

Abstract
Loss of gamma-sarcoglycan (γ-SG) induces muscle degeneration and signaling defects in response to mechanical load, and its absence is common to both Duchenne and limb girdle muscular dystrophies. Growing evidence suggests that aberrant signaling contributes to the disease pathology; however, the mechanisms of γ-SG-mediated mechanical signaling are poorly understood. To uncover γ-SG signaling pathway components, we performed yeast two-hybrid screens and identified the muscle-specific protein archvillin as a γ-SG and dystrophin interacting protein. Archvillin protein and message levels were significantly upregulated at the sarcolemma of murine γ-SG-null (gsg−/−) muscle but delocalized in dystrophin-deficient mdx muscle. Similar elevation of archvillin protein was observed in human quadriceps muscle lacking γ-SG. Reintroduction of γ-SG in gsg−/− muscle by rAAV injection restored archvillin levels to that of control C57 muscle. In situ eccentric contraction of tibialis anterior (TA) muscles from C57 mice caused ERK1/2 phosphorylation, nuclear activation of P-ERK1/2 and stimulus-dependent archvillin association with P-ERK1/2. In contrast, TA muscles from gsg−/− and mdx mice exhibited heightened P-ERK1/2 and increased nuclear P-ERK1/2 localization following eccentric contractions, but the archvillin–P-ERK1/2 association was completely ablated. These results position archvillin as a mechanically sensitive component of the dystrophin complex and demonstrate that signaling defects caused by loss of γ-SG occur both at the sarcolemma and in the nucleus.
Introduction
Skeletal muscle mechanotransduction is mediated in part by the dystrophin glycoprotein complex (DGC), an assembly of proteins that also maintains the integrity of the sarcolemma by linking the cytoskeleton and the extracellular matrix (1,2). Mutations in genes encoding DGC components cause several forms of muscular dystrophy including limb girdle muscular dystrophies (LGMD) types 2C-F from mutation of α-, β-, γ- and δ-SG subunits of the sarcoglycan (SG) complex, as well as Duchenne muscular dystrophy (DMD) from mutation of dystrophin. Upon loss of dystrophin in DMD patients and in the mdx mouse model for the disease, there is a secondary loss of the entire DGC including the sarcoglycans. However, with primary mutations in any of the sarcoglycans, only the presence of the SG complex is compromised, with retention of dystrophin and other DGC components (3,4). This holds true for the LGMD2C γ-SG-null mouse model (gsg−/−), where there is secondary reduction or absence of the other three sarcoglycan subunits. Dystrophin loss in skeletal muscle causes compromised force-generating capacity and contraction-induced damage with sarcolemmal tearing and fiber degeneration (5,6), which are thought to be primary contributors to the progressive loss of muscle and its replacement with fat and fibrotic tissue (3). In contrast, there is a different progression to disease in LGMD2C, where skeletal muscles from gsg−/− mice display little contraction-induced damage early in life, yet still develop a similarly severe pathology (6,7), indicating that γ-SG deficiency alone is sufficient to induce several dystrophic symptoms. Based on this separation between mechanical fragility and dystrophic pathology, we and others have asserted that loss of the SG complex disrupts proper load sensing in muscle, which then leads to muscle disease.
Previous work in our lab demonstrated that sarcolemmal localization of γ-SG and phosphorylation of its tyrosine 6 residue is essential for normal extracellular signal-regulated kinase 1 and 2 (ERK1/2) signaling in muscle subjected to eccentric contraction (ECC) (8) and that loss of γ-SG also uncouples the response of the p70S6 kinase pathway to passive stretch (9). These results support that γ-SG is an important component of the normal signaling profile in response to mechanical perturbation. However, the mechanisms by which γ-SG mediates mechanical signal transduction are unknown. Because of the importance of γ-SG as a mechanosensor in the absence of any known endogenous enzymatic activity and the fact that the sequence of its intracellular domain is also critical for normal ERK1/2 signaling, we hypothesized that γ-SG must transmit load-induced signals to downstream pathways via the association with binding partners.
Based on this premise, we performed a yeast two-hybrid assay to identify potential binding partners for the intracellular domain of γ-SG, and an intriguing candidate, archvillin, emerged. Archvillin is a 250-kDa muscle-specific isoform of supervillin, a known actin- and myosin-II-binding protein (10,11). Although a series of yeast two-hybrid and proteomics analyses have identified interactors for non-muscle supervillin isoform 1 (NP_003165.2) (12–16), interaction partners have not been reported for the differentially spliced, muscle-specific insert of amino acids 276–669 in archvillin, which are encoded by coding exons 3, 4 and 5 (10). Thus, even though archvillin is abundantly expressed in cardiac and skeletal muscle (17), its role in striated muscle has not been identified. Co-localization at the sarcolemma with dystrophin in hamster skeletal muscle (10) suggests that archvillin may serve some function in the DGC, but this has not been confirmed. A related isoform, smooth muscle archvillin (SmAV), has been shown to be an ERK scaffolding protein that associates with ERK in a stimulus-dependent manner in aortic tissue stimulated with the alpha agonist phenylephrine (18,19). However, ERK association with the striated muscle archvillin has not been demonstrated to date. Given the known sarcolemmal localization of archvillin combined with its identification in yeast two-hybrid screens with γ-SG and dystrophin, as well as the ability of the smooth muscle isoform to associate with ERK, the goal of this study was to determine whether archvillin is part of the mechanical signaling machinery in skeletal muscle.
Results
Archvillin interacts with γ-SG and dystrophin
To identify binding partners of γ-SG, we performed a yeast two-hybrid assay in which we cloned the intracellular domain of human γ-SG into a pGBKT7 bait plasmid and screened it against a normalized human library. Prey fragments of bait/prey positive interaction colonies were PCR-amplified and identified using the BLAST algorithm and included archvillin, the β-sarcoglycan subunit of the sarcoglycan complex, as well as vesicle trafficking proteins (Table 1 and Supplementary Material, Table S1). We identified two identical prey clones encoding the extreme C-terminus of archvillin (aa 2093–2214) (Fig. 1A), and this fragment was re-transformed into the prey yeast strain for confirmation of the interaction with γ-SG by two methods (Fig. 1B and C). First, we performed a binding assay in which yeast containing either the γ-SG/pGBKT7 bait plasmid or empty pGBKT7 plasmid were re-mated with yeast containing the archvillin prey sequence identified in the initial yeast two-hybrid screen, assuring that the mated yeast contained both bait and prey plasmids. Mated yeast were plated on selective dropout media plates containing x-α-gal, where positive interaction would activate β-galactosidase expression and yield blue colonies. Yeast mated containing the γ-SG/pGBKT7 plasmid yielded blue colonies, whereas yeast mated containing the negative control empty pGBKT7 plasmid did not grow any colonies on the most stringent quadruple dropout plates (Fig. 1B). Second, co-immunoprecipitation (co-IP) with anti-γ-SG was performed on skeletal muscle extracts from wild-type (C57) and gsg−/− mice, followed by immunoblotting with anti-archvillin or anti-γ-SG. In samples from C57 mice, bands for both γ-SG and archvillin were observed following co-IP (Fig. 1C, lane 3), whereas co-IP could not pull down archvillin in samples lacking γ-SG even though there was abundant archvillin in the lysate (Fig. 1C, lane 2, 4). Biochemical confirmation of the archvillin interaction with γ-SG was attempted, but the experiments were unsuccessful owing to insolubility of the tagged γ-SG cytoplasmic domain (data not shown). Thus, the interaction between the intracellular portion of γ-SG and the C-terminus of archvillin identified by the yeast two-hybrid screen was sufficient to afford physical association between these proteins in skeletal muscle, but whether this was a direct interaction between the domains of these proteins or an indirect association via intermediate proteins is unknown.
Table 1.
