


Abstract
The human genome is pervasively transcribed, producing thousands of non-coding RNA transcripts. The majority of these transcripts are long non-coding RNAs (lncRNAs) and novel lncRNA genes are being identified at rapid pace. To streamline these efforts, we created LNCipedia, an online repository of lncRNA transcripts and annotation. Here, we present LNCipedia 3.0 (http://www.lncipedia.org), the latest version of the publicly available human lncRNA database. Compared to the previous version of LNCipedia, the database grew over five times in size, gaining over 90 000 new lncRNA transcripts. Assessment of the protein-coding potential of LNCipedia entries is improved with state-of-the art methods that include large-scale reprocessing of publicly available proteomics data. As a result, a high-confidence set of lncRNA transcripts with low coding potential is defined and made available for download. In addition, a tool to assess lncRNA gene conservation between human, mouse and zebrafish has been implemented.
INTRODUCTION
Over the past decade long non-coding RNAs (lncRNAs) have emerged as a large class of functional non-coding RNAs (ncRNAs) (1). Defined as ncRNA transcripts longer than 200 nucleotides, lncRNAs have been shown to function mainly as transcriptional regulators by interaction with other biomolecules, such as proteins (2–4) and microRNAs (5). They are involved in a wide range of processes including cardiac development (6), dosage compensation (7,8) and cancer (2,9–10). Several specialist databases concerning lncRNA have been developed. Well-known examples are lncRNAdb, which focuses on lncRNAs with described functions (11), and NONCODE (12,13). In addition to these general lncRNA databases, databases that describe specific lncRNA subclasses have been compiled as well. LncRNAdisease contains lncRNAs with published disease associations (14) while lncRNAs targeted by microRNAs can be found in DIANA-LncBase (15).
Distinguishing coding from ncRNA sequences is an important step, both in the ncRNA and the protein research field. Classic approaches are based on either open reading frame (ORF) length, ORF conservation or structural protein domains (16). Recent computational methods make use of more complex features or machine learning approaches. Notable examples are the Coding-Potential Calculator (CPC), Coding-Potential Assessment Tool (CPAT) and PhyloCSF. CPC utilizes a support vector machine trained on features that describe long, high-quality ORFs with sequence similarity (BLASTX) to known proteins (17). CPAT is a logistic regression model that only uses sequence-derived features, such as ORF size, codon and hexamer usage bias (18). In contrast to CPC and CPAT, PhyloCSF employs codon substitution frequencies in whole-genome multi-species alignments and maximum likelihood trees to distinguish between coding and non-coding loci (19).
ORF length is either directly or indirectly used in all these computational prediction methods yet ORFs yielding short peptides (<100 amino acids) are difficult to predict. The discovery of functional peptides shorter than 100 amino acids, like the Drosophila gene tarsal-less (tal), thus raised the possibility that several lncRNAs are actually misclassified protein-coding genes encoding micropeptides (20,21). As small ORFs can also occur by chance in long transcripts, many well-described lncRNAs harbor non-functional ORFs (22). In addition to small ORFs, the in silico prediction of coding ORFs is further complicated by the existence of non-canonical (non-AUG) start codons (23).
Experimental procedures to detect translated ORFs and their products have been developed as well. One such method is referred to as ribosome profiling and is based on deep sequencing of ribosome-protected mRNA fragments. Although many ncRNAs show ribosome occupancy, by using initiation-specific translation inhibitors in combination with ribosome profiling, researchers were able to map translation initiation sites (TIS) with base pair resolution and improve the detection of true ORFs (23,24). Other researchers were able to use the periodicity of ribosome movement on the mRNA to define actively translated ORFs (25). In addition to ribosome profiling, mass spectrometry has been applied in the search for novel peptides arising from lncRNAs (26,27). Several authors report small numbers of (micro) peptides arising from lncRNAs using either ribosome profiling or mass spectrometry. The debate on the putative function and total number of these peptides is still ongoing (26–28).
