


Abstract
HAMAP (High-quality Automated and Manual Annotation of Proteins—available at http://hamap.expasy.org/) is a system for the automatic classification and annotation of protein sequences. HAMAP provides annotation of the same quality and detail as UniProtKB/Swiss-Prot, using manually curated profiles for protein sequence family classification and expert curated rules for functional annotation of family members. HAMAP data and tools are made available through our website and as part of the UniRule pipeline of UniProt, providing annotation for millions of unreviewed sequences of UniProtKB/TrEMBL. Here we report on the growth of HAMAP and updates to the HAMAP system since our last report in the NAR Database Issue of 2013. We continue to augment HAMAP with new family profiles and annotation rules as new protein families are characterized and annotated in UniProtKB/Swiss-Prot; the latest version of HAMAP (as of 3 September 2014) contains 1983 family classification profiles and 1998 annotation rules (up from 1780 and 1720). We demonstrate how the complex logic of HAMAP rules allows for precise annotation of individual functional variants within large homologous protein families. We also describe improvements to our web-based tool HAMAP-Scan which simplify the classification and annotation of sequences, and the incorporation of an improved sequence-profile search algorithm.
INTRODUCTION
Falling costs and continuing technological advances in DNA sequencing have led to an explosion in the number of available whole genome sequences from all branches of the tree of life, opening up exciting new possibilities for research into the evolution and function of biological systems. However as the number of protein-coding gene sequences continues to grow exponentially, the tiny fraction of experimentally characterized sequences continues to shrink—this despite the best efforts of groups such as the Enzyme Function Initiative (1) and COMBREX (2) to accelerate the rate of functional characterization through combined computational and experimental approaches. This growing gap highlights a need for automated systems that can effectively leverage the available experimental information to provide precise functional annotation for the tens of millions of predicted protein sequences that will probably never be characterized (3).
One such system is HAMAP (High-quality Automated and Manual Annotation of Proteins), which provides automatic classification and functional annotation of protein sequences based on their homology to characterized templates (4). HAMAP is based on a collection of expert curated protein family profiles, which are used to determine family membership of protein sequences, and annotation rules, which specify the appropriate annotation for family members. HAMAP rules permit the annotation of protein sequences to the same level of detail and quality as manually curated UniProtKB/Swiss-Prot records, annotating protein and gene names, function, catalytic activity, cofactors, subcellular location, protein–protein interactions, as well as sequence features such as the presence of specific domains, motifs and functionally important sites (such as ion-, substrate- and cofactor-binding sites, catalytic residues and post-translational modifications). Annotations are provided in the form of the human-readable UniProtKB text format and using UniProt controlled vocabularies and terms from the Gene Ontology (GO) (5). As well as the annotations themselves, HAMAP rules also specify the conditions under which these annotations may be applied, such as a requirement for key functional residues (identified by structural or other experimental studies). Such conditions can reduce the incidence of erroneous annotation, particularly in large, functionally diverse families—errors that tend to persist in public sequence databases (6–8).
HAMAP forms one component of the UniProt UniRule system that provides annotation for the unreviewed component of the UniProt Knowledgebase UniProtKB/TrEMBL (9). HAMAP family profiles and annotation rules are created (and updated) concurrently with the curation of experimentally characterized templates into UniProtKB/Swiss-Prot, by the same expert curators. This ensures that the family profiles accurately reflect the properties of trusted protein family members, that target sequences are annotated to the quality standards of UniProtKB/Swiss-Prot, and that updates to UniProtKB/Swiss-Prot records are subsequently recorded in HAMAP rules (and propagated to homologous UniProtKB/TrEMBL records). In addition to UniProtKB, HAMAP also provides protein family annotation for Ensembl Genomes (10) as well as a number of other genome annotation pipelines (11,12).
In the remainder of this article we describe developments in HAMAP since our last report in the Database Issue of Nucleic Acids Research. We also provide examples of how the careful manual curation of HAMAP profiles and associated rules can generate precise functional annotation for individual members of large and functionally diverse protein families.