Candidate binding partners for human γ-SG intracellular domain aa 1–35 and hAV muscle-specific insert 1 aa 277–669.
| Bait | Interactors | Accession number |
Number of clones |
||
|---|---|---|---|---|---|
| Protein | Nucleotide | Total | Individual | ||
| γ-SG aa 1–35 | Archvillin (SVIL)a | NP_003165.2 | NM_003174.3 | 2 | 1 |
| Coatomer subunit delta isoform 2 (COPD2) | NP_001135753.1 | NM_001142281.1 | 2 | 1 | |
| Beta-sarcoglycan (SGCB) | NP_000223.1 | NM_000232.3 | 1 | 1 | |
| PDZ and LIM domain protein 2 isoform 2 (PDLIM2) | NP_067643.3 | NM_021630.5 | 1 | 1 | |
| hAV aa 227–669b | Nexilin (NEXN) | NP_001165780 | NM_001172309.1 | 15 | 6 |
| Myosin binding protein C (MYBPC1) | NP_996555 | NM_206819 | 8 | 6 | |
| Dystrophin (DMD) | NP_004001 | NM_004010 | 8 | 5c | |
| Utrophin (UTRN) | NP_009055 | NM_007124 | 2 | 2 | |
| Basic helix-loop-helix family member e40 (BHLHE40) | NP_003661 | NM_003670 | 2 | 1 | |
| Lamin A/C (LMNA) | NP_733822 | NM_170708 | 2 | 1 | |
| A kinase anchor protein 9 (AKAP9) | NP_005742 | NM_005751 | 1 | 1 | |
| Acyl-CoA binding domain containing 3 (ACBD3) | NP_476507 | NM_022735 | 1 | 1 | |
| Collagen, type VI, alpha 3 (COL6A3) | NP_001120959 | NM_057166 | 1 | 1 | |
| Filamin C, gamma (FLNC) | NP_006603 | NM_001127487 | 1 | 1 | |
| Kelch-like family member 7 (KLHL7) | NP_061334 | NM_018846 | 1 | 1 | |
| Kinesin family member 1C (KIF1C) | NP_073572 | NM_006612 | 1 | 1 | |
| Protein inhibitor of activated STAT, 1 (PIAS1) | NP_057250 | NM_016166 | 1 | 1 | |
| Trio Rho guanine nucleotide exchange factor (TRIO) | NP_009049 | NM_007118 | 1 | 1 | |
Positive interactors were identified in two yeast two-hybrid screens. Names, protein and nucleic acid accession numbers and the numbers of total and unique clones for each interactor are shown.
aArchvillin was confirmed as a γ-SG aa 1–35 interactor by secondary assays.
bhAV interactors were confirmed by secondary assays. See Supplementary Material, Table S1 for complete list of candidate interactors from the γ-SG yeast two-hybrid screen.
cTwo of the identified dystrophin clones had incomplete sequencing results and were thus included in the count of total clones, but not in the count of individual, distinct preys.
Figure 1.

Archvillin binds γ-SG and dystrophin. (A) Schematic of interacting domains in human archvillin for dystrophin and γ-SG. The dystrophin preys all contain spectrin-like repeats 11–12; black bar indicates this predicted binding region. The clones of archvillin preys interacting with γ-SG were identical, encoding aa 2093–2214. Sequences of interest within human archvillin are indicated: blue-shading, muscle-specific differentially spliced sequences encoded by coding exons 3, 4, 5 [aa 277–669; yeast two-hybrid (Y2H) bait sequence] or coding exon 9 (aa 750–781); red bars, predicted docking motifs for binding to ERK1/2 or other MAP kinases; yellow bars, F-actin-binding regions; purple shading, nebulin-binding domain; green shading, Y2H prey sequence recovered with the γ-SG bait. Schematics of dystrophin and γ-SG showing the locations of actin-binding domains 1 and 2 (ABD1, ABD2, orange), numbered spectrin-like repeats 1–24, the cysteine-rich domain and the carboxy-terminus (COOH) of dystrophin, along with the transmembrane domain and extracellular domain of γ-SG. The locations of potential ERK-binding sites were predicted by The Eukaryotic Linear Motif resource for Functional Sites in Proteins using the default motif probability cutoff of 100 (http://elm.eu.org/, last accessed on 22 January 2015) (20). (B) Confirmation of γ-SG-archvillin interaction. Yeast containing either γ-SG/pGBKT7 or empty pGBKT7 plasmid were mated with yeast containing the archvillin prey sequence (AV/pGADT7) identified in the yeast two-hybrid screen. White colonies indicate the presence of both pGBKT7 and pGADT7 plasmids in diploid yeast. Blue colonies indicate β-galactosidase gene activation from positive bait/prey interaction on DDO and QDO plates. Empty pGBKT7 and AV/pGADT7 matings afforded colony growth on DDO plates, confirming that mated yeast contained both plasmids, but not on QDO plates (negative control). (C) Co-IP of archvillin and γ-SG. C57 and gsg−/− TA muscle lysates were immunoprecipitated with anti-γ-SG, and blots were probed with anti-archvillin and anti-γ-SG. Lanes 1 and 2: archvillin, γ-SG and tubulin in lysates (input). Archvillin co-precipitated with γ-SG in C57 muscle (lane 3), whereas no signal could be seen in gsg−/−negative control IP (lane 4) or in negative control IPs without primary antibody (lanes 5 and 6). (D) Archvillin-specific sequence encoded by coding exons 3, 4 and 5 binds to dystrophin spectrin repeats 10–12 in vitro. Immunoblots of recombinant His-tagged DMD spectrin repeats 10–12 co-sedimented with the indicated GST fusion proteins. Blots of bound and unbound fractions were probed with anti-His; Ponceau stain of bound fractions shows amounts of GST fusion proteins, as well as the bound His-DMD SR 10-12 (aa 1361–1686) in Lane 3. Lane 1: His-DMD-10-12 input; Lane 2: GST (∼26 kDa); Lane 3: GST-AV-345 (doublet at 74–76 kDa) and His-DMD-10-12 (∼45 kDa) and Lane 4: GST-AV-1-171 (∼48 kDa).
We also used yeast two-hybrid screening and co-sedimentation assays to show a direct interaction between archvillin and dystrophin (Table 1 and Fig. 1D). In a separate yeast two-hybrid screen, the bait was the differentially spliced first muscle-specific sequence (aa 277–669) of human archvillin. Thirty unique prey clones from 14 identified interactors were confirmed from this screen (Table 1). Five unique dystrophin clones were identified that spanned rod domains 8–13 (aa 1239–1724), as well as two unique clones spanning rod domains 9–13 of the dystrophin ortholog utrophin (aa 1284–1715), with rod domains 11–12 present in all dystrophin and utrophin clones (Fig. 1A). Other prominent prey fragments from the screens with the archvillin bait were from nexilin and myosin binding protein C, which also play a role in maintaining sarcomeric integrity (Table 1). We confirmed the archvillin–dystrophin interaction by co-sedimenting purified recombinant His-tagged dystrophin spectrin repeat domains 10–12 (His-DMD-10-12) with purified GST-tagged archvillin protein fragments. His-DMD-10-12 was pulled down with the muscle-specific archvillin sequences encoded by exons 3–5 (GST-AV-277-669), but did not co-sediment with either GST-AV-1-171 or GST alone (Fig. 1D). Thus, based on two independent yeast two-hybrid screens and subsequent co-IP/co-sedimentation experiments, the muscle-specific insert of archvillin binds to dystrophin, and its interaction with γ-SG impacts its association with the DGC. These interactions are schematized in Figure 1A.
Archvillin levels depend on DGC integrity
Dystrophic muscle presents with an upregulation of membrane-associated proteins involved in maintaining the cytoskeleton/ECM link and signaling; this includes integrins, which are thought to function as a compensatory mechanism (8,21,22). We examined the expression of archvillin in quadriceps, tibialis anterior (TA), extensor digitorum longus (EDL), soleus, diaphragm and heart muscles from C57, gsg−/−, δ-SG-null (dsg−/−) and mdx mice. Archvillin levels were consistently elevated in gsg−/− and dsg−/− muscles and comparable with C57 levels in mdx muscles (Fig. 2A). Western blot quantification and quantitative real-time PCR showed that archvillin protein and mRNA were ∼2.5-fold higher in gsg−/− quadriceps muscle compared with C57 muscle, but were not significantly different in mdx muscle (Fig. 2B and C). Archvillin localization was first observed in hamster skeletal muscle primarily along the sarcolemma, supporting the interaction with dystrophin (10). To determine whether archvillin localization was altered in dystrophic muscle, we immunostained for archvillin in EDL muscles from C57, gsg−/− and mdx mice. Similar to previous observations, archvillin was localized in patches along the sarcolemma in C57 muscle. In gsg−/− muscle, archvillin was enriched at the sarcolemma and outlined nearly the entire circumference of the muscle fibers. In mdx cross-sections, archvillin was virtually undetectable along the sarcolemma (Fig. 2D). Thus, localization of archvillin to the sarcolemma depends upon the presence of dystrophin, but does not require γ-SG. However, the compensatory increases of archvillin are apparent only in the absence of the SG complex.
