin (bottom panel) levels were detected by immunoblotting.
Figure 8.

Rhodopsin protein, but not mRNA, levels are significantly diminished in P23H mice. (A) Retinal protein lysates were collected from Rho+/+, RhoP23H/+, and RhoP23H/P23H mice at P15. Rhodopsin was detected by immunoblotting. GAPDH protein levels served as a loading control (B) Rhodopsin mRNA levels from Rho+/+, RhoP23H/+, and RhoP23H/P23H mice at P12 were measured by quantitative PCR. Filled circles represent rhodopsin mRNA levels in individual mouse. Horizontal red bars represent mean rhodopsin mRNA levels from each experimental cohort of mice. NS: statistically not significant. (C) Retinas from Rho+/+ and RhoP23H/P23H mice at P10 were sectioned and rhodopsin (red) and calnexin (green) were visualized by confocal microscopy. DAPI counterstain highlighted nuclei. Under equivalent confocal microscopy detection settings, strong rhodopsin protein staining was evident in the developing OS of Rho+/+ but only a scant amount of rhodopsin protein staining was visualized in the presumptive OS layer in RhoP23H/P23H photoreceptors. Rhodopsin protein did not colocalize with calnexin in either Rho+/+ or RhoP23H/P23H photoreceptors. (D) Retinal protein lysates were collected from Rho+/+, RhoP23H/+, and RhoP23H/P23H mice at P5. Rhodopsin and GNAT1 were detected by immunoblotting. HSP90 protein levels served as a loading control. (E) P23H rhodopsin was expressed in HEK293 cells. The cells were treated with 10 μM kifunensine for 20 hours. Cells were also treated with bortezomib 10 μM as a control for proteasomal-mediated degradation (F) HEK293 cells expressing P23H rhodopsin were treated with 1.25, 2.5, 3.75, 5, or 7.5 μM of eeyarestatin I for 20 hours (Lane 2 to 6). (E and F) Rhodopsin protein was detected by immunoblotting. GAPDH protein levels served as a loading control. Significant cell toxicity was observed with some drug treatment conditions (indicated with *).
Loss of rhodopsin protein in RhoP23H/P23H photoreceptors was apparent as early as P5 (Figure 8D). Loss of P23H rhodopsin protein was not due to photoreceptor cell death, as we observed no frank degeneration of photoreceptor compartment morphology, pyknotic nuclei, or any ONL thinning at this early age (Figure 1 and Table S1). Furthermore, we found normal protein levels of rod photoreceptor-specific transducin α-subunit (GNAT1) at P5[52], indicating that photoreceptors still generated other rod specific proteins (Figure 8D). We conclude that loss of rhodopsin protein occurs shortly after photoreceptors are formed. Furthermore, the loss of rhodopsin protein precedes the onset of morphologic defects arising in photoreceptor compartments, obvious photoreceptor cell death, and retinal degeneration. ERAD was directly involved in rhodopsin protein degradation, as we prevented the degradation of P23H rhodopsin upon application of ERAD inhibitors, kifunensine, an inhibitor of ER mannosidase I, [53,54] or eeyarastatin I, an inhibitor of p97/VCP ATPase activity, [55,56], in vitro (Figure 8E, 8F).
Chronic ER stress-induced proapoptotic Chop does not contribute to P23H rhodopsin-induced retinal degeneration
Uncorrectable ER stress ultimately triggers cell death, most notably by activating the PERK signaling pathway of the UPR which phosphorylates eIF2α leading to strong induction of ATF4 transcriptional activator and its downstream proapoptotic Chop gene [17,1]. ER stress-inducing toxins, tunicamycin and thapsigargin, strongly induced Chop, and overexpression of P23H rhodopsin in heterologous cell types or atop native wild-type rhodopsin in transgenic mice also upregulated Chop [26,57]. Furthermore, Chop−/− mice were resistant to cell death and damage induced by ER protein misfolding [58–60]. These findings support a model whereby P23H rhodopsin expression in photoreceptors leads to strong Chop induction that drives photoreceptor cell death in vivo. However, our analysis of RhoP23H/P23H retinas revealed low steady-state levels of P23H rhodopsin in photoreceptors (Figure 8A, 8D) and no obvious aggregation or retention of P23H rhodopsin within the ER (the RIS/perinuclear compartment) of photoreceptors (Figure 8C). Furthermore, ER dilation and fragmentation, ultrastructural abnormalities seen with irreparable ER stress, were not observed in the RIS of RhoP23H/P23H photoreceptors, (Figure 3C). These findings raise an alternative scenario whereby the robust degradation of P23H rhodopsin by induction of ERAD in photoreceptors effectively eliminated misfolded rhodopsin from the ER. In this model, ER stress is alleviated, and therefore, Chop would not be induced nor causally contribute to photoreceptor cell death.