Here, we report on LNCipedia 3.0, the latest version of our publically available lncRNA database. In version 3.0, our major improvement is the evaluation of protein-coding potential with state-of-the-art algorithms and data sets. As such we have generated a high-confidence data set that excludes lncRNAs with possible protein-coding potential. In addition, a new tool to assess the conservation of lncRNA genes has been implemented. The database content has been updated and now contains over five times the number of transcripts compared to the first version.
MATERIALS AND METHODS
Locus conservation
The upstream and downstream protein-coding genes that flank a human lncRNA gene are queried in the public Ensembl (29) MySQL database (version 73). For both genes, the orthologs in mouse and zebrafish are obtained using the Ensembl Compara API (version 73). If any pair of orthologs are neighboring genes, the locus is reported as conserved.
PhyloCSF
Whole-genome alignments of 46 species are obtained from the UCSC website (30) and processed using the PHAST (31) package (version 1.3) to obtain the required input format for PhyloCSF (19). To validate our workflow, we benchmarked PhyloCSF with transcripts annotated in Ensembl (version 75). Transcripts with biotype ‘lincRNA’ or ‘antisense’ (20 320 transcripts) serve as negative set while transcripts with biotype ‘protein_coding’ and an annotated coding sequence (36 959 transcripts) serve as positive set.
TIS
Ribosome profiling sequencing data of HEK-293 cells treated with cycloheximide (CHX) and lactimidomycin (LTM) were processed (24). Two technical replicates of both treatments were pooled (Bioproject http://www.ncbi.nlm.nih.gov/bioproject/PRJNA171327: runs SRR618770 and SRR618771 for CHX and runs SRR618772 and SRR618773 for LTM).
The reads were first clipped to remove their 3′ cloning adaptor sequence using the FASTX-Toolkit (fastx_clipper tool). Unclipped and clipped reads shorter than 25 nt were discarded. The remaining reads were mapped using the RNA-seq STAR aligner (32), sequentially using indices based on the following sequences: (i) Phix genome (widely used as a quality control for Illumina sequencing runs), (ii) Homo sapiens rRNA (Refseq IDs NR_003285.2, NR_003286.1, NR_003287.1, NR_023363.1) and (iii) the human reference genome (downloaded from the igenomes repository http://support.illumina.com/sequencing/sequencing_software/igenome.ilmn, using the H. sapiens genome build GRCh37 and Ensembl annotation version 70). The human STAR index was built taking into account the splice site annotation from Ensembl. Only uniquely mapped reads that are between 28 and 35 nt long were retained. Footprint alignments were assigned to a specific P-site nucleotide based on the fragment length (the 5′ offset is set to respectively 12, 13 or 14 for profiles with length ≤ 30 nt, 31–33 nt, or ≥ 34 nt (23)).
PRoteomics IDEntifications (PRIDE) reprocessing
The processing pipeline consists of three major modules. The first module is based on the PRIDE automated spectrum annotation pipeline (pride-asap) (33), and is used to reverse engineer the original search parameters from submitted data. The key parameters extracted by pride-asap in this stage are the allowable mass errors, the post-translational modifications (PTMs) to consider, and the enzyme used. Recent developments in this module have greatly improved the PTM inference by considering the modifications found in the PSI-mod (34) and Unimod (35) databases, as well as the frequency of occurrence of these modifications. Two thresholds are calculated based on this information, with the first one serving as a lower threshold to exclude very low abundance modifications while the second threshold is used to determine whether a sufficiently abundant modification is to be considered as either variable or fixed. A second development has been the impromptu determination of the protease used in the original experiment. Instead of assuming the use of trypsin, the pride-asap module now calculates the most likely enzyme based on all reported peptide sequences reported in PRIDE for that experiment. Overall, these updates to the module allow a reduction in search space to consider, providing faster processing times and leaving less room for false-positive matches.