Figure 2.

Archvillin expression is altered in dystrophic muscle. (A) Archvillin protein levels in quadriceps (Quad), TA, EDL, soleus (Sol), diaphragm (Dia) and heart (Hrt) muscle of 12- to 14-week-old C57/Bl6, gsg−/−, dsg−/− and mdx mice. Increased archvillin is apparent in all striated muscles from gsg−/− and dsg−/− mice. (B) Quantification of archvillin protein in quadriceps confirms 2.7- to 4-fold increase of archvillin in gsg−/− and dsg−/− mouse muscles. *P < 0.05; **P < 0.01 versus C57; †P < 0.05 versus mdx by one-way ANOVA with Bonferroni post hoc test, n = 3. (C) Quantitative RT–PCR for archvillin expression shows significant upregulation in gsg−/− quadriceps muscles, as compared with C57 and mdx muscles. Bars represent fold change; means ± SEM relative to C57. *P < 0.05; **P < 0.01 versus C57; †P < 0.05 versus mdx by one-way ANOVA with Bonferroni post hoc test; n = 3–5. (D) Immunostaining of EDL muscles shows that archvillin is localized primarily along the sarcolemma in control C57 muscle, where it is present with increased intensity when γ-SG is absent in gsg−/− muscle. Archvillin localization is disrupted by the absence of dystrophin in mdx muscle. Dotted line indicates γ-SG staining on an adjacent section. Bars: 25 μm. (E) Archvillin protein levels in quadriceps muscles from normal healthy control, LGMD2C, LGMD2E and DMD patient biopsies. Increased archvillin is apparent in biopsies with primary sarcoglycan deficiency, whereas DMD patients with primary dystrophin deficiency have reduced archvillin expression. All dystrophic biopsies demonstrate elevated P-ERK activation versus normal. Panels are from one membrane with one lane removed for clarity. (F) Quantification of archvillin protein in human quadriceps confirms ∼4-fold increase of archvillin in LGMD2C patients. Relative archvillin values represent archvillin/GAPDH relative to normal patients. *P < 0.05 versus normal; †P < 0.05 versus DMD by one-way ANOVA with Bonferroni post hoc test, n = 2–3; LGMD2E, n = 1 (not included in one-way ANOVA). See Supplementary Material, Table S2 for gene mutations and age at time of biopsy of human patient biopsies.
To determine whether archvillin levels were also altered in diseased human muscle, we examined quadriceps muscle biopsies from LGMD2C, LGMD2E and DMD patients with primary deficiency in γ-SG, β-SG and dystrophin, respectively. Immunoblotting showed increased archvillin in LGMD2C biopsies, similar to observations in gsg−/− muscle, whereas DMD muscle expressed slightly lower archvillin relative to normal biopsies (Fig. 2E and F). Therefore, in both mouse and human muscle samples, archvillin levels increased in the absence of γ-SG but were normal or reduced when dystrophin was missing. We have previously shown that reintroduction of γ-SG in gsg−/− muscle by recombinant adeno-associated virus (AAV) injection restores normal ERK1/2 signaling (8). To assess the dependence of archvillin on the presence of γ-SG, we injected 3-week-old gsg−/− mice with AAV expressing γ-SG and harvested tissue for analysis 1 month post-injection. AAV injection restored γ-SG expression to C57 levels in the injected limb and normalized archvillin expression levels (Fig. 3A and B). Thus, the level of archvillin in muscle appears inversely proportional to the presence of γ-SG.
Figure 3.

γ-SG restoration normalizes archvillin expression. (A) Immunoblots of TA muscles with (+) and without (−) injection of AAV expressing human γ-SG show that archvillin expression decreases with restoration of γ-SG expression (lane 6). (B) Western blot quantification shows archvillin levels significantly decrease in gsg−/− TA muscles with γ-SG AAV injection. Bars represent means ± SEM relative to C57 L. *P < 0.05; **P < 0.01 versus C57 corresponding limb; †P < 0.05 versus. gsg−/− R-; ‡P < 0.01 versus AAV gsg−/− L- by two-way ANOVA and Bonferroni post hoc test, n = 4–5.
Archvillin association with P-ERK is stimulus dependent
Mechanical perturbation evokes transient increases in P-ERK in skeletal muscle (23), and aberrant ERK signaling is common to many mouse models of muscular dystrophies (24–26). Increased basal P-ERK is not only a phenotype of diseased murine muscle, but in probing our biopsy samples from human patients, we found that P-ERK was also elevated in all of the LGMD and DMD samples compared with biopsies from healthy subjects (Fig. 2E). Thus, the identification of potential mediators of ERK signaling may provide insight into disease mechanisms. Interestingly, the smooth muscle archvillin isoform, which lacks the differentially spliced sequences encoded by SVIL exons 3, 5 and 9, has been shown to bind ERK in aortic tissue in a stimulus-dependent manner (18,19). Candidate ERK docking sites exist in archvillin (Fig. 1A), raising the potential for a similar response to occur for this protein.
To determine whether archvillin also associates with ERK, we subjected TA muscles to a series of ECCs in situ during which the muscle was simultaneously stimulated to contract and lengthened to induce maximal strain on the membrane. Measurements of maximal isometric tetanic force showed that gsg−/− and mdx muscle generated less force compared with C57 muscle (Fig. 4A). However, following ECC stimulation, the loss of force production in gsg−/− muscles was comparable with that in C57 muscles, whereas mdx muscles displayed a significant drop in force generation following ECCs (Fig. 4B). This is consistent with studies performed on isolated muscles from the gsg−/− mouse (7) and distinguishes the functional phenotypes of gsg−/− and mdx muscles.
Figure 4.

Archvillin associates with ERK1/2 in a stimulation- and sarcoglycan-dependent manner. (A) Dystrophic gsg−/− and mdx TA muscles generate less force than C57 muscles when subjected to in situ isometric mechanical stimulation. *P < 0.001 versus C57 by one-way ANOVA with Bonferroni post hoc test, n = 5–7. (B) Mice with primary γ-SG deficiency do not show compromised force-generating capacity following in situ ECC mechanical injury, whereas mdx mice show a significant increase in force drop. *P < 0.001 versus C57; †P < 0.01 versus. gsg−/− by one-way ANOVA with Bonferroni post hoc test, n = 5–7. (C) Immunoprecipitation assay of TA muscles with (+) and without (−) in situ ECC stimulation (Stim). Immunoprecipitation with total ERK1/2 (T-ERK1/2) antibody shows that archvillin associates with ERK1/2 in stimulated C57 muscle (lane 2), but this association is lost in gsg−/− and mdx muscle. Without stimulation, archvillin does not associate with ERK1/2 in any strain, indicating a stimulation-dependent, sarcoglycan-dependent interaction. Immunoblotting for γ-SG demonstrates that it does not associate tightly with ERK1/2. Tubulin serves as a loading control for the inputs. (D) Immunoprecipitation with phospho-specific anti-P-ERK1/2 shows that archvillin associates with P-ERK1/2 only in stimulated C57 muscle (lane 2). Results were consistent from three independent experiments.