To address these competing models of Chop’s role in the photoreceptor cell death seen in P23H rhodopsin mice, we measured Chop mRNA levels at time points spanning the entire period of retinal degeneration in heterozygote RhoP23H/+ and homozygous RhoP23H/P23H mice. In both RhoP23H/+ and RhoP23H/P23H mice, we saw no significant increase in Chop mRNA levels compared to age-matched wild-type mice at all ages (Figure 9A, 9B). We also found no increase in ATF4 protein in either RhoP23H/+ or RhoP23H/P23H mice and only mild elevation in phosphorylated eIF2α protein levels (Figure 9C). By contrast, all of these markers were strongly induced by a strong ER toxin, thapsigargin (Tg), in vitro (Figure 9C). These findings indicate that PERK signaling is weakly activated in vivo in photoreceptors expressing P23H rhodopsin, and PERK signaling does not rise to a threshold to trigger its downstream proapoptotic ATF4-CHOP circuit. Consistent with this interpretation, when we examined retinal degeneration in RhoP23H/+Chop−/− mice, we found no significant change in ONL thickness, a proxy for photoreceptor cell numbers, compared to RhoP23H/+Chop+/+ mice (Figure 9D). We conclude that photoreceptor cell death induced by P23H rhodopsin in vivo occurs independently of CHOP.
Figure 9.

Chop is not induced in retinas of P23H rhodopsin mice, and loss of Chop does not affect P23H-induced retinal degeneration. (A–B) Chop mRNA levels were quantified in the retinas of RhoP23H/+ and RhoP23H/P23H mice and are shown relative to levels in age-matched wild-type mice at indicated ages. (C) PERK downstream targets, phospho-eIF2α and ATF4, were analyzed by immunoblotting. Retinas from three Rho+/+, RhoP23H/+ or RhoP23H/P23H mice were collected at P15, pooled, and lysed. MEF cells were treated with thapsigargin (500 nM) for indicated times. Then, 10 μg of MEF total lysates or 40 μg of total retinal lysates were loaded onto the SDS-PAGE. HSP90 protein levels served as a loading control. (D) Mean ONL thicknesses of wild-type Rho+/+Chop+/+, RhoP23H/+Chop+/+, Rho+/+Chop−/−, RhoP23H/+Chop+/−, and RhoP23H/+Chop−/− from perfused enucleations were quantified at P95. Error bars represent SDs based on measurements from at least three mice of each genotype. For RhoP23H/+Chop−/−, only two mice were analyzed.
DISCUSSION
Our studies reveal an unexpected asymmetry in the use of the UPR by photoreceptor cells expressing P23H rhodopsin where we find strong IRE1 signaling but minimal PERK signaling. How does P23H rhodopsin trigger IRE1 so well with lesser effect on PERK? Direct interaction between IRE1’s ER luminal domain and misfolded protein is an important step in activation of IRE1 [61]. One possibility could be that the misfolded P23H rhodopsin protein preferentially interacts with IRE1 but not PERK in the ER lumen of photoreceptors, and therefore the IRE1 pathway is more activated. This possibility appears unlikely given the strong homology between the luminal domains of IRE1 and PERK [62]. A second possibility is that PERK is actively repressed in photoreceptors such as by inhibition of its cytosolic kinase by binding of p58IPK or Nck proteins [63,64]. A third possibility is that photoreceptors eliminate misfolded P23H rhodopsin so efficiently that the ambient levels of ER stress never rise to the threshold needed to trigger strong PERK signaling and its induction of the ATF4-CHOP proapoptotic circuit. The ER of photoreceptors is highly specialized toward the production of rhodopsin, because rhodopsin is expressed in massive quantities only by rod photoreceptors [65,43]. To cope with continuous rhodopsin protein production throughout life, the ER of photoreceptor cells must be exquisitely optimized toward folding rhodopsin as well as recognizing and removing any damaged/misfolded rhodopsin. In support of this model, we observed no obvious aggregates or retention of misfolded rhodopsin in the ER of photoreceptors and no ultrastructural alterations in ER morphology in P23H rhodopsin photoreceptors. By contrast, mutant rhodopsin aggregates are consistently found in the ER of heterologous cell types possibly because the ER of these cells is not as well equipped to process rhodopsin as the ER of native photoreceptor cells.
