


Abstract
Dr.VIS is a database of human disease-related viral integration sites (VIS). The number of VIS has grown rapidly since Dr.VIS was first released in 2011, and there is growing recognition of the important role that viral integration plays in the development of malignancies. The updated database version, Dr.VIS v2.0 (http://www.bioinfo.org/drvis or bminfor.tongji.edu.cn/drvis_v2), represents 25 diseases, covers 3340 integration sites of eight oncogenic viruses in human chromosomes and provides more accurate information about VIS from high-throughput deep sequencing results obtained mainly after 2012. Data of VISes for three newly identified oncogenic viruses for 14 related diseases have been added to this 2015 update, which has a 5-fold increase of VISes compared to Dr.VIS v1.0. Dr.VIS v2.0 has 2244 precise integration sites, 867 integration regions and 551 junction sequences. A total of 2295 integration sites are located near 1730 involved genes. Of the VISes, 1153 are detected in the exons or introns of genes, with 294 located up to 5 kb and a further 112 located up to 10 kb away. As viral integration may alter chromosome stability and gene expression levels, characterizing VISes will contribute toward the discovery of novel oncogenes, tumor suppressor genes and tumor-associated pathways.
INTRODUCTION
Viral integration into host chromosomes plays a key role in viral infection (1,2) and tumorigenesis (3–8). The role of oncogenic viruses in cancer pathogenesis is mediated through mutagenic integration into the host genome as one of important mechanisms (2). Viral integration sites (VIS) have been observed adjacent to oncogenes, at chromosomal fragile sites, scaffold/matrix attachment regions and repeat/satellite sequence-rich regions (9,10). Moreover, chromosomal rearrangements, including deletions and insertions of viral and host genes, are often linked to tumor development and progression (11–13). For instance, Ojesina et al. have demonstrated that the relationship between human papillomavirus (HPV) integration and increased expression of adjacent genes is a widespread phenomenon in primary cervical carcinomas by whole-exome sequencing analysis of 115 cervical carcinoma-normal paired samples, transcriptome sequencing of 79 cases and whole-genome sequencing of 14 tumor-normal pairs (14). The expression levels of oncogenes at HPV integration sites, including MYC, SERPINB4, GLI2 and NR4A2, are shown to be significantly higher than those without virus integrations (14). Furthermore, several hepatitis B virus (HBV) integration sites are located in the genomes of hepatocellular carcinoma (HCC) patients, which contain numerous oncogenes such as TP53, TERT, CCNE1 and MLL4 (15,16). Therefore, a better understanding of VISes and their adjacent DNA features will be of tremendous importance in unraveling the unique mechanisms underlying the pathogenesis of malignancy as well as identifying novel anticancer targets.
With recent advances in high-throughput deep sequencing techniques, whole-genome and whole-exome sequencing have been widely and successfully used to search for VISes (16,17). This has generated massive amounts of data, which need to be analyzed with new and appropriate user-friendly analytical tools and databases. Dr.VIS was first released in 2011 as a database of human disease-related VISes (18). Since then, the number of known VISes has grown rapidly. In this updated version of Dr.VIS, Dr.VIS v2.0, the number of collected VISes has reached 3340. The revised database aims to provide a platform to facilitate both bioinformatic and experimental research. Dr.VIS v2.0 also provides a convenient search option, enabling the efficient recovery of oncogenes, regulatory elements in flanking sequences, related publications and other information.
DATA ANNOTATION AND COLLECTION METHOD
Data collection and annotation for Dr.VIS v2.0 was carried out in a similar fashion as for version 1.0 (18). In recent times, the growth of papers reporting VISes has been rapid. To keep up with this, we systematically collected newly described VISes and curated related information (Figure 1). We first carried out a PubMed literature search using a list of keywords pertaining to virus integration, including ‘virus integration’, ‘virus integration site’, ‘virus integration sequence’, ‘virus integration tumor’ and ‘cancer and disease’. We then extracted more VISes keywords from this literature and filtered the downloaded files using these keywords. The filtered entries were subsequently confirmed by manual curation (Figure 1).