The second module handles the peptide-to-spectrum matching, relying on SearchGUI (36) to automatically run multiple search engines in parallel; in this case OMSSA (37) and X!Tandem (38). SearchGUI is configured to use the target/decoy approach (39), where both the original (target) sequence database is searched, but also a reversed (decoy) version of that database. Matches from the latter can then be used to determine a false discovery rate (FDR) (39).
The third and final module uses PeptideShaker (http://peptide-shaker.googlecode.com) and the compomics-utilities library (40) to collect, process and analyze the results generated by SearchGUI.
RESULTS
LNCipedia 3.0 content
LNCipedia 1.0 (41) combined sequences and annotation from three different public resources, namely, Ensembl (29,42), Human body map lincRNAs (43) and the lncRNA database (11). In LNCipedia version 3.0, we have complemented these resources with four additional public data sets (Table 1). Two of these data sets are obtained from databases (44,45), and two from lncRNA research articles describing RNA sequencing workflows and reporting on novel lncRNAs (46,47). As with LNCipedia 1.0, redundant transcripts are merged into the same record. The result of this extension and integration of sources is that LNCipedia 3.0 represents a more than 5-fold increase in transcript content over version 1.0 (Figure 1). The majority of these transcripts (80%) is found in new loci and as such give rise to novel lncRNA genes.
Table 1. Overview of data sources contributing to lncRNA content in LNCipedia 3.0.
Figure 1.
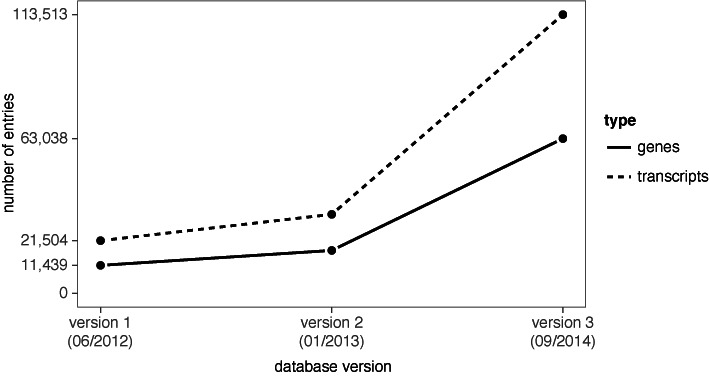
LNCipedia has grown substantially since its first release. The first version (41) was based on sequences and annotation from three different sources and was made available to the public in 2012. For the 2013 release of LNCipedia (unpublished), no additional sources were used, but the different sources were updated to the most recent version. For version 3.0 of LNCipedia, both new sources were added and existing sources were updated.
In LNCipedia 1.0 we introduced a universal lncRNA nomenclature to overcome the confusion caused by the use of different identifiers by different authors and databases. As was suggested by others, we named lncRNAs after neighboring protein-coding genes on the same strand (48). In LNCipedia 3.0, we hold true to this strategy. Existing genes are expanded when novel transcripts have overlapping exons and new genes are created when a transcript does not share exonic sequence with any existing gene.
Locus conservation
The identification of orthologous lncRNAs is an important step for animal modeling and functional research across species. Conservation of gene order is a straightforward metric often used in comparative genomics. We applied the concept of gene order conservation to determine the orthologous locus of a lncRNA in another species. Using the Ensembl Compara API, we have assessed the conservation in the order of the flanking protein-coding genes. Currently, orthologs for non-coding genes are not as well annotated as for protein-coding genes, flanking non-coding genes were therefore not taken into account. When the order is conserved in mouse or zebrafish we report the locus as conserved. In this way, we find locus conservation for 55% of the human lncRNA genes in mouse, and for 27% in zebrafish (Figure 2). The majority of the conserved loci in zebrafish are also conserved in mouse, as one would expect. While locus conservation is no proof for the functional conservation of the lncRNA itself, it may serve a first step in finding the orthologous lncRNA.
Figure 2.