Heterozygous RhoP23H/+ mice closely model the genetic dosage and slow retinal degeneration presentation seen in patients with the P23H mutation, in whom decades typically pass before vision loss becomes detrimental [27]. In RhoP23H/+ mice, IRE1 and ERAD are also activated in photoreceptors as in RhoP23H/P23H mice. However, after an early wave of photoreceptor cell death during the first postnatal month after birth, photoreceptor numbers stabilize at ~50% the number of photoreceptors found in wild-type mice (Figures 2, 4A). Our analysis of RhoP23H/+ERAI+/− mice (Figure 5) revealed coincident peak induction of the IRE1 reporter during this period as well. These findings suggest that IRE1 signaling is perpetually activated in RhoP23H/+ photoreceptor cells throughout life and may maintain a fine proteostatic balance within the cell whereby P23H rhodopsin (synthesized from the mutant allele) is removed, whereas wild-type rhodopsin (produced by the remaining normal rhodopsin allele) is properly folded, exported to the ROS, and vision is retained. In this situation, RhoP23H/+ mimics the rhodopsin haplo-insufficiency seen in Rho+/− mice. Mild ROS and RIS photoreceptor compartment disorganization is seen in Rho+/− mice [21]. However, rhodopsin haplo-insufficiency does not cause photoreceptor cell death or vision loss in Rho+/− mice or people [21,20,66,67]. So, why are heterozygous RhoP23H/+ photoreceptors prone to cell death, albeit it at a slow protracted pace?
One possibility is that the small amount of P23H rhodopsin that exits the ER and escapes ERAD exerts a cytotoxic effect in photoreceptor cells, not seen in cell lines. In photoreceptors, this residual P23H rhodopsin concentrates in misshapen discs within stunted rod outer segment in RhoP23H/P23 mice whereas wild-type rhodopsin is found in normally stacked discs in the rod outer segment in Rho+/+ mice [44]. It is unclear if subcellular mislocalization of P23H rhodopsin is causally linked to the disorganization of discs or dysgenesis of the ROS compartment. However, disruption of rod outer segment structure or function would be deleterious for photoreceptor viability. Another factor affecting why heterozygous RhoP23H/+ photoreceptors are more prone to death as they age is that exogenous insults that trigger ER stress in the retina could exacerbate the ER stress already induced by P23H rhodopsin. Numerous environmental factors including phototoxicity, nutrient imbalances, infections, inflammation, and toxins (e.g., tobacco) have been shown to trigger ER stress in the eye [68–71]. Addition of environmentally induced ER stressors on top of P23H rhodopsin-induced ER stress could upset the proteostatic balance maintained by the UPR signaling network leading to photoreceptor cell death.
Our analysis of misfolded rhodopsin reveals how its preferential activation of different programs and modules of the UPR, in particular ERAD and IRE1, may drive the pathophysiology of photoreceptor cell death and retinal degeneration arising from misfolded rhodopsin-induced ER stress. Other neurodegenerative diseases associated with misfolded proteins also cause ER stress, but the UPR signaling pathways and transcriptional, translational, or post-translational UPR programs most relevant to each disease may also vary depending on the specific misfolded protein, morphologies, and functions found in the targeted neuronal cell types.
Supplementary Material
Acknowledgments
We thank J. Han, N. Hiramatsu, C. Sigurdson, W. C. Smith, S. Tsang, and L. Wiseman for helpful suggestions and reagents. These studies were supported by NIH grants EY001919, P30EY002162, and EY020846 and VA Merit award BX002284. W.-C. Chiang received postdoctoral support from the Fight-for-Sight Foundation.
Abbreviations used in this paper
- ERAD
ER-associated protein degradation
- IRE1
inositol-requiring enzyme 1
- ONL
outer nuclear layer
- PERK
PKR-like endoplasmic reticulum kinase
- qPCR
quantitative real-time PCR
- RP
retinitis pigmentosa
- RIS
rod inner segment
- ROS
rod outer segment
- UPR
unfolded protein response
- XBP-1
X-box-binding protein 1
Footnotes
The authors declare no conflict of interest.
