


Abstract
NCBI RefSeq genome collection http://www.ncbi.nlm.nih.gov/genome represents all three major domains of life: Eukarya, Bacteria and Archaea as well as Viruses. Prokaryotic genome sequences are the most rapidly growing part of the collection. During the year of 2014 more than 10 000 microbial genome assemblies have been publicly released bringing the total number of prokaryotic genomes close to 30 000. We continue to improve the quality and usability of the microbial genome resources by providing easy access to the data and the results of the pre-computed analysis, and improving analysis and visualization tools. A number of improvements have been incorporated into the Prokaryotic Genome Annotation Pipeline. Several new features have been added to RefSeq prokaryotic genomes data processing pipeline including the calculation of genome groups (clades) and the optimization of protein clusters generation using pan-genome approach.
INTRODUCTION
As of October 2014 the RefSeq prokaryotic genome dataset (1,2) contains more than 28 000 genomes from over 5000 species representing a wide range of organisms. They include many important human pathogens, but also organisms that are of interest for non-medical reasons, biodiversity, epidemiology and ecology. Prokaryotic genomes differ in size, genome organization (variable number of chromosomes and plasmid) and nucleotide composition. There is almost a 20-fold range of genome sizes, spanning from the ultra-small 45-kb archaeal genome of Candidatus Parvarchaeum acidiphilum recently obtained from the mine drainage metagenome project (3) to the largest (14.7 Mb) strain of Sorangium cellulosum, alkaline-adaptive epothilone producer (4). The RefSeq prokaryotic genome collection represents assembled genomes with different levels of quality and sampling density. Largely because of the interest in human pathogens and advances in sequencing technologies (5), there are rapidly growing sets (Figure 1) of very closely related genomes representing variations within the species.
Figure 1.
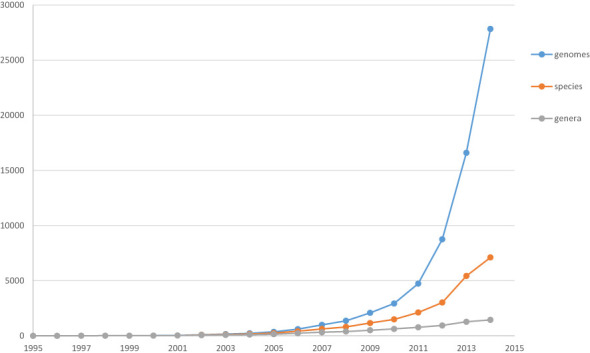
Growth of genomes, species and genera: rapid growth of the number of isolates with relatively slow growth of new genera. Note that the data does not include assemblies from environmental studies where the number of novel species is growing much faster.
The genome annotation submitted to GenBank is done by different methods and is often inconsistent even in closely related genomes with a good reference such as Escherichia coli (6). Many bacterial genomes are submitted to GenBank without annotation. In order to provide consistent annotation and support pan-genome analysis of bacterial populations RefSeq has changed the scope of prokaryotic genome project to include all genomes submitted to public archives that pass minimum quality control (2).
GENOME PROCESSING PIPELINE
How to define a genome?
From the very beginning genome sequencing efforts were aimed at representing the diversity of microbial organisms and sequencing one complete genome from each species. For many years a genome record in GenBank was uniquely identified by NCBI Taxonomy ID. Next Generation Sequencing (NGS) changed the way the genome sequencing is done. A genome is represented not by a single complete chromosome but by a collection of contigs and scaffolds—a genome assembly. Many genome sequencing projects aim to represent a population by sequencing hundreds and even thousands of bacterial genomes.