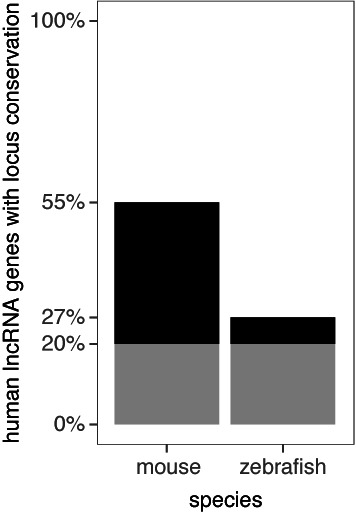
Many lncRNA loci are conserved in mouse or zebrafish. Locus conservation is a novel tool to determine the orthologous locus of a human lncRNA in another species. When the order of the flanking protein-coding genes is conserved in another species, the lncRNA locus is considered conserved. The majority of the conserved loci in zebrafish are also conserved in mouse, this fraction is depicted in gray.
Protein-coding potential
For collection of lncRNA transcript sequences, we rely on public data sets that are often contaminated with small numbers of transcripts harboring coding ORFs (25,26). While we already presented several measures to assess this problem (41), we further expanded these with state-of-the-art tools and included additional lncRNA transcript data sets. One such measure is the PhyloCSF (19) score. We have benchmarked PhyloCSF using Ensembl transcripts and we have determined 41 as an optimal threshold for the PhyloCSF score resulting in a precision of 95% and sensitivity of 91% (Supplemental Material and Figures). From the empirical cumulative distribution (Figure 3a) it is apparent that LNCipedia most likely contains a considerable fraction of protein-coding sequences. When applying our pre-computed cutoff, these transcripts add up to about 26% of the collection. Figure 3c shows the distribution of these putative coding transcripts among the different sources used for LNCipedia. It is clear that some lncRNA data sets suffer more from contamination of coding sequences than others. Strikingly, nearly 50% of Refseq annotated non-coding sequences are predicted to be coding according to the PhyloCSF score cutoff. It is no surprise that the lowest number of coding sequences is observed in Cabili et al. and Hangauer et al. as these studies applied PhyloCSF as a filter in their workflow.
Figure 3.
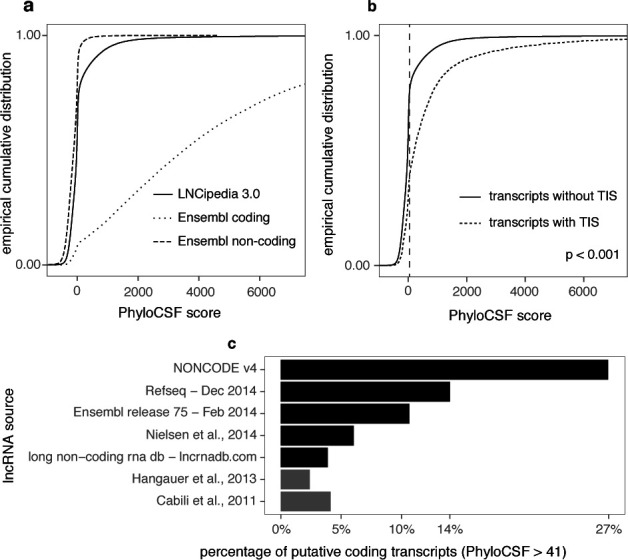
Different methods suggest contamination of coding sequences in lncRNA data sets. (a) PhyloCSF benchmarking and score distributions. We can observe a considerable difference between the score distributions of coding and non-coding transcripts in the Ensembl data set. In addition, while the great majority of LNCipedia is presumably non-coding, it also contains a fraction of transcripts with a PhyloCSF score in the coding range. (b) Transcripts with a TIS have a significantly higher PhyloCSF score (Mann–Whitney U test) compared to other transcripts. (c) Several public lncRNA resources suffer from considerable contamination with protein-coding sequences. The percentage of transcripts with PhyloCSF score greater than 41 is shown for the different sources in LNCipedia 3.0. Two sources already filtered with PhyloCSF are depicted in gray. In the case of RefSeq, only entries with property “biomol_ncrna_lncrna” were considered.
