

Abstract
The National Center for Biotechnology Information's (NCBI) Gene database (www.ncbi.nlm.nih.gov/gene) integrates gene-specific information from multiple data sources. NCBI Reference Sequence (RefSeq) genomes for viruses, prokaryotes and eukaryotes are the primary foundation for Gene records in that they form the critical association between sequence and a tracked gene upon which additional functional and descriptive content is anchored. Additional content is integrated based on the genomic location and RefSeq transcript and protein sequence data. The content of a Gene record represents the integration of curation and automated processing from RefSeq, collaborating model organism databases, consortia such as Gene Ontology, and other databases within NCBI. Records in Gene are assigned unique, tracked integers as identifiers. The content (citations, nomenclature, genomic location, gene products and their attributes, phenotypes, sequences, interactions, variation details, maps, expression, homologs, protein domains and external databases) is available via interactive browsing through NCBI's Entrez system, via NCBI's Entrez programming utilities (E-Utilities and Entrez Direct) and for bulk transfer by FTP.
INTRODUCTION
The Gene database is a resource of the National Center for Biotechnology Information (NCBI) that centralizes gene-related information into individual records (1). Many different types of gene-specific data are connected to the record including sequence accessions, nomenclature, genomic location and organization, publications, gene products and their attributes, expression, interactions, pathways, homology, variation and its phenotypic consequences and useful links to databases both internal and external to NCBI. From its debut in March 1999 as LocusLink (2), which represented 9112 genes of a single taxon (human), Gene has grown to represent more than 16 million genes for 13 000 taxa in 2014, including viruses, prokaryotes and eukaryotes (Table 1). Inclusion in Gene is based on several criteria, but typically the content corresponds to gene features annotated on genomic sequences represented in NCBI's Reference Sequence (RefSeq) database (3). A unique, database identifier, the GeneID, is assigned to each gene annotation and is tracked over time as genome annotation is updated. Extensive connections between Gene and many other NCBI databases are available based on the GeneID. Note that an annotated genome sequence is not required for inclusion in Gene; a GeneID may be created to represent information such as a phenotype supplied by a model organism database but not yet represented by sequence, for genes of an organism scheduled for future sequencing or for genes that are not yet represented in a draft genome assembly.
Table 1. The number of current records per taxa in Gene.
| Taxa | Number of taxaa | Number of genes |
|---|---|---|
| Archaea | 197 | 406 922 |
| Bacteria | 3124 | 8 496 574 |
| Eukaryota | 5602 | 7 236 920 |
| Viroids | 2 | 4 |
| Viruses | 4217 | 209 402 |
aCounts taken on September 10, 2014.
The content of Gene is derived from both automated dataflows and curation by RefSeq staff. The starting point is typically the extraction of gene-specific information from a publicly available, annotated genome sequence. The gene is assigned a category (e.g. protein coding, non-coding RNA (ncRNA), pseudogene, ribosomal RNA (rRNA), unknown) and added value is provided by connecting the information captured from each gene feature with information from collaborating databases (Table 2), public users and literature review, in particular Gene References into Function (GeneRIFs). When new information is available from any of these sources, the record is updated. Most updates to Gene are processed daily.
Table 2. Gene's connections to public resources.