NCBI has developed several databases (7) (BioSample, BioProject, Assembly) to better manage and link the information about the project, organism, sample and genome sequences. The users of GenBank and RefSeq data can sometimes be confused with many different identifiers introduced by NCBI and ask for a single unique identifier that can be used to retrieve a collection of sequences representing a single genome. BioProject ID can no longer define a genome for many multi-isolate and multi-species projects. Historically, BioPorject (former Genome Project) database was design to register and track genome sequencing projects, capture metadata, assign unique locus-tag prefix for the annotation. A single BioProject used to represent a single genome of one organism. More recently the scope of project types has been expanded to include transcriptome, exome, proteome and other non-genome projects and allow multi-isolate and multi-species project types. In the current data model a single BioPorject can contain hundreds of genomes from the clinical isolates or population studies.
Taxonomy ID can no longer define a genome since unique taxid is no longer assigned for individual strains and isolates (8). The collection of DNA sequences of an individual sample (isolate) will be represented by unique BioSample ID and if raw sequence reads are assembled and submitted to GenBank they will get a unique assembly accession.
Genome representation
The source of the genomic sequence in the RefSeq collection is a primary sequence record in the International Nucleotide Sequence Database Consortium, INSDC, public archives (9). Genomic sequences (nucleotide) in prokaryotic RefSeqs are identical copies of the underlying primary INSDC records. All assemblies with full representation of the genome of a single organism that pass validation quality are taken into RefSeq. Genome assembly quality validation criteria are described in details in (2). Metagenome assemblies usually represent not a single organism but rather a composition of bacterial population. Metagenome assemblies are not taken into RefSeq, however, that policy may change as the technologies and methods evolve. Some genomes submitted to GenBank are not taken into RefSeq collection because of the problems with the completeness, coverage and quality of assembly.
Many genomes assemblies coming from single cell sequencing technology give only partial representation of DNA in a cell, ranging from 10 to 90%.
Assemblies with partial genome representation can be found in Entrez Assembly database http://www.ncbi.nlm.nih.gov/assembly/ by using the following query:
Archaea[orgn] or bacteria[orgn] and ‘partial genome representation’[properties]
Genome assemblies from mixed cultures, hybrid organisms and chimeras submitted to GenBank are not accepted into RefSeq because they do not represent an organism. These anomalous assemblies can be found in Entrez Assembly database by using the following query:
Archaea[orgn] or bacteria[orgn] and ‘anomalous’[properties]
Genome representation can be validated by comparative analysis if other genomes are available in closely related groups (species or genus). Some assemblies can be filtered out after the evaluation of the annotation results as described in the Genome Annotation Pipeline section.
Reference pan-genome
Historically, prokaryotic organisms were organized by classical taxonomic ranking system (species, genus, family, order, class and phylum). Unlike eukaryotes, prokaryotes do not have a clear definition of species. Delineation of prokaryotic species was originally based on phenotypic information, pathogenicity and environmental observations. Sequenced-based methods (10–12) organizing genomes in a closely related group for the purpose of downstream analysis. Selected markers, limited to single copy ribosomal proteins conserved in all archaea and bacteria, are predicted in every genome to overcome problems with the genome annotations (missing and/or incorrect annotations) and to normalize the marker dataset. To reduce the redundancy predicted ribosomal proteins are clustered with 85% identity and 85% coverage using BLAST (13); non-redundant representatives are selected from each cluster to create a reference marker set. The set of reference markers is available at the special reports File Transfer Protocol (FTP) site: ftp://ftp.ncbi.nlm.nih.gov/genomes/GENOME_REPORTS/MARKERS/.
Marker predictions are performed by aligning reference protein markers against full genome assemblies. Assemblies with at least 17 markers are passed to the next step. Genome distance is calculated as an average of pairwise protein distances of markers shared in a pair of genomes. Agglomerative hierarchical clustering trees are built within phylum-level groups. Clades at the species level are calculated using a species-aware algorithm. The genome groups (clades) serve as basic target units for downstream analysis. Large groups (more than 20 members) of related genomes corresponding approximately to the taxonomic species level are represented as pan-genomes (Table 1).