Muscle lysates were immunoprecipitated with anti-ERK1/2 and probed for archvillin, ERK1/2 and γ-SG. Archvillin associated with ERK1/2 in stimulated TA muscles from C57 mice, but not in the non-stimulated contralateral control limb. However, this association was eliminated in stimulated TA muscles from gsg−/− or mdx mice, even though there was archvillin present in the lysates of all muscle samples (Fig. 4C). Interestingly, there was no direct interaction between ERK1/2 and γ-SG, supporting that ERK1/2 associated with archvillin, but only when γ-SG was present. We extended this evaluation to distinguish between archvillin association with phosphorylated or non-phosphorylated ERK1/2, employing the same procedures, but with an antibody specific to P-ERK1/2. The ECCs produced a robust increase in P-ERK1/2 in TAs from all three mouse lines. However, only stimulated C57 muscles displayed an association between P-ERK1/2 and archvillin, whereas stimulation of dystrophic muscles failed to cause this interaction (Fig. 4D). Taken together, P-ERK1/2 associates with archvillin in a stimulation- and γ-SG-dependent manner.
Nuclear P-ERK1/2 is elevated in dystrophic muscle
A central component of P-ERK1/2 activity is to phosphorylate transcription factors in the nucleus, which then positively regulates target genes (27). Because we have previously established aberrant ERK1/2 signaling in dystrophic muscles, we determined whether this also altered the distribution of P-ERK1/2 between cytosolic and nuclear fractions from C57, gsg−/− and mdx muscles. Fractioned lysates from muscles subjected to ECC and from non-stimulated control muscles were probed for P-ERK1/2. Eccentric contractions of TA muscles caused a dramatic increase in P-ERK1/2 that was most pronounced in the dystrophic muscles (Fig. 5). Not only was P-ERK1/2 elevated in the cytosolic fraction of muscles subjected to ECCs, there was enhanced localization of P-ERK1/2 in the nuclear fraction in both gsg−/− and mdx muscles, suggesting that the aberrant signaling observed in whole muscle lysates led to increased nuclear accumulation. Of note was the increase of lamin A/C in the nuclear extracts of the dystrophic muscles. We performed additional probes with a second nuclear marker (histone 3), which revealed that lamin A/C was elevated, as opposed to an increase in nuclear content in these tissues, suggesting some compensatory expression of lamin A/C associated with these dystrophies. In sum, the aberrant P-ERK1/2 signaling in gsg−/− and mdx muscle was associated with enhanced localization of this signaling molecule in nuclear fractions.
Figure 5.

Nuclear P-ERK1/2 is elevated in dystrophic muscle following stimulation. Immunoblotting of cytosolic and nuclear fractions of TA muscle with (+) and without (−) in situ ECC stimulation (Stim). Stimulation led to elevated P-ERK1/2 in both fractions, which was augmented in dystrophic muscles. Nuclear accumulation of P-ERK1/2 was most pronounced in gsg−/− and mdx muscles with stimulation. Lamin A/C was also elevated in nuclear fractions from dystrophic muscles. Tubulin and histone 3 were used as loading controls and as cytosolic and nuclear markers, respectively.
Discussion
Growing evidence suggests that signaling defects associated with the loss of DGC components contribute to the pathology of muscular dystrophy. In our previous work, γ-SG was shown to be a mediator of ERK1/2 signaling in response to mechanical perturbation in skeletal muscle (8). However, the proteins involved in γ-SG-mediated signaling are unknown, as the SG complex possesses no known enzymatic activity. In the present study, we have identified the muscle-specific protein archvillin as a γ-SG- and dystrophin-interacting protein through two independent screens and biochemical assays. These results position archvillin as an intriguing component of the mechanical signal transduction pathway associated with the DGC.
The proteins encoded by the Svil gene (supervillin, archvillin and smooth muscle archvillin) are involved in many cellular processes, predominantly through their association with the actin cytoskeleton. Archvillin has several associations with the sarcolemma and cytoskeleton including binding to F-actin and nonmuscle myosin (11), localization with dystrophin at costameres (10), as well as association with the C-terminus of nebulin (28). We show here that the N-terminal muscle-specific insert of archvillin binds to dystrophin and also may interact with the same domains of its ortholog utrophin. The dual interactions of archvillin with both dystrophin and γ-SG suggest a strong association with the DGC that appears to be independent of nebulin binding.
Given this association, we did not anticipate the alterations in archvillin levels and localization observed in the absence of γ-SG and/or dystrophin. Because archvillin interacts with nebulin and has multiple F-actin-binding domains, one would predict that its localization near the membrane would be preserved in the absence of members of the DGC. However, in the absence of dystrophin in mdx tissue, there was diminished concentration of archvillin at the sarcolemma, suggesting that dystrophin is required for archvillin to remain at the membrane. The fact that archvillin is de-localized, but not de-stabilized or degraded in mdx muscle, is likely aided by the many additional binding partners for archvillin in the cell.
More surprising to us was the increased expression and accumulation of archvillin at the sarcolemma in the absence of γ-SG. Loss of the SG complex also results in a compensatory increase in the integrin complex (22,29), and so one possibility is that increased archvillin levels are associated with integrin-associated proteins. While the related protein, supervillin, regulates integrin function, there is no known direct binding of either protein to integrin, though supervillin links to focal adhesions through binding to LIM domain proteins TRIP6 and LPP (15). Ultimately, we cannot discount that the increased archvillin in gsg−/− or in LGMD2C patients is correlated with integrin levels, but even so, the stimulation and mechanically dependent ERK binding to archvillin is completely eliminated in these muscles regardless of the 3-fold increase in the protein levels. This implicates the SG complex in mediating archvillin–ERK association in response to mechanical perturbation.
Smooth muscle archvillin, a highly homologous isoform of archvillin, has been implicated as an ERK scaffolding protein that binds ERK in a stimulus-dependent manner (18,19,30). Archvillin also contains predicted ERK docking motifs, two of which are located within the first muscle-specific insert (20), suggesting that it also has the potential to bind ERK and mediate its activation in skeletal muscle, where there is significant strain imposed on the sarcolemma. To investigate a potential archvillin–ERK1/2 interaction, we subjected muscles to an in situ ECC protocol, which elicits dramatic P-ERK1/2 activation. We show here that archvillin also associates with ERK1/2, specifically with P-ERK1/2, in C57 muscle upon ECC. However, this association is lost in gsg−/− and mdx muscle, indicating a γ-SG and dystrophin-dependent archvillin–P-ERK1/2 association. Complementary to this, we also observed increased nuclear P-ERK1/2 in stimulated gsg−/− and mdx muscles, presenting the possibility that loss of the archvillin–ERK1/2 association in dystrophic muscle may contribute to the elevated nuclear P-ERK1/2, and consequently, activation of downstream nuclear targets. Because there was also an increase in cytosolic P-ERK1/2 in stimulated dystrophic muscle, we cannot distinguish between a model where there is a proportional increase in nuclear translocation of P-ERK1/2, and the loss of the P-ERK1/2 interaction is simply a parallel occurrence, or if indeed the loss of the archvillin–ERK1/2 association is contributing to aberrant P-ERK1/2 signaling. Ideally, ablation of archvillin would help to evaluate the contribution of archvillin to P-ERK1/2 signaling and to the development of muscle disease, but attempts at deleting the Svil gene have been unsuccessful. Targeting the muscle-specific insert may preserve the vital functions of supervillin in the rest of the body and afford examination of archvillin-specific properties.
The potential for archvillin to play a role in mechanical signal transduction is high given the known functions of this protein family. In addition to the ERK docking properties of SmAV, non-muscle supervillin isoforms control stress-dependent thrombus formation of platelets (31), promote cell survival by suppressing p53 protein expression (32) and increase the rapid recycling of integrins and other motile processes (14,16,33–35). The isoform switch from non-muscle supervillin to archvillin occurs early in the differentiation of C2C12 murine skeletal myoblasts (36) and suggests a muscle-specific role for the protein, which is supported by its necessity to afford myotube formation of these cells (10,28,37). While these properties could be associated solely with actin cytoskeleton binding, the additional mechanically sensitive ERK interaction with archvillin may contribute to its actions in muscle.