ANNOTATION AND CONTENT
Refining HAMAP family profiles for increased specificity of functional annotation
HAMAP defines family membership of protein sequences using generalized profiles derived from manually curated multiple sequence alignments (MSAs) of trusted members (4,13). Precise functional annotation requires the careful definition of isofunctional protein families and functionally important residues—excluding other functional categories and closely related families curated in UniProtKB/Swiss-Prot. During curation of the multiple sequence alignment erroneous sequences and misaligned positions are corrected where necessary (described in (4), complete workflow ftp://ftp.expasy.org/databases/hamap/SOP_HAMAP_profile_creation.pdf included as supplementary file S1). Profiles are generated using the pftools package (available at http://web.expasy.org/pftools/) as described in (14,15). The specificity of the resulting profile may be modulated through the use of different pseudocounts, which assign scores to amino acid residues that have not been observed in the sequence alignments used to construct the profile (16). The values of these scores are derived from the PAM (Point Accepted Mutation) (17) and BLOSUM (BLOcks SUbstitution Matrix) (18) amino acid scoring matrices, which cover a wide range of evolutionary distances. Matrices tailored to shorter evolutionary distances will more strongly penalize substitutions that have not been observed, producing profiles that more faithfully reflect the observed diversity in the alignment—and which may better separate closely related subfamilies. There are of course limitations to this approach, and it is not always possible to generate HAMAP profiles that discriminate between very closely related sequences—one example, concerning certain subfamilies of sirtuins, is described below. The process of HAMAP family profile generation is iterative, and curators may modify the seed alignment, the profile construction parameters, and the threshold score for trusted family members until a profile with satisfactory specificity and sensitivity is achieved—based on the annotation of the matching UniProtKB/Swiss-Prot records. The parameters used for final profile generation are stored together with the seed alignment, so that profiles can be regenerated as needed.
HAMAP is continually updated, and HAMAP profiles and families may be modified, extended, or split as results from new phylogenetic analyses and experimental characterization data become available. A case in point is provided by the sirtuin family of proteins, whose members were thought to act exclusively as protein deacetylases (19,20). Phylogenetic analyses (using methods described in 21–25) suggest five families of sirtuins—classes I, II, III, IV and U (17) (see Figure 1). Class III sirtuins, including the human SIR5 protein (UniProtKB/Swiss-Prot record Q9NXA8), were recently found to exhibit both protein demalonylase and protein desuccinylase activity (26,27). The class III sirtuin of Escherichia coli (CobB, P75960) also functions as a protein desuccinylase (28), while that of Plasmodium falciparum (Sir2A, Q8IE47) hydrolyses medium and long chain fatty acyl groups from lysine residues (29), suggesting an ancient divergence of function in evolution. Specificity for these relatively bulky substrates may be conferred by a larger hydrophobic pocket and substrate-binding residues (Tyr-102 and Arg-105 in human SIR5) common to all class III sirtuins from all kingdoms of life (20,30). As part of the normal HAMAP workflow, all characterized sirtuin protein records in UniProtKB/Swiss-Prot were first updated (31). The existing HAMAP family profile for bacterial sirtuins (profile MF_01121) was modified to specifically match only the class III sirtuins, and new family profiles were created for classes II and U (profiles MF_01967 and MF_01968 respectively). HAMAP annotation rules for class III sirtuins were created that allow specific annotation of protein function and sequence features for both prokaryotic and eukaryotic sequences (rules MF_01121 and MF_03160 respectively). Class I and IV subfamilies are not currently treated by HAMAP, as these are further divided into subclasses (Ia, Ib, Ic and IVa, IVb, respectively), where each subclass contains multiple paralogs per species. Such complex duplications may be better addressed using methods that explicitly consider evolutionary history in the form of a phylogenetic tree. Other resources such as Pfam provide broad coverage of sirtuin family proteins (with a single signature PF02146) while a more restricted PIRSF signature (PIRSF037938) currently covers only the sirtuin subclass Ib members.