Table 1. Top 10 most abundant clades.
| Clade name (species, genus) | # Genomes | # CDS | median_cds | # Core clusters |
|---|---|---|---|---|
| Escherichia–Shigella | 1502 | 7 594 943 | 4990 | 3220 |
| Citrobacter–Salmonella | 527 | 2 334 839 | 4511 | 3393 |
| Staphylococcus aureus | 445 | 1 195 744 | 2672 | 2066 |
| Streptococcus | 334 | 714 947 | 2150 | 1223 |
| Brucella | 283 | 886 682 | 3120 | 1704 |
| Helicobacter pylori | 268 | 433 955 | 1631 | 1200 |
| Streptococcus agalactiae | 254 | 523 389 | 2038 | 1595 |
| Acinetobacter | 212 | 796 523 | 3785 | 2755 |
| Neisseria | 194 | 402 822 | 1997 | 1540 |
| Leptospira interrogans | 186 | 778 660 | 4062 | 3024 |
The ‘pan-genome’ concept has been introduced by Tettelin et al. in 2005 (14). The pan-genome has been defined as a super-set of all genes in all the strains of a species. A pan-genome includes the ‘core’ genes that are present in nearly all strains, ‘mobile’ (or ‘accessory’) genes present in two or more strains and finally ‘unique genes’ specific to a single strain (see Figure 2).
Figure 2.
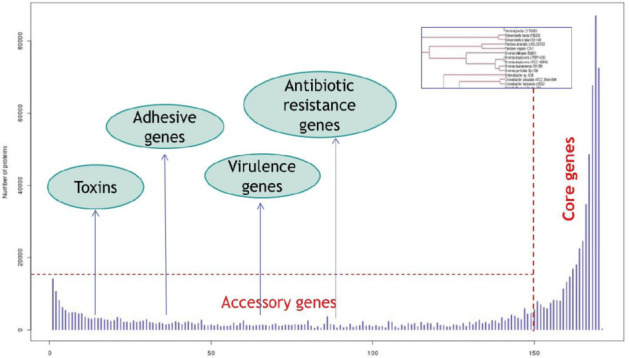
Shigella–Escherichia coli pan-genome: core and mobile components.
Protein clusters within each clade are calculated with hierarchical clustering algorithm; pairwise distance between proteins is defined as modified BLAST score that takes into account alignment score and length.
The pairwise distance between genome assemblies within each clade is further refined by megablast alignment (13). Genomes with more than 95% identity are grouped together, and a representative ‘centroid’ genome for each group is calculated. The illustration of short proximity genome groups for E. coli strains is provided on Figure 3.
Figure 3.
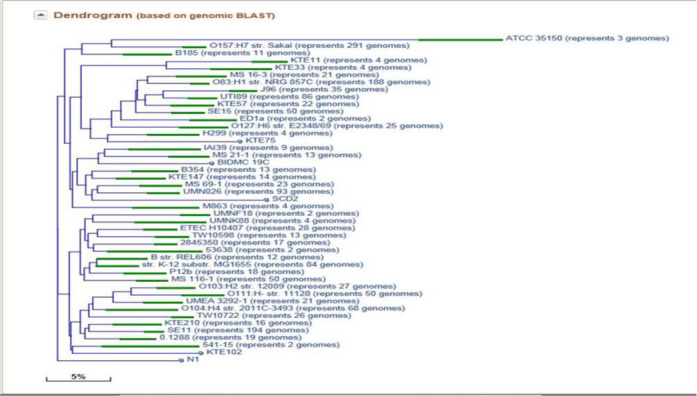
Escherichia coli genomes grouped by BLAST similarity distance; each green box represents a short proximity genome group.
Large-scale genome surveillance projects already generate thousands of whole genome sequences of clinical and environmental isolates and the number is rapidly growing. Organizing closely related genome in short proximity groups allows to reduce the redundancy in pan-genome analysis.