| Data category | Source | Species | Update frequency |
|---|---|---|---|
| Official nomenclature | HUGO Gene Nomenclature Committee (HGNC) | Human | Daily |
| Mouse Gene Nomenclature Committee (MGNC) | Mouse | Daily | |
| Rat Gene Nomenclature Committee | Rat | Bimonthly | |
| Zebrafish Nomenclature Committee (ZNC) | Zebrafish | Weekly | |
| Chicken Gene Nomenclature Consortium (CGNC) | Chicken | Periodically | |
| FlyBase | Drosophila melanogaster | Data release | |
| MaizeGDB | Zea mays | Periodically | |
| SGD | Saccharomyces cerevisiae | Data release | |
| Xenbase | Xenopus tropicalis, Xenopus laevis | Weekly | |
| The Arabidopsis Information Resource | Arabidopsis | Data release | |
| WormBase | Caenorhabditis elegans | Data release | |
| GeneRIF | Index Section, NLM (and public users) | All | Weekly/Daily |
| HuGE Navigator | Human | Bimonthly | |
| GO terms | Gene Ontology | Several | Weekly |
| Disease Names | OMIM | Human | Daily |
| Interactions | BIND/BOND | Several | Static |
| BioGRID | Several | Monthly | |
| EcoCyc | Escherichia coli | Data release | |
| HPRD | Human | Static | |
| KEGG pathway | BioSystems | Several | Weekly |
| REACTOME | REACTOME | Several | Data release |
WEB ACCESS
The root path to Gene is www.ncbi.nlm.nih.gov/gene/. If a GeneID is known, it can be appended to the root path. Otherwise, Gene is indexed for searching via NCBI's Entrez system (http://www.ncbi.nlm.nih.gov/books/NBK184582/); a global query of the NCBI web site provides a direct link to search results in Gene. A database user may also be directed to Gene from a different NCBI database by a Gene sensor, which recognizes a gene symbol in a database query and provides a link to Gene records with that symbol. Gene sensors are tailored to each NCBI resource being queried. For example, a PubMed query for the gene symbol ‘CFTR’ returns the Gene sensor in addition to publication results. The Gene sensor includes links to view the publications stored in the Gene database for the gene, links to view the Gene record for select organisms and a link to see related test data in the Genetic Testing Registry (GTR) (4).
FTP, E-UTILITIES AND E-DIRECT
A comprehensive database extraction is available for FTP transfer at ftp://ftp.ncbi.nlm.nih.gov/gene/. An extensive README file describes the site contents, which are updated daily with the exception of a small number of special reports. In addition to information on Gene record content, several files are provided to facilitate mapping GeneID identifiers to RefSeq accessions, Ensembl IDs, PubMed IDs and more. The data are also available programmatically using E-Utilities (www.ncbi.nlm.nih.gov/books/NBK25501/) or at the UNIX command line using Entrez Direct (www.ncbi.nlm.nih.gov/books/NBK179288/).
RECORD FORMAT
The Full Report display option is the default setting for an individual Gene record (Figure 1). It integrates text, graphics, keywords and links to other databases, providing the entry point where significant information about a gene can be readily retrieved. The report itself is divided into collapsible sections beginning with the Summary, which includes nomenclature, taxonomic information, a list of other database identifiers and RefSeq status. The right sidebar includes a Table of contents for easy navigation to different sections of the full report, a Related information section with a comprehensive listing of other NCBI databases or reports that reference the gene, and other links to relevant public resources as well as options for providing feedback and corrections. The entire right hand column can now be hidden using the top right ‘Hide sidebar’ button if horizontal screen real-estate is limited. More detail about many sections of the Full Report is described in the Changes to the Full Report section, below.
Figure 1.
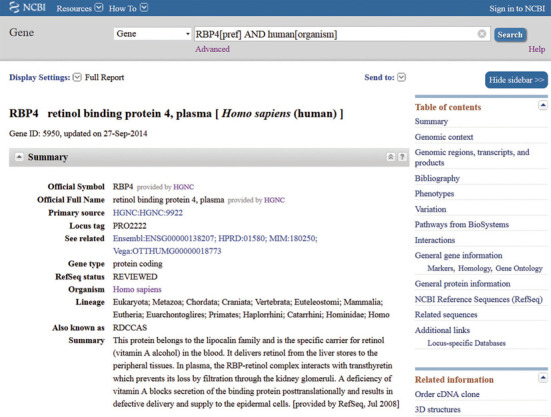
Upper portion of Full Report display in Gene. The record for the human RBP4 gene is shown. The complete report is divided into collapsible sections listed in the Table of contents; only the Summary section is shown. Other report formats are available from the Display settings menu. The ‘Hide sidebar’ button removes the right hand Discovery column from the display. Complete documentation (http://www.ncbi.nlm.nih.gov/books/NBK3841/) is available by following the Help link.