References
- 1.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334 (6059):1081–1086. doi: 10.1126/science.1209038. 334/6059/1081 [pii] [DOI] [PubMed] [Google Scholar]
- 2.Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334 (6059):1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonca N, Liu X, Locasale JW, Simpson KW, Duhamel GE, Kersten S, Yates JR, 3rd, Long Q, Qi L. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A. 2014;111 (5):E582–591. doi: 10.1073/pnas.1318114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fritz JM, Dong M, Apsley KS, Martin EP, Na CL, Sitaraman S, Weaver TE. Deficiency of the BiP cochaperone ERdj4 causes constitutive endoplasmic reticulum stress and metabolic defects. Mol Biol Cell. 2014;25 (4):431–440. doi: 10.1091/mbc.E13-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26 (15):5688–5697. doi: 10.1128/MCB.00779-06. 26/15/5688 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eura Y, Yanamoto H, Arai Y, Okuda T, Miyata T, Kokame K. Derlin-1 deficiency is embryonic lethal, Derlin-3 deficiency appears normal, and Herp deficiency is intolerant to glucose load and ischemia in mice. PLoS ONE. 2012;7 (3):e34298. doi: 10.1371/journal.pone.0034298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller JM, Deinhardt K, Rosewell I, Warren G, Shima DT. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Biophys Res Commun. 2007;354 (2):459–465. doi: 10.1016/j.bbrc.2006.12.206. [DOI] [PubMed] [Google Scholar]
- 8.Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87 (3):391–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 9.Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90 (6):1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 10.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol Cell. 2009;35 (5):551–561. doi: 10.1016/j.molcel.2009.08.021. S1097-2765(09)00601-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415 (6867):92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107 (7):881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 13.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23 (21):7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, 3rd, Su AI, Kelly JW, Wiseman RL. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell reports. 2013;3 (4):1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397 (6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 16.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13 (3):184–190. doi: 10.1038/ncb0311-184. ncb0311-184 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15 (5):481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rattner A, Sun H, Nathans J. Molecular genetics of human retinal disease. Annual review of genetics. 1999;33:89–131. doi: 10.1146/annurev.genet.33.1.89. [DOI] [PubMed] [Google Scholar]
- 19.Palczewski K. G protein-coupled receptor rhodopsin. Annual review of biochemistry. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolo M, Makino CL, Sidman RL. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A. 1999;96 (2):736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, Sieving PA, Sheils DM, McNally N, Creighton P, Erven A, Boros A, Gulya K, Capecchi MR, Humphries P. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15 (2):216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- 22.Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;277 (37):34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- 23.Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88 (19):8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33 (20):6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- 25.Chiang WC, Messah C, Lin JH. IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol Biol Cell. 2012;23 (5):758–770. doi: 10.1091/mbc.E11-08-0663. mbc.E11-08-0663 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, LaVail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318 (5852):944–949. doi: 10.1126/science.1146361. 318/5852/944 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakami S, Maeda T, Bereta G, Okano K, Golczak M, Sumaroka A, Roman AJ, Cideciyan AV, Jacobson SG, Palczewski K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem. 2011;286 (12):10551–10567. doi: 10.1074/jbc.M110.209759. M110.209759 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olsson JE, Gordon JW, Pawlyk BS, Roof D, Hayes A, Molday RS, Mukai S, Cowley GS, Berson EL, Dryja TP. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992;9 (5):815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- 29.LaVail MM, Battelle BA. Influence of eye pigmentation and light deprivation on inherited retinal dystrophy in the rat. Exp Eye Res. 1975;21 (2):167–192. doi: 10.1016/0014-4835(75)90080-9. [DOI] [PubMed] [Google Scholar]