Dataflow
The RefSeq genome processing pipeline consists of three major components: collecting the genome assemblies and preforming quality control; organizing genomes into pan-genomes and close genome proximity groups; using pan-genome data for the annotation pipeline.
The genomic data are dynamic: hundreds of new genomes and assembly updates are submitted to NCBI each day. Every six months we create a snapshot of all live genome assemblies and their nucleotide sequence components (chromosomes, scaffolds and contigs) in an internal Genome Collection database with a date stamp. The assemblies are filtered by quality and passed to the processing script. Marker-based groups (clade) are calculated as described above. Protein clusters are used to create pan-genome data structures; clades are further refined by generating megablast close genome proximity groups. NCBI Annotation Pipeline utilize the pan-genome dataset for the normalized annotation. Genomes that pass annotation quality control are added to RefSeq prokaryotic genome collection. See Figure 4 for details.
Figure 4.
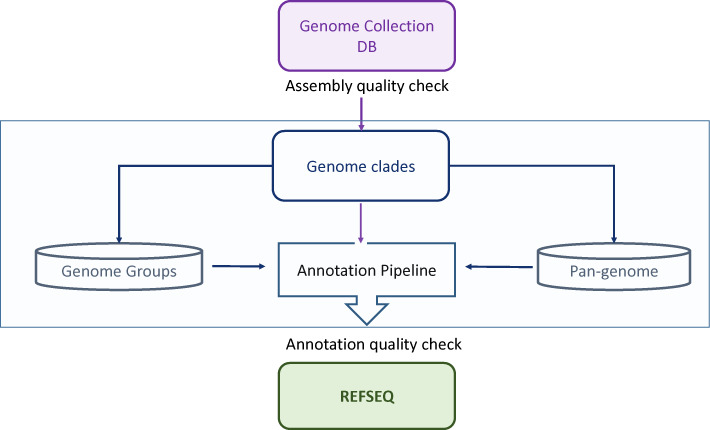
RefSeq prokaryotic genome processing: pan-genome and genome close proximity groups.
GENOME ANNOTATION PIPELINE
NCBI Prokaryotic Genome Annotation Pipeline is designed to annotate bacterial and archaeal genomes (chromosomes and plasmids). Genome annotation is a multi-level process that includes prediction of protein-coding genes, as well as other functional features such as structural RNAs, tRNAs, small RNAs, pseudogenes, control regions, direct and inverted repeats, insertion sequences, transposons and other mobile elements. NCBI has developed an automatic annotation pipeline that combines ab initio gene prediction algorithms with homology-based methods (15). The annotation process combines a gene calling algorithm with similarity-based gene detection approach. The pipeline currently predicts protein-coding genes, structural RNAs (5S, 16S, 23S), tRNAs and small non-coding RNAs. The pipeline utilizes the pre-calculated pan-genome data if available and relies on ab initio prediction for rare organisms. Our approach relies on good normalized structural and functional annotation of the members of pan-genome. By aligning a set of species-specific CORE genes upfront we ensure the annotation of genes that most likely should be present in genome annotation since they have been found in the majority of other genomes of that species. Consistent annotation by protein homology allows to improve the CORE set in pan-genome structure. All RefSeq genomes are annotated by NCBI pipeline except for the Reference genomes manually curated by community and NCBI staff (2). We welcome collaborations with authoritative groups outside NCBI who are willing to provide and maintain the regular updates of the sequence and annotations of the reference genomes.
USE BY THE COMMUNITY
NCBI databases and services are used by more than 2 million people a day online. NCBI also provides programmatic access to many of our data resources, allowing computer programs to run Entrez searches or retrieve sequence records without direct human assistance.
The NCBI Prokaryotic Genome Annotation pipeline is offered as a service to GenBank submitters and is used for consistent annotation of RefSeq genomes. To date, ∼5000 bacterial genomes submitted to Genbank have been annotated by the pipeline at the request of the users. Users have the option to review and edit the annotation prior to release if they choose. In addition, over 16 000 microbial genomes, both complete and draft, have been annotated for RefSeq. A consistent set of annotation results generated by a common process using trackable references is essential for both individual reseachers and large data centers. Researchers can also download data by FTP.