Another measure to assess protein-coding potential is the use of ribosome profiling to map TIS. When we map the TIS observed in HEK-293 (24) to LNCipedia entries, we find 4154 trancripts with at least one TIS. Of note, these transcripts have significantly higher PhyloCSF scores (Figure 3b), which is a good validation of both methods.
PRIDE
Similar to the rapid growth of LNCipedia, the submission of mass spectrometry data to the PRIDE repository has flourished as well (49). While these increased collections of lncRNAs and mass spectrometry data provide even more means to detect potentially coding lncRNAs, they also require much more compute power to process. The only way to analyze these data in a timely fashion is to make use of parallelization on a compute cluster or through grid computing (50). We have therefore set up such a grid environment based on dedicated hardware running a collection of Linux virtual machines, allowing us to re-analyze the full human complement of PRIDE in under a week.
At the time of writing, the pipeline has been run on 2493 PRIDE experiments, containing 39 463 035 fragmentation mass spectra and covering all 68 annotated human tissues in the public repository. This resulted in a total of 8 064 657 peptide-to-spectrum matches (PSMs), of which 747 305 were matched to lncRNAs in LNCipedia (393 859 matched the target database and 353 446 matched the decoy database). Of these PSMs, 18 929 target sequences (representing 2040 transcripts, from 1770 genes) had an identification confidence higher than 90% (in contrast to only 2001 decoy sequences that had such a high confidence). Of note, the estimation of the FDR remains a complex issue in these very broad searches (51,52), and care should be taken to interpret these results. Indeed, as supplementary Figures S1 and S2 illustrate, while the confidence compares reasonably well with the estimated FDR, especially at higher confidences (higher than 90%), the evolution of the FDR toward the higher confidences is very different between the UniProtKB-SwissProt-derived identifications and the lncRNA matches.
No significantly higher PhyloCSF score was found for transcripts containing PSMs with identification confidence higher than 90%. In addition, no significant overlap is observed between the set of transcripts identified in PRIDE and the sets containing TIS and smORFs. This observation illustrates the very unique nature of the PRIDE analysis and strongly suggests its ability to detect coding potential not predicted by other methods.
HIGH-CONFIDENCE SET
Since LNCipedia contains a non-negligible number of putative coding transcripts, we propose a filtering strategy to create a stringent or high-confidence data set. Four groups of putative coding transcripts are removed (Figure 4, Supplementary Figure S3). The first group consists of 253 lncRNAs containing small ORFs (smORFs) (25). Bazzini et al. developed an approach to detect smORFs using ribosome profiling whereby the periodicity of ribosome movement on actively translated ORFs is used to distinguish coding from non-coding sequences. A second approach to apply ribosome profiling in the quest for novel coding RNAs has been described by Lee et al. (24). Using LTM, a ribosome inhibitor specific to initiating ribosomes, TIS were mapped in HEK-293 cells. Note that 4127 lncRNA transcripts containing at least one TIS are thus withdrawn. While these transcripts have a good change to give rise to peptides, it is important to consider that a negative result does not guarantee the opposite. The transcript may not be expressed or translated in the sample. The next filtering step is based on PhyloCSF (19). As discussed earlier, this algorithm can distinguish between coding and non-coding sequences with high accuracy. As such, 27 293 transcripts with a PhyloCSF score higher than 41 are discarded. Finally, the 2040 PSM containing transcripts from the PRIDE reprocessing pipeline are excluded as well. The resulting set of 80 216 transcripts (71% of LNCipedia 3.0) representing 48 028 genes (76%) is referred to as ‘high-confidence set’ and is available for download on the LNCipedia website.
Figure 4.