Figure 1.
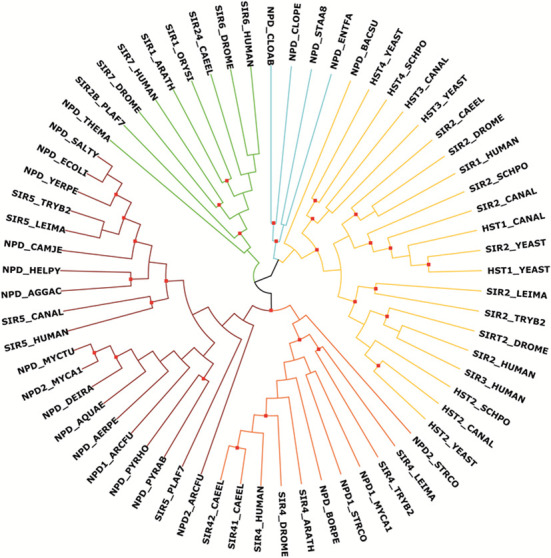
Maximum likelihood cladogram of the sirtuin superfamily. Maximum likelihood (ML) analyses of selected sirtuin family members resulted in 12 trees with two distinct topologies for the main classes I-IV and U, suggesting either classes II and III or classes II and VI to be sister clades. The tree topology with highest branch support is shown. Branches are colored according to families: class I = dark yellow, class II = orange, class III = red, class IV = green, class U = cyan. Branches with aLRT SH-like support values of 0.9 or higher are marked by a red dot. Methods: 65 sirtuin protein family members from 33 species were aligned with MAFFT (21) (version 7; parameters: L-INS-i, JTT200). From the alignment, we selected manually homologous regions using the alignment editor Jalview (22); three data models were created with a length of 238, 220 and 193 amino acids, respectively. The best fitting model of protein evolution was determined with ProtTest (23) (version 3.2; parameters: fixed BIONJ tree calculated under the JTT model of amino acid substitution; rate variation; amino acid frequencies to be the LG model plus gamma distribution). Maximum likelihood (ML) phylogenies and ML consensus trees from 100 bootstrap replicates were inferred with PhyML (24) (version 3.0) and RAxML (25) (version 7.2.8). The tree was visualized with Archaeopteryx (https://sites.google.com/site/cmzmasek/home/software/archaeopteryx). Protein sequences and multiple sequence alignments are provided in supplementary file S2.
HAMAP allows specific functional annotation within homologous protein families
The rule syntax used by HAMAP (described in http://hamap.expasy.org/unirule/unirule.html) allows for control statements that specify conditions–such as the occurrence of specific residues or motifs–for the application of annotation. These control statements provide a flexible means of fine-tuning the annotation of individual members of protein families, illustrated here using the 6-phosphofructokinase (PFK) family. PFK is a key regulatory enzyme of glycolysis that is present in all three domains of life. Despite this high level of conservation the enzyme has a remarkable evolutionary history, featuring a high rate of horizontal gene transfer and substitution in its active site (32). These substitutions have a profound impact on enzyme function; PFK family members with a glycine (G) at the active site catalyze the phosphorylation of D-fructose 6-phosphate to fructose 1,6-bisphosphate using adenosine triphosphate (ATP) (in the first committed step of glycolysis), while those with aspartate (D) use inorganic phosphate (PPi) as the phosphoryl donor in a reversible reaction that occurs in both glycolysis and gluconeogenesis (32–34).