List of the Reference Genomes can be found at http://www.ncbi.nlm.nih.gov/genome/browse/reference/.
CURATED GENOMIC REGIONS
Primary nucleotide records which include annotated proteins with experimental evidence are being made into manually curated reference sequences. These records may include a region of a microbial chromosome, a complete or partial plasmid or mobile element such as a transposon. This provides a more complete record of experimentally confirmed protein functions in RefSeq, which in turn will be used for annotation of new genomes. Curated genomic regions often contain experimentally verified genes and or genes that are hard to predict automatically (short peptides, highly variable genes and control regions). Our future plan is to utilize this manually curated regions by aligning them to same species genome during the annotation process.
Example: NG_034195, partial sequence of plasmid pMYSH6000 in Shigella flexneri containing genes involved in the type III secretion system:

TARGETED LOCI PROJECT
The Targeted Loci Project initiated in 2009 is a database of molecular markers used for phylogenetic analyses and identification for bacteria, archaea and fungi. The initial project consisted of 16S ribosomal RNA from bacterial and archaeal type strains and has expanded to include 23S and 5S ribosomal RNA from bacterial and archaeal genomes as well as 18S and 28S ribosomal RNA and ITS (internal transcribed spacer) DNA sequences from fungi.
Ribosomal RNAs in prokaryotes and the ITS region in fungi have become widely used molecular markers for species identification and phylogenetic analysis. In prokaryotes the 16S rRNA sequence in particular has become a standard molecular marker for the description of a new species. While these marker sequences have become widely used, the quality of the sequence data and the associated meta-data being submitted to INSDC databases varies considerably. Recognizing the importance of access to high quality data for these markers, NCBI has expanded its Targeted Loci Project to provide an up to date source of curated data.
The Targeted Loci Project maintain sets of reference sequences from type strains whenever possible as the type strains are considered the exemplar of the species. This work involved an exhaustive review and update to the underlying taxonomy database which was used in conjunction with NCBI's type strain Entrez filter to retrieve candidate sequences. The sequence data and their associated taxonomy/meta-data have been reviewed and corrected to include the most up to date information. The sequence data was reviewed to validate the data and the associated annotation, in many cases corrections were incorporated. If a sequence failed validation or could not be accurately validated, it was excluded. These reference sequences can now be used as ‘gold standards’ for the analysis of existing and new rRNA sequences.
Reference sets of targeted loci sequences from type strains are regularly used in GenBank submission process for the validation and correction of taxonomic classification. Reference sequences of 5S, 16S, 23S structural RNAs are used in the NCBI annotation pipeline for finding structural RNAs by homology.
The complete dataset of Targeted Loci Project is available at:
http://www.ncbi.nlm.nih.gov/refseq/targetedloci/ along with customized BLAST databases at:
http://www.ncbi.nlm.nih.gov/blast/Blast.cgi?PAGE_TYPE=BlastSearch&BLAST_SPEC=TargLociBlast.
DATA ACCESS
New genomes processed for RefSeq, and made public in Entrez and added to FTP directories daily.
The complete list of prokaryotic genomes is available in Entrez Genome browser
Link: http://www.ncbi.nlm.nih.gov/genome/browse/
The text version of the table can be downloaded from the FTP site
ftp://ftp.ncbi.nlm.nih.gov/genomes/GENOME_REPORTS/
Genome FTP
NCBI has redesigned the ‘genomes FTP site’ to expand the content and facilitate data access through an organized predictable directory hierarchy with consistent file names and formats. The updated site provides greater support for downloading assembled genome sequences and/or corresponding annotation data. The new FTP site structure provides a single entry point to access content representing either GenBank or RefSeq data. More detailed information can be found at http://www.ncbi.nlm.nih.gov/genome/doc/ftpfaq/
RefSeq release
The RefSeq release FTP site is organized by major taxonomic groups. The microbial RefSeq sequence data is available at ftp://ftp.ncbi.nih.gov/refseq/release/bacteria.