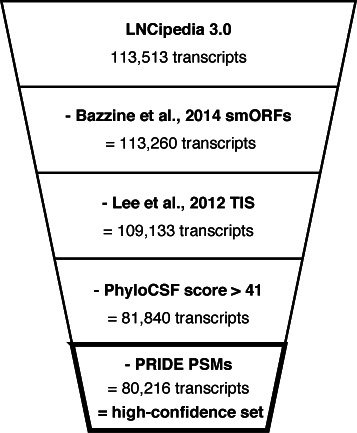
Transcripts with a likely coding potential are removed in the definition of a high-confidence set. Transcripts containing small ORFs (25), TIS (24), PhyloCSF score greater than 41 or PSMs with an identification confidence higher than 90% are excluded.
CONCLUSION AND FUTURE DIRECTION
With over 90 000 new transcripts, LNCipedia content increased 5-fold since its first publication in 2012. This makes it to our knowledge the largest publicly available human lncRNA resource. Furthermore, we improved the evaluation of coding potential with state-of-the-art algorithms, published data sets and an improved PRIDE reprocessing pipeline. In addition, we have developed a locus conservation analysis tool, which can aid in the search for lncRNA orthologs or prioritarization of lncRNAs for animal studies.
As in the previous years, LNCipedia will be updated when new lncRNA data sets are available. With the arrival of a new human reference genome (GRCh38), an important improvement to the database will be remapping chromosomal positions to this new reference genome. We will also continue to automatically run searches against the ever-growing contents of the PRIDE database on a routine basis. Furthermore, we will improve the specificity of the PRIDE searches by taking possible contamination from viral sequences into account.
In conclusion, LNCipedia 3.0 provides significant improvements over the previous version in terms of data content and data annotation.
AVAILABILITY
LNCipedia 3.0 can be accessed trough a web interface at www.lncipedia.org. Exports are available in FASTA, GFF, GTF or BED format for both the entire lncRNA collection and the high-confidence set. In addition, Integrative Genome Viewer (IGV) users have the option of loading an IGV optimized data set directly in the application. As in version 1.0, the database can be queried by chromosomal position or (partial) sequence. We encourage the lncRNA research community to contribute to LNCipedia by submitting newly discovered lncRNAs and by adding PubMed literature records to existing entries using the web interface.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to acknowledge Jasper Anckaert, Stephanie Letellier and Justine Nuytens for their contribution in the development of this LNCipedia version.
FUNDING
Multidisciplinary Research Partnership ‘Bioinformatics: From Nucleotides to Networks’ Project of Ghent University [01MR0310W to P.V. and K. Vandepoele]; Fund for Scientific Research Flanders [FWO; to P.M. and G.M.]; The European Union 7th Framework Program ‘PRIME-XS’ [262067 to L.M.]; Ghent University [to K. Verheggen]. Funding for open access charge: Multidisciplinary Research Partnership ‘Bioinformatics: From Nucleotides to Networks’ Project of Ghent University [01MR0310W].
Conflict of interest statement. None declared.