HAMAP defines 8 PFK families in line with the currently accepted classification of PFKs (32,35) (Table 1). Several of the eight HAMAP families include both PPi-dependent and ATP-dependent members, suggesting that phosphoryl-donor specificity may have changed at multiple times during the evolution of the PFK superfamily. Figure 2 illustrates how this functional variation within families is treated by HAMAP using annotation rule MF_01976, which describes members of the mixed substrate PFK group III subfamily. The precise annotation that is applied to members of this family depends on the nature of the active site residue (D104 in the experimentally characterized template of Amycolatopsis methanolica—UniProtKB/Swiss-Prot record Q59126). Case statements within the rule specify the correct protein name, catalytic activity (including EC number), function, keywords, GO terms and other annotations for family members bearing either D or G at their active site. Sequences having neither of these residues are annotated as generic 6-phosphofructokinases of unknown substrate-specificity. The example of PFK illustrates how a single residue may determine substrate specificity and enzyme function, but HAMAP rule syntax also allows conditional annotation based on the combination of multiple residues or sequence motifs. The methylthioadenosine (MTA) phosphorylases are one example, where conserved amino acid substitutions in the substrate binding pocket convert the substrate specificity of this enzyme from 6-aminopurine (EC 2.4.2.28) to 6-oxopurine nucleosides (EC 2.4.2.44 and EC 2.4.2.1) (described in MF_01963).
Table 1. The PFK family of proteins in HAMAP.

Figure 2.
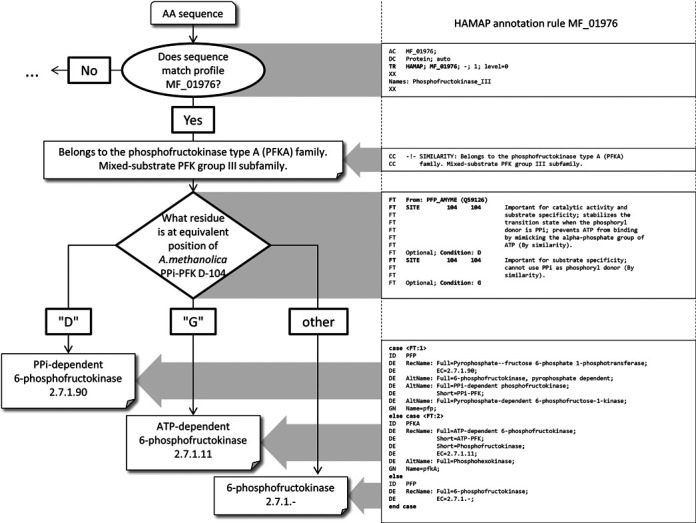
HAMAP annotation rule MF_01976 for mixed-substrate PFK group III family. The right hand panel shows snippets of the annotation rule MF_01976 including conditions used to specify site-specific annotations propagated to target sequences. If a protein sequence matches the HAMAP family profile MF_01976, then appropriate annotations for all members of that family (such as family membership) are attached to the sequence. For the annotation of sequence features, the target sequence is aligned to the seed alignment and the active site residue from the template sequence mapped to the target sequence. The nature of the residue at the equivalent position in the target sequence determines which of the possible conditional annotations will be attached to the sequence.
HAMAP statistics
Since our last publication in the NAR Database Issue 2013, we have added 203 new family profiles and 278 new annotation rules to HAMAP. As of 3 September 2014, HAMAP contains 1983 family classification profiles and 1998 annotation rules (a single HAMAP family profile may be associated with multiple HAMAP annotation rules, where each rule applies to a distinct taxonomic group). Through the UniRule pipeline, HAMAP provides annotations for 10,874,356 UniProtKB/TrEMBL sequence records (release 2014_08), which is around 13% of all sequence records in UniProtKB/TrEMBL, and 16% of the sequence records of each prokaryotic complete proteome. HAMAP provides 48% of all annotations and 90% of all sequence-specific feature annotations for the UniRule automatic annotation pipeline of UniProt. One of the strengths of HAMAP lies in the granularity and the comprehensiveness of its annotations, with each HAMAP rule providing over 16 annotations per UniProtKB/TrEMBL record on average.