Figure 1.
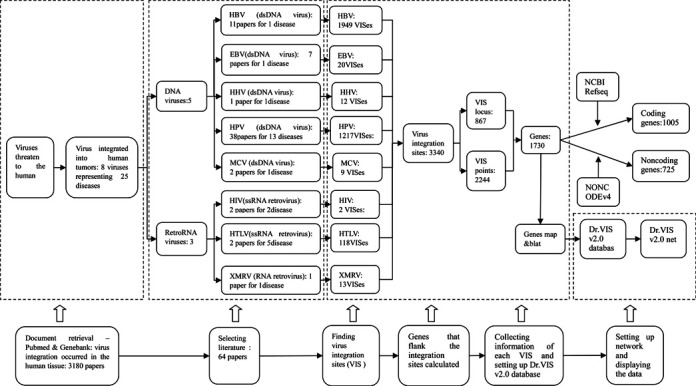
The flow scheme for setting up the Dr.VIS v2.0 and the overview of the database. Abbreviations: HBV, hepatitis B virus; EBV, Epstein–Barr virus; HHV, human herpesvirus ; HPV, human herpesvirus; MCV, Merkel cell polyomavirus; HIV, human immunodeficiency virus; HTLV, human T lymphotropic virus; XMRV, xenotropic murine leukemia virus-related virus.
Around 3180 papers were initially obtained using a list of keywords pertaining to virus integration and tumor. We manually filtered those papers for relevance to human disease-related viruses and got 196 ones. Then a total of 64 papers were selected, in which researchers completed their study using high-throughput deep sequencing or reported exact VISes or junction sequences. Curators intensively read these selected papers in full to extract the VIS characteristics required by the data model. Subsequent manual retrieval and curation were performed from the original literature reporting junction sequences. For all VIS records obtained, basic information related to references and PubMed accession numbers was extracted and entered into the Dr.VIS v2.0 database. All VISes deposited in Dr.VIS v2.0 were sequenced or detected from tumor tissues, non-tumor tissues of patients or cell lines. A process of redundancy elimination was then performed on the entire dataset, including both previously existing and newly collected data. Each VIS was repeatedly checked and given a Dr.VIS accession number (unique VIS ID). The annotations and genomic mapping information of coding genes and non-coding sequences relied on data from original papers, the supplementary materials of papers, the NCBI RefSeq database (http://www.ncbi.nlm.nih.gov/RefSeq/) (19), the NONCODEv4 database (20) or BLAT at the University of California Santa Cruz (UCSC) (http://genome.ucsc.edu/) (21). We next identified the genes that were closest to VISes. GenBank annotations were used to create figures for all VISes to enable visualization of their location in the genome or within a specific DNA fragment, together with regulatory elements in flanking sequences.
DATABASE CONTENT AND STRUCTURE
The purpose of the database is to serve as a knowledge base for experimentally oriented studies and as a resource for medical and bioinformatics applications. The first release of Dr.VIS in 2012 covered 567 natural VISes of five oncogenic viruses representing 11 diseases (18). It was completed before 2012, when next-generation sequencing (NGS) was less widely applied. This updated version, representing 25 diseases, covers 3340 integration sites of eight oncogenic viruses in human chromosomes and provides more accurate information about VISes based on deep sequencing results obtained mainly after 2012. Additionally, data of VISes for three newly identified oncogenic viruses for 14 related diseases have been added to this updated version. Table 1 compares the previous and current versions of Dr.VIS, and demonstrates a 5-fold increase of VIS information in this 2015 Dr.VIS v2.0 update. There are 1949 VISes of HBV representing HCC from 11papers, 1217 VISes of HPV representing 13 diseases from 38 papers, 118 VISes of HTLV representing five diseases from two papers, 20 VISes of EBV representing one tumor from seven papers, 13 VISes of XMRV representing one cancer from one paper, nine VISes of MCV representing one disease from two papers and two VISes of HIV representing two tumor from two papers (Figure 1).