Other Gene report types are available via the Display Settings options (Figure 1). Tabular or Summary formats display multiple query results, and the annotated intron and exon structure represented by RefSeq sequences can be explored using the Gene Table display. The GeneRIF option displays the publication titles, authors and brief descriptive text describing the gene function. Most GeneRIF text is provided by the Medical Subject Headings indexers of the National Library of Medicine, but can also be submitted by public users. XML and ASN.1 formats are also available. More information on each of these formats is provided in the extensive on-line Gene Help documentation (http://www.ncbi.nlm.nih.gov/books/NBK3841/).
NEW FEATURES
Search results
When a search of Gene returns more than one result, they are displayed by default in the new Tabular display option (Figure 2). This format replaces the Summary display as the default format for searches and exposes more data in a compact, structured manner. Reviewing a long list of search results is facilitated by having comparable content aligned. Each row of the Tabular display includes the preferred gene symbol and GeneID, the Description (including the complete gene name and species), the location on a genomic RefSeq for the reference assembly, unofficial symbols and for human only, the Mendelian Inheritance in Man (MIM) number for the gene. The symbol in the Name/GeneID column links to the Full Report display. The Summary format is still available using the Display settings menu.
Figure 2.
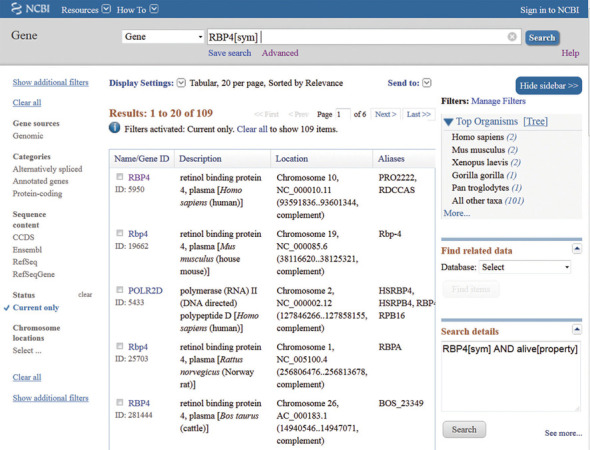
Tabular format view. The Tabular format is the default display setting when a query of Gene returns more than one record. A subset of the 107 records returned from a query using the gene symbol RBP4 is shown. The Name/Gene ID column contains links to each Gene record. Results can be refined further in several ways, including by Organism, by any of the filters in the left column and by user-specified filters managed through MyNCBI. Search result sets can be downloaded using the ‘Send to:’ menu.
For display purposes, columns of the web Tabular display combine several data types. To more easily parse and sort result sets, the format can be changed to Tabular (text) using the Display settings menu. This format includes additional columns and does not combine data types, however, a maximum of 200 records can be returned in this manner. To download all of a larger search result set, use the upper right ‘Send to:’ menu and choose Tabular(text) or another of the available formats.
Filters
To the left of the results table (Figure 2), the Filter sidebar provides a convenient means to refine the search results. It replaces most of the functionality of the previous Limits page. Clicking on a filter activates that filter, which will continue to operate until it is cleared. Searches can be refined by Gene source, Categories, Sequence content, Status, Chromosome locations and all the search fields available in the Advanced Search Builder interface. Only filters that are valid options for the result set are shown. Detailed information on sidebar filters is available (www.ncbi.nlm.nih.gov/books/NBK3841/). The Top Organisms section in the right sidebar provides filtering options by organism for result sets that include records from more than one taxon. Additional filters for Gene (both standard and customized) can be accessed through a user's MyNCBI account.
Advanced search
The Entrez system allows for sophisticated searches of the Gene database. Recent improvements include the ability to query for genes annotated on a specific genome assembly (e.g. GRCh38[assembly name]), genes that have been curated to have potential or known readthrough transcripts (e.g. readthrough[properties]), and genes with matches to VEGA or Ensembl annotation (e.g. ‘matches ensembl’[properties]). Extensive documentation on detailed queries is available (www.ncbi.nlm.nih.gov/books/NBK3841/).