We were intrigued to find that the nuclear fractions from both dystrophic muscle samples displayed increased lamin A/C. Because dystrophies exhibit heightened degeneration and regeneration, as well as increased immune infiltration, the nuclear content is elevated compared with healthy tissue (38). To control for this, we loaded equal protein from each fraction and used histone 3 as a nuclear marker pointed to the specific elevation of lamin A/C. Lamin A/C gene (LMNA) mutations are one cause of Emery-Dreifuss muscular dystrophy, and a mouse model for the LMNA H222P exhibits progressive dystrophic pathology and heightened ERK1/2 phosphorylation in skeletal and cardiac muscle (25,39). Whether or not the elevated lamin A/C in mdx and gsg−/− muscles is a compensatory measure to counter nuclear accumulation of P-ERK1/2 is not known. Further, the potential interaction between lamin A/C and archvillin identified in our yeast two-hybrid screen could implicate archvillin–ERK binding at both the sarcolemma and nucleus. Ultimately, these independent observations raise the potential for a common pathway—aberrant ERK signaling—that contributes to a range of dystrophies. This is substantiated by the upregulated P-ERK1/2 we observed in the LGMD and DMD human samples. Because there is now evidence that pharmacologic ERK inhibition improves muscle function in LMNA H222P EDMD mice (25,26), application of this strategy to other dystrophies is warranted.
A yeast two-hybrid screen using the intracellular domain of γ-SG as bait has been performed previously (40). Surprisingly, no preys emerged in common between our two screens, suggesting that there may be many γ-SG interactors. Because, to our knowledge, none of the prey hits from the previous screen has been pursued, the interactions with archvillin characterized here may represent the first of many new insights into processes involved in the dystrophic phenotype.
Based on our results, we provide a model in which archvillin sarcolemmal expression and localization are dependent on both the SG complex and dystrophin, where gsg−/− muscle has strongly upregulated archvillin levels concentrated at the sarcolemma (Fig. 6). In the absence of both sarcoglycan and dystrophin binding partners in mdx muscle, archvillin is no longer detected at the sarcolemma. The archvillin–P-ERK1/2 interaction is stimulus- and γ-SG-dependent, and we posit the possibility that the absence of the SG complex or the entire DGC may contribute to the aberrant ERK signaling observed in dystrophic muscle by an archvillin-mediated mechanism. Further experiments will be required to determine the contribution of archvillin to ERK signaling, and manipulation of ERK activity will provide insight into its impact on pathology. If archvillin proves to have a substantial impact on ERK signaling in muscle, it may be a viable target for muscle-specific manipulation of ERK activation. Given the role of ERK signaling on cell survival and proliferation, targeting archvillin may normalize the aberrant ERK activation and potential misregulation of its targets in dystrophic muscle. In conclusion, we have implicated archvillin as a new player in mechanical signal transduction in muscle and have shown that its localization and actions require the presence of dystrophin and the sarcoglycans, respectively.
Figure 6.
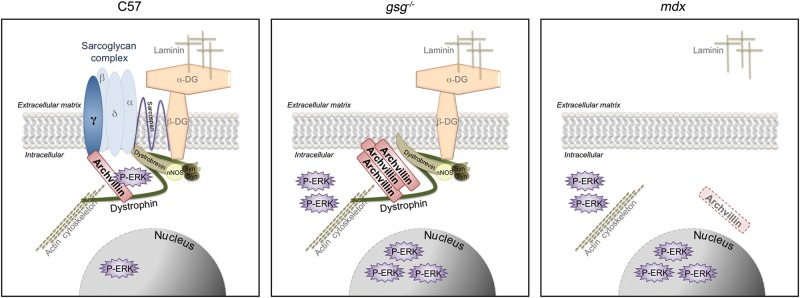
Model of archvillin-γ-SG signaling. Archvillin sarcolemmal localization is dependent on both the sarcoglycan complex and dystrophin. Muscle deficient in the sarcoglycan complex alone in gsg−/− muscle has strongly upregulated archvillin levels concentrated at the sarcolemma. However, in the absence of both sarcoglycan and dystrophin binding partners in mdx muscle, archvillin is no longer detected at the sarcolemma. Archvillin exhibits stimulus- and sarcoglycan-dependent association with P-ERK1/2. Archvillin only associates with P-ERK in C57 muscle subjected to in situ ECC; this association is absent in gsg−/− and mdx muscle with or without mechanical stimulation.
Materials and Methods
Yeast two-hybrid assays
Two independent yeast two-hybrid assays were performed to identify binding partners for the intracellular domain of γ-SG, and binding partners for the muscle-specific insert of archvillin. The Matchmaker 3 system (Clontech, Mountain View, CA, USA) was used to perform the γ-SG yeast two-hybrid assay. The bait plasmid was generated by cloning the cDNA sequence corresponding to the intracellular domain of human γ-SG (NG_008759) (aa 1–35) into the pGBKT7 bait vector containing the DNA-binding domain (BD) of Gal4. The bait plasmid was electroporated into AH109 yeast. The normalized human cDNA library prey, fused to the activation domain of Gal4, was pre-transformed in AH109 yeast. The mated yeast were screened on trp-, leu-, his-, ade-, x-alpha-Gal+ media plates (dropout media for positive selection of bait and prey plasmids and their interaction) and colonies that grew and were blue were considered hits. All appropriate control tests were performed as described in the assay kit. Prey fragments from all positive hits were amplified by PCR, sequenced and identified using the GenBank database (NCBI). A confirmation of binding assay for the archvillin prey was performed as described in the Matchmaker 3 system. In short, AH109 yeast containing either empty pGBKT7 plasmid or the γ-SG/pGBKT7 bait constructs were mated with Y187 yeast containing the archvillin/pGADT7 plasmid prey identified from the original yeast two-hybrid screen. The mated yeast were screened on either double dropout (DDO) trp-, leu-, x-alpha-Gal+ plates or quadruple dropout (QDO) trp-, leu-, his-, ade-, x-alpha-Gal+ media plates.
Archvillin yeast two-hybrid screening was performed by Hybrigenics Services, S.A.S., Paris, France (http://www.hybrigenics-services.com, last accessed on 22 January 2015). The sequence from the differentially spliced coding exons 3, 4 and 5, which encodes a large N-terminal insert (amino acids 276–669) in human supervillin isoform 2 (archvillin, NP_068506) and in supervillin isoform 4 (AGE81989.1), was generated by PCR (10,32). This PCR-amplified sequence was cloned into pB27, a derivative of pBTM116 (41), as a C-terminal fusion to LexA (N-LexA-SVIL-C) and into pB66, a derivative of pAS2ΔΔ (42), as a C-terminal fusion to the Gal4 DNA-binding domain (N-Gal4-SVIL-C). The constructs were verified by sequencing and used as baits for screening a random-primed Human Adult and Fetal Skeletal Muscle cDNA library in pP6, a derivative of pGADGH (43). The LexA bait construct was used to screen 61 million clones (6-fold coverage of the library), using a mating approach with YHGX13 (matα) and L40ΔGal4 (mata) yeast strains, as previously described (42) yielding a total of 17 positive colonies. The Gal4 construct and the same mating approach were used to screen 50 million clones (5-fold library coverage) with HGX13 (matα) and CG1945 (mata) yeast strains, yielding a total of 42 positive colonies. The prey fragments of the positive clones were confirmed for interaction, amplified by PCR, sequenced and identified using the GenBank database (NCBI).
Generation of His-DMD-10-12
An intein-tagged plasmid encoding dystrophin spectrin repeats (aa 1361–1686; pTYB1-DMD-10-12), a kind gift from Dr Elisabeth Le Rumeur (Université de Rennes), was used as template for PCR using the following primers: sense: 5′ GAATTCATGAGGCAAAAGTTGCTTGAACAGAGCATC; antisense: 5′ CTCGAGACTGGTCAAAAGTTTCCATGTGTTTCTGGTA. The resulting fragment was cloned into the pET30a+ vector using the EcoRI and XhoI restriction sites as described previously (15). The vector was verified by sequencing.
Recombinant protein purification
GST fusion proteins and His-DMD-10-12 were purified from Rosetta 2 pLysS (DE3) cells (EMD-Millipore Biosciences, Billerica, MA, USA), grown and extracted as described previously (13). Briefly, cells were induced with 0.2 mm IPTG at 30°C for 18 h and collected by centrifugation before resuspension and extraction. The GST proteins (13) and His-tagged DMD 10-12 (44) were retrieved, as described. His-tagged DMD 10-12 beads were washed five times with 10 ml of wash buffer B (10 mm phosphate buffer, pH 7.5, 60 mm NaCl, 2% glycerol, 0.01% DTT and 20 mm imidazole) (44). Three 1-ml fractions were eluted using wash buffer B containing 250 mm imidazole. All purified proteins were analyzed by SDS–PAGE.