Table 1. A data comparison between Dr.VIS v2.0 and Dr.VIS v1.0.
| Data features | Dr.VIS v1.0 | Dr.VIS v2.0 |
|---|---|---|
| Total number of viruses | 5 | 8 |
| Total number of related diseases | 11 | 25 |
| Samples which VISes are detected from | ||
| Tumor | NA | 2446 |
| Non-tumor | NA | 882 |
| Cell-line | NA | 11 |
| Total number of VISes | 567 | 3340 |
| Total number of VIS points | 197 | 2244 |
| Total number of VIS locus | 370 | 867 |
| Total number of junction sequences | 197 | 551 |
| Total number of human genes involved | 266 | 1730 |
| Coding genes | 247 | 1005 |
| Non-coding genes | 19 | 725 |
| Genes in which VISes located | NA | 1153 |
| Genes involved with a range of 5 kb | NA | 294 |
| Genes involved with a range of 10 kb | NA | 112 |
| Total number of integration sites of viruses | NA | 1453 |
| Total number of virus genes involved | NA | 462 |
| Number of articles annotated | 43 | 64 |
Most VISes deposited in Dr.VIS v2.0 are sequenced or detected from patient samples, including tumor and non-tumor tissues. A total of 2446 VISes are detected from tumors, while 882 VISes are found in non-tumor tissues, and 11 from cell lines. Traditionally, most virus integration breakpoints have been detected by polymerase chain reaction (PCR)-based methods such as Alu-PCR (22). However, the recent rapid development of massive parallel sequencing technology and NGS such as whole-genome sequencing and whole-exon sequencing has introduced new ways of detecting viral integration in the human genome (12,16,17,23). NGS is therefore the most commonly used detection method for VISes in the Dr.VIS v2.0 database. This database covers 2244 precise integration sites, 867 integration regions and 551 junction sequences. Viruses may affect their host chromosome during the integration process, but also may affect their own replication, assembly and integration. For instance, Sung et al. reported that approximately 40% of observed breakpoints were restricted to the 1800-bp region of the HBV genome where the viral enhancer, X gene and core gene are located (12). And Dr.VIS v2.0 contains 1453 breakpoints across viral genomes and 462 viral genes. A total of 2295 VISes are located near 1730 human disease genes, including 1005 coding and 725 non-coding genes. Many VISes (1153) are located within exons or introns of disease genes, with 294 located up to 5 kb from the nearest gene, and an additional 112 located up to 10 kb away.
Cytobands covering VIS can be non-coding sequences or interrupted genes with specific coordinates of subcomponents (such as exons or introns), and must have been approved by the HUGO (Gene Nomenclature Committee). Meanwhile, the genomic location of each integration site in the human genome assembly before 2009 must have been converted to hg19 and must be able to be identified by BLAT from the UCSC database (21). Genes that flank integration sites (5–10 kb) can be further calculated using UCSC Blat (21), the NCBI RefSeq (24) and NONCODEv4 (20).
DATABASE VISUALIZATION
Within the Dr.VIS v2.0 database, basic information is available about each human disease-related VISes. This includes the corresponding human disease, type of sample, method of detection, related virus name, chromosome location, cytoband covering the VIS, integration site, human strand, virus break point, virus-gene/important element, virus strand, virus genome rearrangement, nearest gene integration site, distance to nearest gene from integration site and integrated sequence covering the junction point. Genomic traits of a VIS cluster include the gene distribution and gene distance from the integration site. The integrated sequences covering the junction point are recorded as ‘human genome-viral genome-human genome’. For convenient data organization, VISes representing the above-mentioned basic information are clustered to generate a unique data entry known as a viral integration cluster (or VIS cluster) (Figure 2). Each VIS can also be labeled simultaneously with several tags.
Figure 2.