Changes to the full report
Genomic context
This section (Figure 3) provides the location, the number of exons (for eukaryotic taxa), and a graphic of neighboring genes. For genes annotated by NCBI's Eukaryotic Genome Annotation Pipeline (www.ncbi.nlm.nih.gov/books/NBK169439/), the annotated location of a gene on the reference assembly is now conveniently displayed in table format, with a link to NCBI's Assembly database for more information. To facilitate working with previous assembly versions, the sequence coordinates from the last annotation of the previous assembly version may also be listed. The number of exons represents the union of distinct, non-overlapping exons annotated for all RefSeq transcripts of the gene. Information on the neighboring genes for all genomic placements can be obtained by following the ‘Gene neighbors’ link in the Related Information menu.
Figure 3.
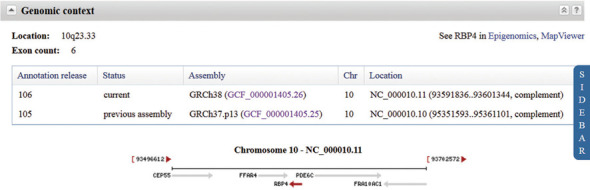
Genomic context section of the Full Report. The section for the human RBP4 gene is shown, with the sidebar collapsed. Information about the location of the gene on the current and previous annotation releases is provided in a table and includes links to NCBI's Assembly database. The graphic displays neighboring genes and their orientation, and each gene symbol links to the corresponding Gene record for easy navigation.
Genomic regions, transcripts and products
This section (Figure 4) provides a detailed, interactive view of the annotated location of a gene and its features on a genomic RefSeq. Multiple coordinate systems (i.e. previous or alternate assemblies and RefSeqGene records; www.ncbi.nlm.nih.gov/refseq/rsg/) may be available from the Genomic Sequence menu for some eukaryotes. Zoom and pan navigation functions are available and many aspects of the display can be customized using the ‘Configure’ button, as described in extensive help documentation (accessible through the ‘?’ icon) as well as the NCBI YouTube channel (www.youtube.com/user/NCBINLM). For example, the default rendering of the Genes track merges all RNA and CDS features together; this can be changed to show all annotated transcripts and proteins using the Genes tab of the Configure Page. This also illustrates a significant change in how vertebrate genes are represented by RefSeq accessions. To provide increased annotation of splice variants, genes may now include a mixture of known RefSeqs (accessions starting with N) and model RefSeqs (accessions starting with X) computed by NCBI's Eukaryotic Genome Annotation Pipeline.
Figure 4.
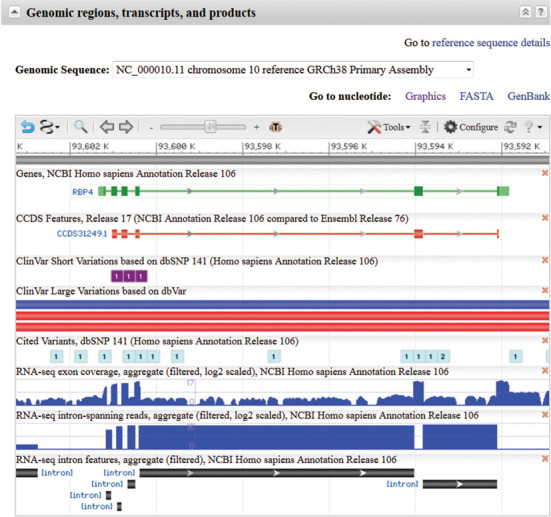
Genomic regions, transcripts and products section of the Full Report. The section for the human RBP4 gene is shown. This is an interactive display of the location of a gene annotated on a genomic RefSeq. More than one genomic RefSeq sequence may be available, and the gene's location on any can be displayed using the drop down menu at the top. Zoom and panning functions are enabled, and the tracks displayed can be configured using the ‘Configure’ button. By default, the Genes track displays a merged rendering of transcripts and coding regions. Complete documentation is available by clicking the ‘?’ icon.