Special reports
MARKERS
CLADES
FUNDING
Funding for open access charge: Intramural Research Program of the National Institutes of Health; National Library of Medicine; National Center for Biotechnology Information; National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1.Pruitt K.D., Tatusova T., Brown G.R., Maglott D.R. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tatusova T, Ciufo S., Fedorov B., O'Neill K., Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014;42:D553–D559. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fujishima K., Sugahara J., Miller C.S., Baker B.J., Di Giulio M., Tomita M., Banfield J.F., Kanai A.A. Novel three-unit tRNA splicing endonuclease found in ultra-small Archaea possesses broad substrate specificity. Nucleic Acids Res. 2011;39:9695–9704. doi: 10.1093/nar/gkr692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han K., Li Z.E., Peng R., Zhu L.P., Zhou T., Wang L.G., Li S.G., Zhang X.B., Hu W., Wu Z.H., et al. Extraordinary expansion of a Sorangium cellulosum genome from an alkaline milieu. Sci. Rep. 2013;3:2101. doi: 10.1038/srep02101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loman N.J., Constantinidou C., Chan J.Z., Halachev M., Sergeant M., Penn C.W., Robinson E.R., Pallen M.J. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat. Rev. Microbiol. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- 6.Poptsova M.S., Gogarten J.P. Using comparative genome analysis to identify problems in annotated microbial genomes. Microbiology. 2010;156:1909–1917. doi: 10.1099/mic.0.033811-0. [DOI] [PubMed] [Google Scholar]
- 7.Barrett T., Clark K., Gevorgyan R., Gorelenkov V., Gribov E., Karsch-Mizrachi I., Kimelman M., Pruitt K.D., Resenchuk S., Tatusova T., et al. BioProject and BioSample databases at NCBI: facilitating capture and organization of metadata. Nucleic Acids Res. 2012;40:D57–D63. doi: 10.1093/nar/gkr1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Federhen S., Clark K., Barrett T., Parkinson H., Ostell J., Kodama Y., Mashima J., Nakamura Y., Cochrane G., Karsch-Mizrachi I. Toward richer metadata for microbial sequences: replacing strain-level NCBI taxonomy taxids with BioProject, BioSample and assembly records. Stand Genomic Sci. 2014;9:1275–1277. doi: 10.4056/sigs.4851102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura Y., Cochrane G., Karsch-Mizrachi I., on behalf of the International Nucleotide Sequence Database Collaboration The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2013;41:D21–D24. doi: 10.1093/nar/gks1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darling A.E., Jospin G., Lowe E., Matsen F.A. 4th, Bik H.M., Eisen J.A. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2014;2:e243. doi: 10.7717/peerj.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mende D.R., Sunagawa S., Zeller G., Bork P. Accurate and universal delineation of prokaryotic species. Nat. Methods. 2013;10:881–884. doi: 10.1038/nmeth.2575. [DOI] [PubMed] [Google Scholar]
- 12.Larsen M.V., Cosentino S., Lukjancenko O., Saputra D., Rasmussen S., Hasman H., Sicheritz-Pontén T., Aarestrup F.M., Ussery D.W., Lund O. Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 2014;52:1529–1539. doi: 10.1128/JCM.02981-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tettelin H., Masignani V., Cieslewicz M.J., Donati C., Medini D., Ward N.L., Angiuoli S.V., Crabtree J., Jones A.L., Durkin A.S., et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial ‘pan-genome’. Proc. Natl. Acad. Sci. U.S.A. 2005;102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.NCBI Handbook. http://www.ncbi.nlm.nih.gov/books/NBK174280/