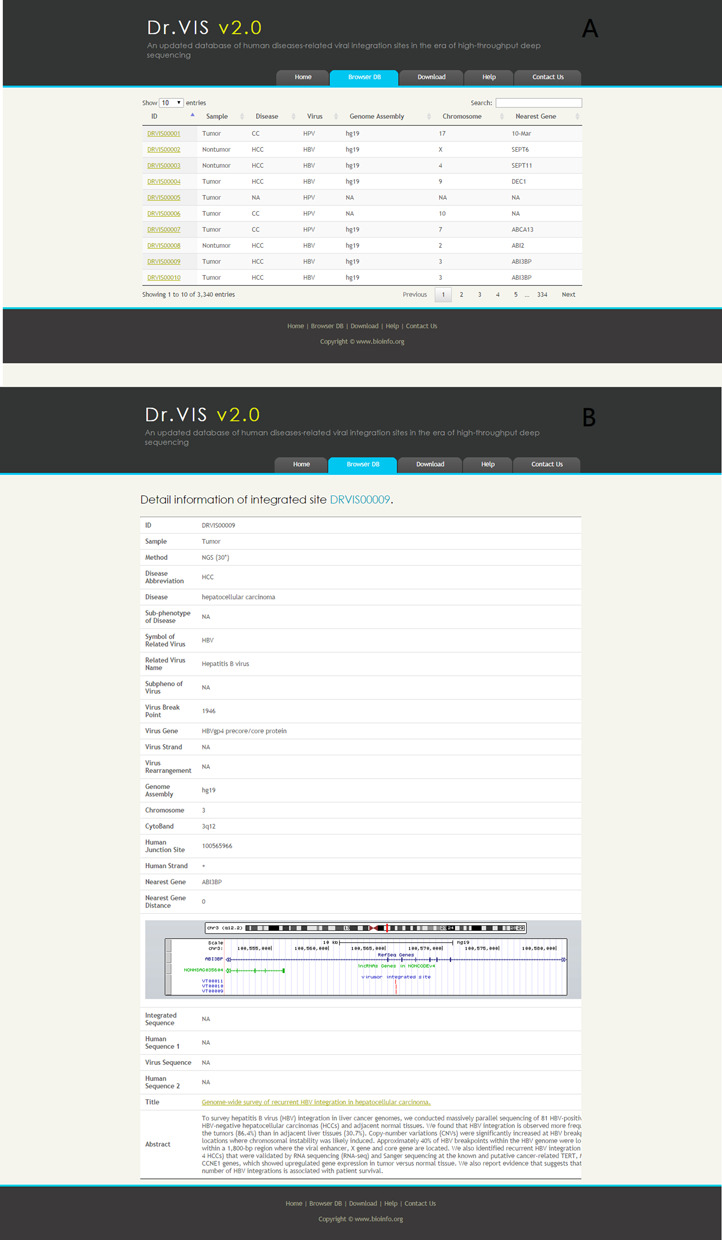
(A) The Dr.VIS v2.0 database window with VIS annotations. (B) The browsing page with detailed information about an integrated site.
Users can query the database through the search interface by providing the virus name, genome assembly, chromosome, human disease, nearest gene, VIS ID or any other descriptive words. By clicking the ‘search’ box in the upper right-hand corner, the page will display matching data including VIS ID, sample, related disease, virus, genome assembly, chromosome, nearest gene, precise integration site in the genome, gene locus and abstract of reference papers (Figure 2). Sequences can be searched using accession numbers found in Dr.VIS v2.0. Search results are also linked to full GenBank entries. A collection of published research articles describing high-throughput investigations on VIS has been provided for the benefit of users. Users could obtain an overview of the landscape of virus–tumor associations, especially in malignant cancers, mainly through the following ways: (i) by analyzing VIS distribution in diseases, viruses, chromosomes and other aspects; (ii) by analyzing the distribution of the genes (coding or non-coding) closest to VISes in diseases, viruses and other aspects. Availability of this information will be useful for both clinicians and researchers, and will enable the identification and verification of new oncogenes, tumor suppressor genes and tumor-associated pathways (25).
The database can be accessed through the following URL: http://www.bioinfo.org/drvis or bminfor.tongji.edu.cn/drvis_v2, which is easily accessible to all users, free of charge and does not require the user to log in. The entire Dr.VIS v2.0 dataset can be directly downloaded from http://www.bioinfo.org/drvis/download.php.