GST pull-down experiments and His immunoblot analysis
Immediately prior to use, the purified His-DMD-10-12 solution in wash buffer B was modified to contain 90 mm NaCl, 2 mm DTT, 10 mg/ml BSA and 1% Tween-20 and clarified by centrifugation at 15 000g for 15 min. After the His-DMD-10-12 supernatant was transferred to a fresh tube, 100 μl aliquots were mixed with 200 μl of GST or GST fusion proteins and incubated for 2 h at 4°C with rotation. Aliquots of glutathione–Sepharose 4B (50 μl slurry volume; GE Healthcare Bio-Sciences, Piscataway, NJ, USA) were washed three times with wash buffer before addition of the GST/His protein mixtures and then incubated at 4°C with agitation for 1.5 h. The Sepharose beads were collected by centrifugation, and the supernatants were saved as unbound fractions. Beads were then washed five times with 500 μl of 0.5× TBST (83.5 mm NaCl, 5 mm Tris, 0.025% Tween-20, pH 7.5); at the second wash, the bead slurry was moved to a fresh tube. Bound fractions were eluted with 100 μl of 10 mm glutathione in wash buffer B. All samples were solubilized in Laemmli Sample Buffer (45), resolved on 12% SDS–PAGE gels, and transferred to 0.45 μm nitrocellulose (Whatman GmBH, Dassel, Germany). Immunoblots were probed using rabbit polyclonal anti-His (Cell Signaling, Beverly, MA, USA) and HRP-conjugated goat anti-rabbit (Jackson ImmunoResearch, West Grove, PA, USA), developed using SuperSignal WestFemto ECL (Thermo Scientific; Rockford, IL, USA) and imaged on a BioRad Gel Doc with Image Lab 4.1 software (BioRad, Hercules, CA, USA). The experiment was repeated twice with comparable results.
Immunohistochemistry
Ten-micrometer frozen cross-sections from the midbelly of EDL muscles were fixed for 10 min in 4% formaldehyde and washed three times with phosphate-buffered saline (PBS), followed by staining using the mouse-on-mouse (M.O.M) staining kit (Vector Laboratories, Burlingame, CA, USA). Sections were blocked for 1 h at room temperature in blocking buffer plus 2% horse serum and 2% BSA in PBS, then incubated overnight at 4°C in M.O.M. diluent with rabbit polyclonal anti-archvillin (Sigma Life Sciences, St. Louis, MO, USA; A1355) and anti-dystrophin (Vector Laboratories; VP-D505) or anti-γ-SG (Vector Laboratories VP-G803), all at 1:10 dilution. Secondary antibodies were applied at 1:500 (Life Technologies, Grand Island, NY, USA; A11034, S32354 and S32356), and slides were mounted using Vectashield with DAPI (Vector). Images were acquired at ×400 on a Leica DMR microscope and Leica DFC300 CCD camera, using Improvision OpenLab software (Perkin Elmer, Waltham, MA, USA).
Immunoprecipitation
Immunoprecipitation experiments were carried out using the Pierce Classic IP kit (Thermo Scientific). Muscle lysates containing 100 or 200 μg total protein were immunoprecipitated with anti-P-ERK1/2 (Cell Signaling; #9101 1:100), anti-ERK1/2 (Cell Signaling; #9107 1:100) or anti-γ-SG (Vector Laboratories; VP-G803 1:20) overnight at 4°C with end-over-end mixing. The immune complex was eluted with non-reducing sample buffer and boiled at 100°C for 5 min before being applied to a SDS–PAGE gel and immunoblotted.
Immunoblotting
Snap-frozen tissues were ground using a mortar and pestle over dry ice and lysed in RIPA buffer (50 mm HEPES pH 7.5, 150 mm NaCl, 5 mm EDTA, 1 mm EGTA, 15 mm p-nitrophenyl phosphate disodium hexahydral, 1% NP-40, 0.1% SDS, 1% deoxycholate and 0.025% sodium azide) with protease and phosphatase inhibitor cocktails added (Sigma). Lysates were incubated on ice for 1 h, vortexing half way through, centrifuged at 16000g for 30 min at 4°C and the supernatants retained. Protein concentration was measured using a Bradford method protein assay kit (Bio-Rad). Lysates were separated by SDS–PAGE on Tris–HCl polyacrylamide gels (Bio-Rad) and transferred to PVDF membranes. Membranes were blocked in 5% milk and 2% BSA in TTBS and then probed with antibodies to the following: anti-archvillin (10) 1:500, anti-P-ERK1/2 (Cell Signaling; #9101 1:1000), anti-total-ERK1/2 (Cell Signaling; #9107 1:1000), anti-γ-SG (Vector Laboratories; VP-G803, 1:300), anti-β-SG (Vector Laboratories; VP-B206 1:500), anti-δ-SG (Vector Laboratories; VP-D501 1:50), anti-histone 3 (Cell Signaling; #9715 1:2000), anti-tubulin (Sigma; T5168 1:20 000), anti-lamin A/C (Cell Signaling; #2032 1:500) and anti-GAPDH (Santa Cruz; sc-32233 1:5000). Membranes were then incubated with HRP-conjugated secondary antibodies (Cell Signaling; #7074, #7076 1:2000) and visualized using ECL Plus reagent (Thermo Scientific) and autoradiography film (BioExpress, Kaysville, UT).
For human samples, 25 pooled, 10-µm frozen sections of quadriceps biopsies from LGMD2 patients, DMD patients and healthy male controls were lysed in RIPA buffer, and 5% of the lysate was used for immunoblotting, as described earlier. Samples were obtained from the Wellstone Muscular Dystrophy Tissue and Cell Repository at the University of Iowa Carver College of Medicine (Iowa City, IA, USA). LGMD patients were between 9 and 17 years, DMD patients between 5 and 8 years and controls between 8 and 13 years at the time of biopsy (Supplemental Material, Table S2).
Quantitative RT–PCR
Total RNA was isolated from frozen quadriceps muscles using TRIzol (Life Technologies). Equal amounts of total RNA from each sample were subjected to single-strand reverse transcription (Applied Biosystems, Foster City, CA-now part of Life Technologies). The resultant cDNA was utilized for quantitative real-time PCR with primers specific for archvillin exon 4: sense—5′ AAAGAGGAGAGTGCTCGCAG 3′; antisense—5′ GCTGGTGACCCTATCAAAGGT 3′, using the Applied Biosystems 7300 Real-Time PCR System and reagents (Power SYBR Green PCR Master Mix, Life Technologies). All samples were loaded in duplicate in 96-well plates. Expression of 18S was used to control for cDNA content.
Animals
All experiments were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. C57Bl/6 (C57), γ-SG-null (gsg−/−), δ-SG-null (dsg−/−) and mdx mice were used. Three-week-old mice were used for viral injections; all other experiments were done with 12- to 14-week-old mice. The gsg−/− mouse lacks γ-SG owing to gene targeting, resulting in an additional loss of β- and δ-SG and a decrease of α-SG (6), and was backcrossed for >10 generations onto the C57Bl/6 strain. The dsg−/− mouse lacks δ-SG also owing to gene targeting, resulting in an additional loss of α-, β- and γ-SG on a 129SvJ/129SvEms-+Ter/J background (46). Mdx mice lacking dystrophin were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and were maintained as a colony of homozygous mutant animals (C57Bl/10 background strain).