WEBSITE
Improvements to the web-interface for HAMAP-Scan
Protein sequences can be classified and annotated using HAMAP through our HAMAP-Scan web service (http://hamap.expasy.org/hamap_scan.html). We provide a single-page, 3-step, dynamic submission form where required fields are clearly marked, and every field is accompanied by a short explanatory text. Each user choice dynamically updates the submission form, such that only necessary fields are displayed. The form allows submission of user sequences (FASTA) and UniProt sequence record identifiers or sequence accessions; users may submit individual sequences or whole proteome sequences. All submitted sequences are returned to the user in UniProtKB format in the order of submission, while protein sequences that have a trusted match to a HAMAP family profile are also annotated by the associated HAMAP rule. All result entries (including entries that are not annotated) contain an additional section with information on matches to HAMAP family profiles, including the profile accession number and identifier, the match quality (trusted or weak), and the match score (with the score difference to the trusted cut-off score of the profile in parenthesis) (Figure 3). HAMAP profiles are also available through InterProScan (36) provided by the InterPro Consortium (37), of which HAMAP is a member.
Figure 3.
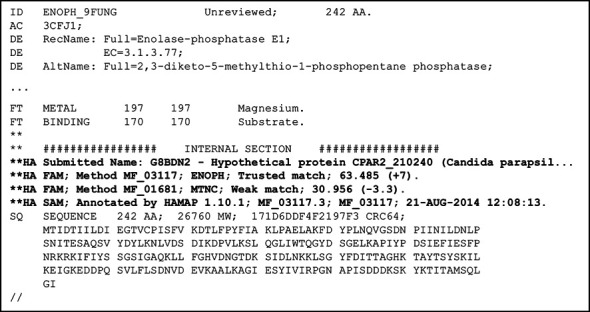
Partial output of a HAMAP-Scan showing the additional information provided next to the actual annotations. The sequence of Candida parapsilosis hypothetical protein CPAR2_210240 (CCE43379.1) was submitted in FASTA format to HAMAP-Scan. The internal section in the output file contains information such as the submitted FASTA header, a trusted match (including the match score and the score difference to the trusted cut-off score) to profile MF_03117 (ENOPH), a weak match to profile MF_01681 (MTNC, the homologous bacterial family), as well as the information that the sequence has consequently been annotated by HAMAP rule MF_03117 associated with profile MF_03117. The full annotation produced for this sequence can be viewed in UniProtKB/TrEMBL record G8BDN2 for C. parapsilosis CPAR2_210240.
Accelerated HAMAP-Scan with pfsearchV3
To facilitate the use of HAMAP-Scan for the classification and annotation of large datasets such as whole proteome sequences we have implemented the improved version of the PROSITE search tool pfsearchV3 (38) for HAMAP. pfsearchv3 uses modern CPU instructions to exploit the capabilities of multicore processors and a new heuristic filter to rapidly score and select possible candidate matches, achieving speeds up to two orders of magnitude faster than the previous version of this algorithm. We plan to make the heuristic score thresholds for HAMAP profiles available to our users in the near future.
CONCLUSION
HAMAP provides accurate and detailed functional annotation for the exponentially growing population of uncharacterized protein sequences in public databases such as UniProtKB/TrEMBL, as well as tools and services for external users. HAMAP profiles allow the definition of isofunctional protein families of whatever size and scope according to current knowledge. HAMAP annotation rules provide fine-grained annotations for family members, based on the presence of specific functional residues (as illustrated here for the PFK families). The creation of family profiles and annotation rules in HAMAP is a manual effort performed by expert curators. Manual curation of the experimental literature in UniProtKB/Swiss-Prot is highly accurate (6), with expert curation of HAMAP profiles and rules specifically designed to avoid over-annotation through the careful definition of isofunctional protein families and functionally important residues. HAMAP annotations can be accessed via UniProtKB, or generated by users for their own protein or proteome sequences via the HAMAP-Scan service on the HAMAP website.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Anne Morgat and Marco Pagni for insightful comments and discussions on the scope and direction of HAMAP. We also thank Brigitte Boeckmann for critical reading of the manuscript and for help with the phylogenetic analysis of the sirtuin protein family.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Swiss Federal Government through the State Secretariat for Education, Research and Innovation; National Institutes of Health [U41HG006104]; Swiss National Science Foundation [JRP09 and JRP13]. Funding for open access charge: Swiss Federal Government through the State Secretariat for Education, Research and Innovation.