REFERENCES
- 1.Mercer T., Dinger M. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 2.Gupta R.A., Shah N., Wang K.C., Kim J., Horlings H.M., Wong D.J., Tsai M.-C., Hung T., Argani P., Rinn J.L., et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Margueron R., Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai M.-C., Manor O., Wan Y., Mosammaparast N., Wang J.K., Lan F., Shi Y., Segal E., Chang H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cesana M., Cacchiarelli D., Legnini I., Santini T., Sthandier O., Chinappi M., Tramontano A., Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Gastroenterology. 2011;147:358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klattenhoff C.A., Scheuermann J.C., Surface L.E., Bradley R.K., Fields P.A., Steinhauser M.L., Ding H., Butty V.L., Torrey L., Haas S., et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Gastroenterology. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penny G.D., Kay G.F., Sheardown S.A., Rastan S., Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379:131–137. doi: 10.1038/379131a0. [DOI] [PubMed] [Google Scholar]
- 8.Lee J.T., Davidow L.S., Warshawsky D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 1999;21:400–404. doi: 10.1038/7734. [DOI] [PubMed] [Google Scholar]
- 9.Panzitt K., Tschernatsch M.M.O., Guelly C., Moustafa T., Stradner M., Strohmaier H.M., Buck C.R., Denk H., Schroeder R., Trauner M., et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology. 2007;132:330–342. doi: 10.1053/j.gastro.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 10.Gibb E.A., Brown C.J., Lam W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer. 2011;10:38. doi: 10.1186/1476-4598-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amaral P.P., Clark M.B., Gascoigne D.K., Dinger M.E., Mattick J.S. lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 2010;39:D146–D151. doi: 10.1093/nar/gkq1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu C., Bai B., Skogerbø G., Cai L., Deng W., Zhang Y., Bu D., Zhao Y., Chen R. NONCODE: an integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 2005;33:D112–D115. doi: 10.1093/nar/gki041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bu D., Yu K., Sun S., Xie C., Skogerbo G., Miao R., Xiao H., Liao Q., Luo H., Zhao G., et al. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2011;40:D210–D215. doi: 10.1093/nar/gkr1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen G., Wang Z., Wang D., Qiu C., Liu M., Chen X., Zhang Q., Yan G., Cui Q. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2012;41:D983–D986. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Reczko M., Maragkakis M., Dalamagas T.M., Hatzigeorgiou A.G. DIANA-LncBase: experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013;41:D239–D245. doi: 10.1093/nar/gks1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinger M.E., Pang K.C., Mercer T.R., Mattick J.S. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput. Biol. 2008;4:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong L., Zhang Y., Ye Z.Q., Liu X.Q., Zhao S.Q., Wei L., Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L., Park H.J., Dasari S., Wang S., Kocher J.-P., Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin M.F., Jungreis I., Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:I275–I282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galindo M.I., Pueyo J.I., Fouix S., Bishop S.A., Couso J.P. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 2007;5:e106. doi: 10.1371/journal.pbio.0050106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crappé J., Van Criekinge W., Menschaert G. Little things make big things happen: a summary of micropeptide encoding genes. EuPA Open Proteom. 2014;3:128–137. [Google Scholar]
- 22.Dinger M.E., Gascoigne D.K., Mattick J.S. The evolution of RNAs with multiple functions. Biochimie. 2011;93:2013–2018. doi: 10.1016/j.biochi.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 23.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S., Liu B., Lee S., Huang S.-X., Shen B., Qian S.-B. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E2424–E2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bazzini A.A., Johnstone T.G., Christiano R., Mackowiak S.D., Obermayer B., Fleming E.S., Vejnar C.E., Lee M.T., Rajewsky N., Walther T.C., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014;33:981–993. doi: 10.1002/embj.201488411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gascoigne D.K., Cheetham S.W., Cattenoz P.B., Clark M.B., Amaral P.P., Taft R.J., Wilhelm D., Dinger M.E., Mattick J.S. Pinstripe: a suite of programs for integrating transcriptomic and proteomic datasets identifies novel proteins and improves differentiation of protein-coding and non-coding genes. Bioinformatics. 2012;28:3042–3050. doi: 10.1093/bioinformatics/bts582. [DOI] [PubMed] [Google Scholar]