- 30.Winkler BS. The electroretinogram of the isolated rat retina. Vision Res. 1972;12 (6):1183–1198. doi: 10.1016/0042-6989(72)90106-x. [DOI] [PubMed] [Google Scholar]
- 31.Michon JJ, Li ZL, Shioura N, Anderson RJ, Tso MO. A comparative study of methods of photoreceptor morphometry. Invest Ophthalmol Vis Sci. 1991;32 (2):280–284. [PubMed] [Google Scholar]
- 32.LaVail MM, Gorrin GM, Repaci MA, Thomas LA, Ginsberg HM. Genetic regulation of light damage to photoreceptors. Invest Ophthalmol Vis Sci. 1987;28:1043–1048. [PubMed] [Google Scholar]
- 33.Faktorovich EG, Steinberg RH, Yasumura D, Matthes MT, LaVail MM. Basic fibroblast growth factor and local injury protect photoreceptors from light damage in the rat. J Neurosci. 1992;12 (9):3554–3567. doi: 10.1523/JNEUROSCI.12-09-03554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spira A, Hudy S, Hannah R. Ectopic photoreceptor cells and cell death in the developing rat retina. Anatomy and embryology. 1984;169 (3):293–301. doi: 10.1007/BF00315634. [DOI] [PubMed] [Google Scholar]
- 35.Hao W, Wenzel A, Obin MS, Chen CK, Brill E, Krasnoperova NV, Eversole-Cire P, Kleyner Y, Taylor A, Simon MI, Grimm C, Reme CE, Lem J. Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet. 2002;32(2):254–260. doi: 10.1038/ng984ng984. [pii] [DOI] [PubMed] [Google Scholar]
- 36.LaVail MM, Sidman RL. C57BL-6J mice with inherited retinal degeneration. Arch Ophthalmol. 1974;91 (5):394–400. doi: 10.1001/archopht.1974.03900060406015. [DOI] [PubMed] [Google Scholar]
- 37.McDonald WH, Tabb DL, Sadygov RG, MacCoss MJ, Venable J, Graumann J, Johnson JR, Cociorva D, Yates JR., 3rd MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid communications in mass spectrometry: RCM. 2004;18 (18):2162–2168. doi: 10.1002/rcm.1603. [DOI] [PubMed] [Google Scholar]
- 38.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. Journal of proteome research. 2003;2 (1):43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 39.Venable JD, Xu T, Cociorva D, Yates JR., 3rd Cross-correlation algorithm for calculation of peptide molecular weight from tandem mass spectra. Analytical chemistry. 2006;78 (6):1921–1929. doi: 10.1021/ac051636h. [DOI] [PubMed] [Google Scholar]
- 40.Caley DW, Johnson C, Liebelt RA. The postnatal development of the retina in the normal and rodless CBA mouse: a light and electron microscopic study. Am J Anat. 1972;133 (2):179–212. doi: 10.1002/aja.1001330205. [DOI] [PubMed] [Google Scholar]
- 41.Sanyal S, Bal A. Comparative light and electron microscopic study of retinal histogenesis in normal and rd mutant mice. Z Anat Entwickl Gesch. 1973;142 (2):219–238. doi: 10.1007/bf00519723. [DOI] [PubMed] [Google Scholar]
- 42.LaVail MM. KINETICS OF ROD OUTER SEGMENT RENEWAL IN THE DEVELOPING MOUSE RETINA. The Journal of Cell Biology. 1973;58 (3):650–661. doi: 10.1083/jcb.58.3.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palczewski K. Chemistry and biology of vision. J Biol Chem. 2012;287 (3):1612–1619. doi: 10.1074/jbc.R111.301150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakami S, Kolesnikov AV, Kefalov VJ, Palczewski K. P23H opsin knock-in mice reveal a novel step in retinal rod disc morphogenesis. Hum Mol Genet. 2014;23 (7):1723–1741. doi: 10.1093/hmg/ddt561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwawaki T, Akai R, Kohno K, Miura M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med. 2004;10(1):98–102. doi: 10.1038/nm970. nm970 [pii] [DOI] [PubMed] [Google Scholar]
- 46.Shimazawa M, Inokuchi Y, Ito Y, Murata H, Aihara M, Miura M, Araie M, Hara H. Involvement of ER stress in retinal cell death. Mol Vis. 2007;13:578–587. v13/a62 [pii] [PMC free article] [PubMed] [Google Scholar]