In situ TA muscle mechanics
In situ muscle function was examined on the TA using a protocol developed from methods described previously (47). Briefly, mice were deeply anesthetized by i.p. injection of ketamine–xylazine (80 and 10 mg/kg), with body temperature maintained at 37°C, and monitored throughout the experiment. For each preparation, the distal tendon of the TA was carefully dissected and tied with 4.0 braided surgical silk. The sciatic nerve was exposed, and all of its branches were cut except for the common peroneal nerve. The foot was secured to a platform and the knee immobilized using a stainless steel pin, with care taken not to interfere with the blood supply to the muscles. The suture from the TA tendon was attached to the lever arm of a 305B dual-mode servomotortransducer (Aurora Scientific, Ontario, Canada). Isometric muscle contractions were then elicited by stimulating the distal part of the sciatic nerve via bipolar electrodes, using supramaximal square-wave pulses of 0.02 ms (701A stimulator; Aurora Scientific). Data acquisition and control of the servomotor were conducted using a Lab-View-based DMC program (version 5.202; Aurora Scientific). Optimal muscle length (Lo) was determined by incrementally stretching the muscle until the maximum isometric twitch force was achieved (Po). Muscle length was measured using digital calipers based on well-defined anatomical landmarks. Three maximum isometric tetanic forces were acquired using a train of 150 Hz, 500 ms supramaximal electrical pulses at the optimal length in the muscles and highest force was recorded. A 2-min resting period was allowed between each tetanic contraction. Following isometric contractions, muscles were subjected to two ECCs with the muscle stimulated at 150 Hz for a total of 300 ms. TA muscles were stretched 24% Lo in the final 200-ms stimulation. The second ECC was administrated after a 10-s rest period under the same parameters. Post-ECC muscle isometric forces were measured after 1, 5 and 15 min. The drop in force between the isometric force during ECC and recovery time served as an index of susceptibility to ECC-induced injury.
Viral constructs and injections
Human γ-SG cDNA was utilized to generate recombinant AAV serotype 2/8 (rAAV) as described previously (8), and expression was regulated by a truncated desmin promoter (48).Vector production was performed at the University of Pennsylvania Vector Core. Solutions containing 1 × 1011 viral particles diluted in 50 μl of PBS were injected into the anterior compartment of the lower hindlimbs of 3-week-old gsg−/− mice, targeting the TA and EDL muscles as described previously. After injection, mice were housed in the animal facility until time of analysis, 1 month post-injection. The TA muscles were dissected and rapidly frozen in liquid nitrogen for biochemical analysis, whereas the EDL muscles were frozen in OCT (Sakura Finetek, Torrance, CA, USA) for immunohistochemical analysis.
Cytosolic/nuclear fractionation
Snap-frozen TA muscles were ground using a mortar and pestle and processed as described previously (49). In short, nuclear fractions were isolated by lysates undergoing a series of centrifugations and resuspensions in STM buffer (250 mm sucrose, 50 mm Tris–HCl pH 7.4, 5 mm MgCl2, protease and phosphatase inhibitors) followed by resuspension in NET buffer (20 mm HEPES pH 7.9, 1.5 mm MgCl2, 0.5 m NaCl, 0.2 mm EDTA, 20% glycerol, 1% Triton X-100, protease and phosphatase inhibitors) and lysing of nuclei by passage through an 18-gauge needle and sonication. Cytosolic fractions were isolated by centrifugation, precipitation in 100% acetone at −20°C for 1 h and resuspension in STM buffer. Processed lysates were then analyzed by immunoblotting as described earlier.
Statistics
All results are shown as the means ± S.E.M. unless stated otherwise. Statistical analyses of the data were performed using one-way or two-way ANOVA followed by Bonferroni post hoc test. P-values of <0.05 were considered statistically significant.
Supplementary Material
Funding
This work was supported by National Institute of Health Grants (U54 AR052646 to E.R.B., R01 GM033048-26S1 to E.J.L.), and by the Department of Cell and Developmental Biology at the University of Massachusetts Medical School. J.M.S. was supported by a National Institutes of Health fellowship awarded to the Pennsylvania Muscle Institute, University of Pennsylvania, Philadelphia, PA AR-052461.
Supplementary Material
Acknowledgements
We thank Steven A. Moore at the University of Iowa Muscular Dystrophy Tissue and Cell Repository for providing control, DMD and LGMD patient biopsies. We also thank Dr Marcela Nunez and her colleagues at Hybrigenics Services, S.A.S. for their assistance and expertise in yeast two-hybrid screening. We also are grateful to Dr Zhiyou Fang (University of Massachusetts Medical School) for the GST-AV-345 constructs and to Dr Nick Menhart (Illinois Institute of Technology, Chicago, IL, USA) and Dr Elisabeth Le Rumeur (Université de Rennes, Rennes, France) for the kind gifts of dystrophin repeat containing plasmids.
Conflict of Interest statement. None declared.
References
- 1.Lapidos K.A., Kakkar R., McNally E.M. (2004) The dystrophin glycoprotein complex. Circ. Res., 94, 1023–1031. [DOI] [PubMed] [Google Scholar]
- 2.Ervasti J.M., Sonnemann K.J. (2008) Biology of the striated muscle dystrophin–glycoprotein complex. In Jeon K.W. (ed), International Review of Cytology: A Survey of Cell Biology. Academic Press, San Diego, CA, Vol. 265, pp. 191–225. [DOI] [PubMed] [Google Scholar]
- 3.Engel A.G., Franzini-Armstrong C. (2004) Myology: Basic and Clinical. McGraw-Hill, Inc., Clumbus, OH. [Google Scholar]
- 4.Durbeej M., Campbell K.P. (2002) Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr. Opin. Genet. Dev., 12, 349–361. [DOI] [PubMed] [Google Scholar]
- 5.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl Acad. Sci. USA, 90, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hack A.A., Ly C.T., Jiang F., Clendenin C.J., Sigrist K.S., Wollmann R.L., McNally E.M. (1998) γ-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J. Cell Biol., 142, 1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hack A.A., Cordier L., Shoturma D.I., Lam M.Y., Sweeney H.L., McNally E.M. (1999) Muscle degeneration without mechanical injury in sarcoglycan deficiency. Proc. Natl Acad. Sci. USA, 96, 10723–10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barton E.R. (2010) Restoration of γ-sarcoglycan localization and mechanical signal transduction are independent in murine skeletal muscle. J. Biol. Chem., 285, 17263–17270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moorwood C., Philippou A., Spinazzola J., Keyser B., Macarak E.J., Barton E.R. (2014) Absence of gamma-sarcoglycan alters the response of p70S6 kinase to mechanical perturbation in murine skeletal muscle. Skelet. Muscle, 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oh S.W., Pope R.K., Smith K.P., Crowley J.L., Nebl T., Lawrence J.B., Luna E.J. (2003) Archvillin, a muscle-specific isoform of supervillin, is an early expressed component of the costameric membrane skeleton. J. Cell Sci., 116, 2261–2275. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y., Takizawa N., Crowley J.L., Oh S.W., Gatto C.L., Kambara T., Sato O., Li X.-d., Ikebe M., Luna E.J. (2003) F-actin and myosin II binding domains in supervillin. J. Biol. Chem., 278, 46094–46106. [DOI] [PubMed] [Google Scholar]
- 12.Nebl T., Pestonjamasp K.N., Leszyk J.D., Crowley J.L., Oh S.W., Luna E.J. (2002) Proteomic analysis of a detergent-resistant membrane skeleton from neutrophil plasma membranes. J. Biol. Chem., 277, 43399–43409. [DOI] [PubMed] [Google Scholar]