- 27.Slavoff S.A., Mitchell A.J., Schwaid A.G., Cabili M.N., Ma J., Levin J.Z., Karger A.D., Budnik B.A., Rinn J.L., Saghatelian A. Peptidomic discovery of short open reading frame–encoded peptides in human cells. Nat. Chem. Biol. 2012;9:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chew G.-L., Pauli A., Rinn J.L., Regev A., Schier A.F., Valen E. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 2013;140:2828–2834. doi: 10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubbard T., Barker D., Birney E., Cameron G., Chen Y., Clark L., Cox T., Cuff J., Curwen V., Down T., et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hubisz M.J., Pollard K.S., Siepel A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief. Bioinformat. 2011;12:41–51. doi: 10.1093/bib/bbq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hulstaert N., Reisinger F., Rameseder J., Barsnes H., Vizcaíno J.A., Martens L. Pride-asap: automatic fragment ion annotation of identified PRIDE spectra. J. Proteom. 2013;95:89–92. doi: 10.1016/j.jprot.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montecchi-Palazzi L., Beavis R., Binz P.-A., Chalkley R.J., Cottrell J., Creasy D., Shofstahl J., Seymour S.L., Garavelli J.S. The PSI-MOD community standard for representation of protein modification data. Nat. Biotechnol. 2008;26:864–866. doi: 10.1038/nbt0808-864. [DOI] [PubMed] [Google Scholar]
- 35.Creasy D.M., Cottrell J.S. Unimod: protein modifications for mass spectrometry. Proteomics. 2004;4:1534–1536. doi: 10.1002/pmic.200300744. [DOI] [PubMed] [Google Scholar]
- 36.Vaudel M., Barsnes H., Berven F.S., Sickmann A., Martens L. SearchGUI: an open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 2011;11:996–999. doi: 10.1002/pmic.201000595. [DOI] [PubMed] [Google Scholar]
- 37.Geer L.Y., Markey S.P., Kowalak J.A., Wagner L., Xu M., Maynard D.M., Yang X., Shi W., Bryant S.H. Open mass spectrometry search algorithm. J. Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 38.Fenyö D., Beavis R.C. A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 2003;75:768–774. doi: 10.1021/ac0258709. [DOI] [PubMed] [Google Scholar]
- 39.Vaudel M., Sickmann A., Martens L. Current methods for global proteome identification. Expert Rev Proteomics. 2012;9:519–532. doi: 10.1586/epr.12.51. [DOI] [PubMed] [Google Scholar]
- 40.Barsnes H., Vaudel M., Colaert N., Helsens K., Sickmann A., Berven F.S., Martens L. Compomics-utilities: an open-source Java library for computational proteomics. BMC Bioinformat. 2011;12:70. doi: 10.1186/1471-2105-12-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volders P.-J., Helsens K., Wang X., Menten B., Martens L., Gevaert K., Vandesompele J., Mestdagh P. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013;41:D246–D251. doi: 10.1093/nar/gks915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flicek P., Ahmed I., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., et al. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cabili M.N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A., Rinn J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M., et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie C., Yuan J., Li H., Li M., Zhao G., Bu D., Zhu W., Wu W., Chen R., Zhao Y. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2013;42:D98–D103. doi: 10.1093/nar/gkt1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen M.M., Tehler D., Vang S., Sudzina F., Hedegaard J., Nordentoft I., Orntoft T.F., Lund A.H., Pedersen J.S. Identification of expressed and conserved human noncoding RNAs. RNA. 2014;20:236–251. doi: 10.1261/rna.038927.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hangauer M.J., Vaughn I.W., McManus M.T. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9:e1003569. doi: 10.1371/journal.pgen.1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P., et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vizcaíno J.A., Côté R.G., Csordas A., Dianes J.A., Fabregat A., Foster J.M., Griss J., Alpi E., Birim M., Contell J., et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verheggen K., Barsnes H., Martens L. Distributed computing and data storage in proteomics: many hands make light work, and a stronger memory. Proteomics. 2014;14:367–377. doi: 10.1002/pmic.201300288. [DOI] [PubMed] [Google Scholar]
- 51.Vaudel M., Burkhart J.M., Sickmann A., Martens L., Zahedi R.P. Peptide identification quality control. Proteomics. 2011;11:2105–2114. doi: 10.1002/pmic.201000704. [DOI] [PubMed] [Google Scholar]
- 52.Colaert N., Degroeve S., Helsens K., Martens L. Analysis of the resolution limitations of peptide identification algorithms. J. Proteome Res. 2011;10:5555–5561. doi: 10.1021/pr200913a. [DOI] [PubMed] [Google Scholar]