- 47.Kunte MM, Choudhury S, Manheim JF, Shinde VM, Miura M, Chiodo VA, Hauswirth WW, Gorbatyuk OS, Gorbatyuk MS. ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2012;53 (7):3792–3800. doi: 10.1167/iovs.11-9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21 (1):81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 49.Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27 (1):53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 50.Urano F. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 51.Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2012;14 (1):93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calvert PD, Krasnoperova NV, Lyubarsky AL, Isayama T, Nicoló M, Kosaras B, Wong G, Gannon KS, Margolskee RF, Sidman RL, Pugh EN, Makino CL, Lem J. Phototransduction in transgenic mice after targeted deletion of the rod transducin α-subunit. Proceedings of the National Academy of Sciences. 2000;97 (25):13913–13918. doi: 10.1073/pnas.250478897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elbein AD, Tropea JE, Mitchell M, Kaushal GP. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. Journal of Biological Chemistry. 1990;265 (26):15599–15605. [PubMed] [Google Scholar]
- 54.Tokunaga F, Brostrom C, Koide T, Arvan P. Endoplasmic Reticulum (ER)-associated Degradation of Misfolded N-Linked Glycoproteins Is Suppressed upon Inhibition of ER Mannosidase I. Journal of Biological Chemistry. 2000;275 (52):40757–40764. doi: 10.1074/jbc.M001073200. [DOI] [PubMed] [Google Scholar]
- 55.Wang Q, Li L, Ye Y. Inhibition of p97-dependent Protein Degradation by Eeyarestatin I. Journal of Biological Chemistry. 2008;283 (12):7445–7454. doi: 10.1074/jbc.M708347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fiebiger E, Hirsch C, Vyas JM, Gordon E, Ploegh HL, Tortorella D. Dissection of the Dislocation Pathway for Type I Membrane Proteins with a New Small Molecule Inhibitor, Eeyarestatin. Molecular Biology of the Cell. 2004;15 (4):1635–1646. doi: 10.1091/mbc.E03-07-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gorbatyuk MS, Knox T, LaVail MM, Gorbatyuk OS, Noorwez SM, Hauswirth WW, Lin JH, Muzyczka N, Lewin AS. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107 (13):5961–5966. doi: 10.1073/pnas.0911991107. 0911991107 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12 (7):982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109 (4):525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pennuto M, Tinelli E, Malaguti M, Del Carro U, D’Antonio M, Ron D, Quattrini A, Feltri ML, Wrabetz L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57 (3):393–405. doi: 10.1016/j.neuron.2007.12.021. S0896-6273(07)01034-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333 (6051):1891–1894. doi: 10.1126/science.1209126. science.1209126 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci U S A. 2006;103 (39):14343–14348. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, Katze MG. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc Natl Acad Sci U S A. 2002;99 (25):15920–15925. doi: 10.1073/pnas.252341799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamani L, Latreille M, Larose L. Interaction of Nck1 and PERK phosphorylated at Y561 negatively modulates PERK activity and PERK regulation of pancreatic β-cell proinsulin content. Molecular Biology of the Cell. 2014;25 (5):702–711. doi: 10.1091/mbc.E13-09-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sung CH, Chuang JZ. The cell biology of vision. J Cell Biol. 2010;190 (6):953–963. doi: 10.1083/jcb.201006020. jcb.201006020 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992;1 (3):209–213. doi: 10.1038/ng0692-209. [DOI] [PubMed] [Google Scholar]
- 67.Liang Y, Fotiadis D, Maeda T, Maeda A, Modzelewska A, Filipek S, Saperstein DA, Engel A, Palczewski K. Rhodopsin Signaling and Organization in Heterozygote Rhodopsin Knockout Mice. Journal of Biological Chemistry. 2004;279 (46):48189–48196. doi: 10.1074/jbc.M408362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kroeger H, Messah C, Ahern K, Gee J, Joseph V, Matthes MT, Yasumura D, Gorbatyuk MS, Chiang WC, Lavail MM, Lin JH. Induction of Endoplasmic Reticulum Stress Genes, BiP and Chop, in Genetic and Environmental Models of Retinal Degeneration. Invest Ophthalmol Vis Sci. 2012;53 (12):7590–7599. doi: 10.1167/iovs.12-10221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang LP, Wu LM, Guo XJ, Li Y, Tso MO. Endoplasmic reticulum stress is activated in light-induced retinal degeneration. J Neurosci Res. 2008;86 (4):910–919. doi: 10.1002/jnr.21535. [DOI] [PubMed] [Google Scholar]
- 70.Salminen A, Kauppinen A, Hyttinen JM, Toropainen E, Kaarniranta K. Endoplasmic reticulum stress in age-related macular degeneration: trigger for neovascularization. Mol Med. 2010;16 (11–12):535–542. doi: 10.2119/molmed.2010.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ambati J, Fowler BJ. Mechanisms of age-related macular degeneration. Neuron. 2012;75 (1):26–39. doi: 10.1016/j.neuron.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.