Conflict of interest statement. None declared.
REFERENCES
- 1.Gerlt J.A., Allen K.N., Almo S.C., Armstrong R.N., Babbitt P.C., Cronan J.E., Dunaway-Mariano D., Imker H.J., Jacobson M.P., Minor W. Enzyme Function Initiative. Biochemistry. 2011;50:9950–9962. doi: 10.1021/bi201312u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anton B.P., Chang Y.C., Brown P., Choi H.P., Faller L.L., Guleria J., Hu Z., Klitgord N., Levy-Moonshine A., Maksad A., et al. The COMBREX project: design, methodology, and initial results. PLoS Biol. 2013;11:e1001638. doi: 10.1371/journal.pbio.1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radivojac P., Clark W.T., Oron T.R., Schnoes A.M., Wittkop T., Sokolov A., Graim K., Funk C., Verspoor K., Ben-Hur A., et al. A large-scale evaluation of computational protein function prediction. Nat. Methods. 2013;10:221–227. doi: 10.1038/nmeth.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pedruzzi I., Rivoire C., Auchincloss A.H., Coudert E., Keller G., de Castro E., Baratin D., Cuche B.A., Bougueleret L., Poux S., et al. HAMAP in 2013, new developments in the protein family classification and annotation system. Nucleic Acids Res. 2013;41:D584–D589. doi: 10.1093/nar/gks1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blake J.A., Dolan M., Drabkin H., Hill D.P., Li N., Sitnikov D., Bridges S., Burgess S., Buza T., McCarthy F., et al. Gene Ontology annotations and resources. Nucleic Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnoes A.M., Brown S.D., Dodevski I., Babbitt P.C. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell M.J., Collison M., Lord P. Can inferred provenance and its visualisation be used to detect erroneous annotation? A case study using UniProtKB. PLoS One. 2013;8:e75541. doi: 10.1371/journal.pone.0075541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilks W.R., Audit B., De Angelis D., Tsoka S., Ouzounis C.A. Modeling the percolation of annotation errors in a database of protein sequences. Bioinformatics. 2002;18:1641–1649. doi: 10.1093/bioinformatics/18.12.1641. [DOI] [PubMed] [Google Scholar]
- 9.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku989. doi:10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kersey P.J., Allen J.E., Christensen M., Davis P., Falin L.J., Grabmueller C., Hughes D.S., Humphrey J., Kerhornou A., Khobova J., et al. Ensembl Genomes 2013: scaling up access to genome-wide data. Nucleic Acids Res. 2014;42:D546–D552. doi: 10.1093/nar/gkt979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darrasse A., Carrere S., Barbe V., Boureau T., Arrieta-Ortiz M.L., Bonneau S., Briand M., Brin C., Cociancich S., Durand K., et al. Genome sequence of Xanthomonas fuscans subsp. fuscans strain 4834-R reveals that flagellar motility is not a general feature of xanthomonads. BMC Genomics. 2013;14:761. doi: 10.1186/1471-2164-14-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oakeson K.F., Gil R., Clayton A.L., Dunn D.M., von Niederhausern A.C., Hamil C., Aoyagi A., Duval B., Baca A., Silva F.J., et al. Genome degeneration and adaptation in a nascent stage of symbiosis. Genome Biol. Evol. 2014;6:76–93. doi: 10.1093/gbe/evt210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gattiker A., Michoud K., Rivoire C., Auchincloss A.H., Coudert E., Lima T., Kersey P., Pagni M., Sigrist C.J., Lachaize C., et al. Automated annotation of microbial proteomes in SWISS-PROT. Comput. Biol. Chem. 2003;27:49–58. doi: 10.1016/s1476-9271(02)00094-4. [DOI] [PubMed] [Google Scholar]
- 14.Bucher P., Karplus K., Moeri N., Hofmann K. A flexible motif search technique based on generalized profiles. Comput. Chem. 1996;20:3–23. doi: 10.1016/s0097-8485(96)80003-9. [DOI] [PubMed] [Google Scholar]