The default display is customized according to the data available for a given organism. For human, the display includes several variation tracks representing short variants from dbSNP (5), longer variants from dbVar (6) and variants of medical relevance from ClinVar (7). A new feature of the display is the addition of aggregate (shown by default) and sample-specific tracks of RNA-seq exon coverage, intron-spanning reads and interpreted intron features. Data are shown based on the subset of RNA-seq data available in the SRA database that was aligned by NCBI's Eukaryotic Genome Annotation Pipeline. Coverage tracks corresponding to reads from individual BioSamples (8) can be loaded from the Expression tab on the Configure Page and provide a visual indication of differential expression in tissues and developmental stages.
Additional tracks are available for some taxa, such as human and mouse, including Consensus CDS (CCDS) features (9). Ensembl (10) gene annotation, paralogous/pseudo gene alignments and additional sequence features such as repeats, CpG islands, a six-frame translation and tracks provided by the Genome Reference Consortium (GRC) (11).
Phenotypes
This section continues to expand support for making connections between variation and phenotype, particularly for human disease. Links to NCBI's Genetic Testing Registry provide information about which test includes the gene and allow a comparison of test providers for any given test. Links to professional guidelines may be provided where variation may be related to a human condition, and as practice guidelines, position statements and recommendations are developed. The Phenotype-Genotype Integrator pieces together information from the National Human Genome Research Institute's (NHGRI) genome-wide association catalog, Gene, NCBI's dbSNP, dbGAP, the Gene-Tissue Expression project (12) and OMIM (13). It allows a researcher to query for variation related to a phenotype, or for a phenotype related to a particular variation or genomic location, and offers tabular and graphic displays to explore the results and an option to download. The remainder of the section may display a) associated conditions with links to more information available in GTR (4), MedGen (www.ncbi.nlm.nih.gov/books/NBK159970/), OMIM (omim.org/) and GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116/), b) evidence of dosage sensitivity as determined by the ClinGen Structural Variant Working Group (www.clinicalgenome.org/data-curation/structural-variant-curation/) and c) information from the NHGRI GWAS Catalog (14).
Variation
This section has been improved for human Gene records. Links to NCBI's new Variation Viewer genome browser resource allow a user to explore variation data in a genomic region in the context of either the GRCh38 or GRCh37.p13 assemblies. The section also includes a convenient link to NCBI's 1000 genomes browser (www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) to explore population variation data from the 1000 Genomes project (15) and links to a subset of variation resources in ClinVar that may be of medical relevance.
Pathways
This section lists the pathways known to include the gene product. Its content is from NCBI's BioSystems (16) database, which centralizes information about pathways from multiple sources, including KEGG (17) and Reactome (18). By following the link to the BioSystem record, all the genes in a pathway can be selected and then viewed in Gene's Tabular display format or saved to a file.
General gene information
This section has been augmented to report more relationship types between genes. A recent addition is the report of orthologous genes calculated by NCBI's Eukaryotic Genome Annotation Pipeline. The process calculates orthology using protein homology and synteny between a genome being annotated by the pipeline and a reference, typically human. ‘Orthologs from Annotation Pipeline’ supplements the content of HomoloGene and provides this type of relationship for newly annotated genomes independently of a HomoloGene build.
In addition, this section now includes information about gene/pseudogene relationships as well as officially named multi-genic regions, such as immunoglobulin loci or a cluster of related genes. For the latter, the ‘Related region members’ link on the Region GeneID (e.g. GeneID 3492) connects to all members, whereas the ‘Related region gene’ link (e.g. GeneID 28444) on each member connects those records to the Region GeneID. The full report of genes and their relationship to other genes is available by FTP: ftp://ftp.ncbi.nih.gov/gene/DATA/gene_group.gz.