- 13.Smith T.C., Fang Z., Luna E.J. (2010) Novel interactors and a role for supervillin in early cytokinesis. Cytoskeleton (Hoboken), 67, 346–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith T.C., Fridy P.C., Li Y., Basil S., Arjun S., Friesen R.M., Leszyk J., Chait B.T., Rout M.P., Luna E.J. (2013) Supervillin binding to myosin II and synergism with anillin are required for cytokinesis. Mol. Biol. Cell, 24, 3603–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takizawa N., Smith T.C., Nebl T., Crowley J.L., Palmieri S.J., Lifshitz L.M., Ehrhardt A.G., Hoffman L.M., Beckerle M.C., Luna E.J. (2006) Supervillin modulation of focal adhesions involving TRIP6/ZRP-1. J. Cell Biol., 174, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takizawa N., Ikebe R., Ikebe M., Luna E.J. (2007) Supervillin slows cell spreading by facilitating myosin II activation at the cell periphery. J. Cell Sci., 120, 3792–3803. [DOI] [PubMed] [Google Scholar]
- 17.Pope R.K., Pestonjamasp K.N., Smith K.P., Wulfkuhle J.D., Strassel C.P., Lawrence J.B., Luna E.J. (1998) Cloning, characterization, and chromosomal localization of human superillin (SVIL). Genomics, 52, 342–351. [DOI] [PubMed] [Google Scholar]
- 18.Gangopadhyay S.S., Kengni E., Appel S., Gallant C., Kim H.R., Leavis P., DeGnore J., Morgan K.G. (2009) Smooth muscle archvillin is an ERK scaffolding protein. J. Biol. Chem., 284, 17607–17615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gangopadhyay S.S., Takizawa N., Gallant C., Barber A.L., Je H.-D., Smith T.C., Luna E.J., Morgan K.G. (2004) Smooth muscle archvillin: a novel regulator of signaling and contractility in vascular smooth muscle. J. Cell Sci., 117, 5043–5057. [DOI] [PubMed] [Google Scholar]
- 20.Dinkel H., Van Roey K., Michael S., Davey N.E., Weatheritt R.J., Born D., Speck T., Kruger D., Grebnev G., Kuban M., et al. (2014) The eukaryotic linear motif resource ELM: 10 years and counting. Nucl. Acids Res., 42, D259–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allikian M.J., Hack A.A., Mewborn S., Mayer U., McNally E.M. (2004) Genetic compensation for sarcoglycan loss by integrin α7β1 in muscle. J. Cell. Sci., 117, 3821–3830. [DOI] [PubMed] [Google Scholar]
- 22.Hodges B.L., Hayashi Y.K., Nonaka I., Wang W., Arahata K., Kaufman S.J. (1997) Altered expression of the alpha7 beta1 integrin in human and murine muscular dystrophies. J. Cell Sci., 110 (Pt 22), 2873–2881. [DOI] [PubMed] [Google Scholar]
- 23.Martineau L.C., Gardiner P.F. (2001) Insight into skeletal muscle mechanotransduction: MAPK activation is quantitatively related to tension. J. Appl. Physiol., 91, 693–702. [DOI] [PubMed] [Google Scholar]
- 24.Barton E.R. (2006) Impact of sarcoglycan complex on mechanical signal transduction in murine skeletal muscle. Am. J. Physiol. Cell Physiol., 290, C411–C419. [DOI] [PubMed] [Google Scholar]
- 25.Muchir A., Kim Y.J., Reilly S.A., Wu W., Choi J.C., Worman H.J. (2013) Inhibition of extracellular signal-regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutation. Skelet. Muscle, 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu W., Iwata S., Homma S., Worman H.J., Muchir A. (2014) Depletion of extracellular signal-regulated kinase 1 in mice with cardiomyopathy caused by lamin A/C gene mutation partially prevents pathology before isoenzyme activation. Hum. Mol. Genet., 23, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roux P.P., Blenis J. (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev., 68, 320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee M.-A., Joo Y.M., Lee Y.M., Kim H.S., Kim J.-H., Choi J.-K., Ahn S.-J., Min B.-I., Kim C.-R. (2008) Archvillin anchors in the Z-line of skeletal muscle via the nebulin C-terminus. Biochem. Biophys. Res. Commun., 374, 320–324. [DOI] [PubMed] [Google Scholar]
- 29.Burkin D.J., Wallace G.Q., Nicol K.J., Kaufman D.J., Kaufman S.J. (2001) Enhanced expression of the alpha7 beta1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol., 152, 1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y., Reznichenko M., Tribe R.M., Hess P.E., Taggart M., Kim H., DeGnore J.P., Gangopadhyay S., Morgan K.G. (2009) Stretch activates human myometrium via ERK, caldesmon and focal adhesion signaling. PLoS ONE, 4, e7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edelstein L.C., Luna E.J., Gibson I.B., Bray M., Jin Y., Kondkar A., Nagalla S., Hadjout-Rabi N., Smith T.C., Covarrubias D., et al. (2012) Human genome-wide association and mouse knockout approaches identify platelet supervillin as an inhibitor of thrombus formation under shear stress. Circulation, 125, 2762–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang Z., Luna E.J. (2013) Supervillin-mediated suppression of p53 protein enhances cell survival. J. Biol. Chem., 288, 7918–7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang Z., Takizawa N., Wilson K.A., Smith T.C., Delprato A., Davidson M.W., Lambright D.G., Luna E.J. (2010) The membrane-associated protein, supervillin, accelerates F-actin-dependent rapid integrin recycling and cell motility. Traffic, 11, 782–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crowley J.L., Smith T.C., Fang Z., Takizawa N., Luna E.J. (2009) Supervillin reorganizes the actin cytoskeleton and increases invadopodial efficiency. Mol. Biol. Cell, 20, 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhuwania R., Cornfine S., Fang Z., Kruger M., Luna E.J., Linder S. (2012) Supervillin couples myosin-dependent contractility to podosomes and enables their turnover. J. Cell Sci., 125, 2300–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senetar M.A., Moncman C.L., McCann R.O. (2007) Talin2 is induced during striated muscle differentiation and is targeted to stable adhesion complexes in mature muscle. Cell Motil. Cytoskel., 64, 157–173. [DOI] [PubMed] [Google Scholar]
- 37.Lim M.J., Seo Y.H., Choi K.J., Cho C.H., Kim B.S., Kim Y.H., Lee J., Lee H., Jung C.Y., Ha J., et al. (2007) Suppression of c-Src activity stimulates muscle differentiation via p38 MAPK activation. Arch. Biochem. Biophys., 465, 197–208. [DOI] [PubMed] [Google Scholar]
- 38.Evans N.P., Misyak S.A., Robertson J.L., Bassaganya-Riera J., Grange R.W. (2009) Immune-mediated mechanisms potentially regulate the disease time-course of duchenne muscular dystrophy and provide targets for therapeutic intervention. PM R, 1, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arimura T., Helbling-Leclerc A., Massart C., Varnous S., Niel F., Lacene E., Fromes Y., Toussaint M., Mura A.M., Keller D.I., et al. (2005) Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet., 14, 155–169. [DOI] [PubMed] [Google Scholar]
- 40.Blandin G., Marchand S., Charton K., Daniele N., Gicquel E., Boucheteil J.B., Bentaib A., Barrault L., Stockholm D., Bartoli M., et al. (2013) A human skeletal muscle interactome centered on proteins involved in muscular dystrophies: LGMD interactome. Skelet. Muscle, 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vojtek A.B., Hollenberg S.M. (1995) Ras-Raf interaction: two-hybrid analysis. Meth. Enzymol., 255, 331–342. [DOI] [PubMed] [Google Scholar]
- 42.Fromont-Racine M., Rain J.C., Legrain P. (1997) Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat. Genet., 16, 277–282. [DOI] [PubMed] [Google Scholar]
- 43.Bartel P., Chien C.T., Sternglanz R., Fields S. (1993) Elimination of false positives that arise in using the two-hybrid system. Biotechniques, 14, 920–924. [PubMed] [Google Scholar]
- 44.Takizawa N., Schmidt D.J., Mabuchi K., Villa-Moruzzi E., Tuft R.A., Ikebe M. (2003) M20, the small subunit of PP1M, binds to microtubules. Am. J. Physiol. Cell Physiol., 284, C250–C262. [DOI] [PubMed] [Google Scholar]
- 45.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 46.Hack A.A., Lam M.Y., Cordier L., Shoturma D.I., Ly C.T., Hadhazy M.A., Hadhazy M.R., Sweeney H.L., McNally E.M. (2000) Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J. Cell Sci., 113 (Pt 14), 2535–2544. [DOI] [PubMed] [Google Scholar]
- 47.Dellorusso C., Crawford R.W., Chamberlain J.S., Brooks S.V. (2001) Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J. Muscle Res. Cell Motil., 22, 467–475. [DOI] [PubMed] [Google Scholar]
- 48.Li Z., Marchand P., Humbert J., Babinet C., Paulin D. (1993) Desmin sequence elements regulating skeletal muscle-specific expression in transgenic mice. Development, 117, 947–959. [DOI] [PubMed] [Google Scholar]
- 49.Dimauro I., Pearson T., Caporossi D., Jackson M.J. (2012) A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res Notes, 5, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.