- 15.Sigrist C.J., Cerutti L., Hulo N., Gattiker A., Falquet L., Pagni M., Bairoch A., Bucher P. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002;3:265–274. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- 16.Luthy R., Xenarios I., Bucher P. Improving the sensitivity of the sequence profile method. Protein Sci. 1994;3:139–146. doi: 10.1002/pro.5560030118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dayhoff M.O., Schwartz R., Orcutt B.C. Atlas of protein sequence and structure. 1978;5:345–358. [Google Scholar]
- 18.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauve A.A., Wolberger C., Schramm V.L., Boeke J.D. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 20.North B.J., Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004;5:224. doi: 10.1186/gb-2004-5-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J. Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darriba D., Taboada G.L., Doallo R., Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 25.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., Yang K., et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.012658. M111 012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du J., Zhou Y., Su X., Yu J.J., Khan S., Jiang H., Kim J., Woo J., Kim J.H., Choi B.H., et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colak G., Xie Z., Zhu A.Y., Dai L., Lu Z., Zhang Y., Wan X., Chen Y., Cha Y.H., Lin H., et al. Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol. Cell. Proteomics. 12:3509–3520. doi: 10.1074/mcp.M113.031567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu A.Y., Zhou Y., Khan S., Deitsch K.W., Hao Q., Lin H. Plasmodium falciparum Sir2A preferentially hydrolyzes medium and long chain fatty acyl lysine. ACS Chem. Biol. 2011;7:155–159. doi: 10.1021/cb200230x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frye R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 31.Poux S., Magrane M., Arighi C.N., Bridge A., O'Donovan C., Laiho K. Expert curation in UniProtKB: a case study on dealing with conflicting and erroneous data. Database (Oxford) 2014 doi: 10.1093/database/bau016. bau016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bapteste E., Moreira D., Philippe H. Rampant horizontal gene transfer and phospho-donor change in the evolution of the phosphofructokinase. Gene. 2003;318:185–191. doi: 10.1016/s0378-1119(03)00797-2. [DOI] [PubMed] [Google Scholar]
- 33.Moore S.A., Ronimus R.S., Roberson R.S., Morgan H.W. The structure of a pyrophosphate-dependent phosphofructokinase from the Lyme disease spirochete Borrelia burgdorferi. Structure. 2002;10:659–671. doi: 10.1016/s0969-2126(02)00760-8. [DOI] [PubMed] [Google Scholar]
- 34.Chi A., Kemp R.G. The primordial high energy compound: ATP or inorganic pyrophosphate? J. Biol. Chem. 2000;275:35677–35679. doi: 10.1074/jbc.C000581200. [DOI] [PubMed] [Google Scholar]
- 35.Muller M., Lee J.A., Gordon P., Gaasterland T., Sensen C.W. Presence of prokaryotic and eukaryotic species in all subgroups of the PP(i)-dependent group II phosphofructokinase protein family. J. Bacteriol. 2001;183:6714–6716. doi: 10.1128/JB.183.22.6714-6716.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones P., Binns D., Chang H.Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunter S., Jones P., Mitchell A., Apweiler R., Attwood T.K., Bateman A., Bernard T., Binns D., Bork P., Burge S., et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuepbach T., Pagni M., Bridge A., Bougueleret L., Xenarios I., Cerutti L. pfsearchV3: a code acceleration and heuristic to search PROSITE profiles. Bioinformatics. 2013;29:1215–1217. doi: 10.1093/bioinformatics/btt129. [DOI] [PMC free article] [PubMed] [Google Scholar]