FUTURE DIRECTIONS
In the past, Gene was committed to representing annotated genes for all RefSeq genomes; however, the explosion in sequencing for bacteria, viruses and some eukaryotic pathogenic organisms necessitates changes to this data model. The RefSeq project is now defining reference and representative genomes to use as a standard baseline for comparison while continuing to provide genome annotation for all bacterial strains, including disease outbreak isolates, to support surveillance and testing needs (19). Reflecting this change, Gene in the future will focus on content for reference and representative RefSeq genomes for prokaryotes, reducing intra-species record redundancy. NCBI's prokaryotic annotation pipeline is exploring methods to provide GeneID cross references when annotating non-reference/representative genomes to continue to provide access to relevant gene information for those RefSeq genomes. The model for higher eukaryotes is unchanged, and content in Gene is expected to increase as additional higher eukaryotes become available in RefSeq.
In addition, Gene staff are working to add gene and transcript expression information based on RNA-seq data, streamline the Reference Sequence section, incorporate additional curated content for higher eukaryotes (2) and expand the number of annotation tracks available in the graphical display. Future plans include adding Ensembl annotation tracks for more species and incorporating annotation that has been submitted to GenBank and its partners in the International Nucleotide Sequence Database collaboration. These additions will facilitate comparison of different annotation sources to the evidence-based RefSeq annotation produced by NCBI's Eukaryotic Genome Annotation Pipeline.
FEEDBACK
We welcome feedback concerning the interface to Gene and the contents of the database. Please use one of the Feedback options on the right side of any Gene record.
FUNDING
Intramural Research Program of the National Institutes of Health, National Library of Medicine. Funding for open access charge: Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1.Maglott D., Ostell J., Pruitt K.D., Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39:D52–D57. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maglott D.R., Katz K.S., Sicotte H., Pruitt K.D. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M., et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubinstein W.S., Maglott D.R., Lee J.M., Kattman B.L., Malheiro A.J., Ovetsky M., Hem V., Gorelenkov V., Song G., Wallin C., et al. The NIH genetic testing registry: a new, centralized database of genetic tests to enable access to comprehensive information and improve transparency. Nucleic Acids Res. 2013;41:D925–D935. doi: 10.1093/nar/gks1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lappalainen I., Lopez J., Skipper L., Hefferon T., Spalding J.D., Garner J., Chen C., Maguire M., Corbett M., Zhou G., et al. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2013;41:D936–D941. doi: 10.1093/nar/gks1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrett T., Clark K., Gevorgyan R., Gorelenkov V., Gribov E., Karsch-Mizrachi I., Kimelman M., Pruitt K.D., Resenchuk S., Tatusova T., et al. BioProject and BioSample databases at NCBI: facilitating capture and organization of metadata. Nucleic Acids Res. 2012;40:D57–D63. doi: 10.1093/nar/gkr1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farrell C.M., O'Leary N.A., Harte R.A., Loveland J.E., Wilming L.G., Wallin C., Diekhans M., Barrell D., Searle S.M., Aken B., et al. Current status and new features of the Consensus Coding Sequence database. Nucleic Acids Res. 2014;42:D865–D872. doi: 10.1093/nar/gkt1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flicek P., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Church D.M., Schneider V.A., Graves T., Auger K., Cunningham F., Bouk N., Chen H.C., Agarwala R., McLaren W.M., Ritchie G.R., et al. Modernizing reference genome assemblies. PLoS Biol. 2011;9:e1001091. doi: 10.1371/journal.pbio.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Consortium T.G. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amberger J., Bocchini C.A., Scott A.F., Hamosh A. McKusick's Online Mendelian Inheritance in Man (OMIM) Nucleic Acids Res. 2009;37:D793–D796. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geer L.Y., Marchler-Bauer A., Geer R.C., Han L., He J., He S., Liu C., Shi W., Bryant S.H. The NCBI BioSystems database. Nucleic Acids Res. 2010;38:D492–D496. doi: 10.1093/nar/gkp858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croft D., Mundo A.F., Haw R., Milacic M., Weiser J., Wu G., Caudy M., Garapati P., Gillespie M., Kamdar M.R., et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42:D472–D477. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatusova T., Ciufo S., Fedorov B., O'Neill K., Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014;42:D553–D559. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]