CONCLUSION
The identification of novel cancer-associated viruses and understanding the genomic effects of known viruses on the human genome is technically complex and incomplete (26). Virus integration sites were reported to be found to be distributed randomly or not uniformly over the whole genome (9,14). However, it may not be the fact. Some researchers found that the integration hotspots of human genome were located in oncogenes, such as TERT, MLL4, CCNE1 and so on (13,22), and fragiles or other special structures of human chromosome (12). This strategic virus integration may activate oncogenes, corrupt tumor suppressors, or impose cis-regulatory effects on the expression of downstream genes, form chimeric human fusion genes and thereby dysregulate the transcription network through some pathways in tumors (12). Meanwhile, viral breakpoints may be strategic and facilitate virus insertion (12). As a result, cancers may come out. Therefore, robust analysis of characterizing VISes, their adjacent DNA features and their associations with human diseases will contribute toward the discovery of novel oncogenes, tumor suppressors and tumor-associated pathways.
Dr.VIS v2.0 is one of the most comprehensive databases of viral integration and human diseases, and is developed to enable biological scientists to explore their data in a more systems-oriented manner. Compared with the original Dr.VIS, the new version is a step toward a more integrated knowledge database, with expansion of the total number of viruses, related diseases, VISes and nearest genes. Dr.VIS v2.0 is also user-friendly and is of enormous value for the analysis of VIS and related malignancies. As new VISes are progressively discovered, we will continue to update the Dr. VIS v2.0 database. Submissions of new VISes are invited and should be sent to zhaoht@pumch.cn.
Acknowledgments
We thank Dr Yan Wu for carefully reading our manuscript.
Footnotes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.
FUNDING
Capital Special Research Project for Health Development [2014-2-4012]; Training Program of the Major Research Plan of the National Natural Science Foundation of China [91229120]; International Science and Technology Cooperation Projects [2010DFB33720]; Program for New Century Excellent Talents in University [NCET-11-0288]; National Natural Science Foundation of China [30970623]; Natural Science Foundation of Shanghai [12ZR1421500]; Shanghai Municipal Health Bureau Scientific Research Task [20114182]; National Natural Science Foundation of China [81101955]. Funding for open access charge: International Science and Technology Cooperation Project [2010DFB33720].
Conflict of interest statement. None declared.
REFERENCES
- 1.Martin D., Gutkind J.S. Human tumor-associated viruses and new insights into the molecular mechanisms of cancer. Oncogene. 2008;27(Suppl. 2):S31–S42. doi: 10.1038/onc.2009.351. [DOI] [PubMed] [Google Scholar]
- 2.Khoury J.D., Tannir N.M., Williams M.D., Chen Y., Yao H., Zhang J., Thompson E.J., Network TCGA, Meric-Bernstam F., Medeiros L.J. Landscape of DNA virus associations across human malignant cancers: analysis of 3775 cases using RNA-Seq. J. Virol. 2013;87:8916–8926. doi: 10.1128/JVI.00340-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brechot C., Pourcel C., Louise A., Rain B., Tiollais P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature. 1980;286:533–535. doi: 10.1038/286533a0. [DOI] [PubMed] [Google Scholar]
- 4.Paterlini-Bréchot P., Saigo K., Murakami Y., Chami M., Gozuacik D., Mugnier C., Lagorce D., Bréchot C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–3916. doi: 10.1038/sj.onc.1206492. [DOI] [PubMed] [Google Scholar]
- 5.Murakami Y., Saigo K., Takashima H., Minami M., Okanoue T., Bréchot C., Paterlini-Bréchot P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut. 2005;54:1162–1168. doi: 10.1136/gut.2004.054452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minami M., Daimon Y., Mori K., Takashima H., Nakajima T., Itoh Y., Okanoue T. Hepatitis B virus-related insertional mutagenesis in chronic hepatitis B patients as an early drastic genetic change leading to hepatocarcinogenesis. Oncogene. 2005;24:4340–4348. doi: 10.1038/sj.onc.1208628. [DOI] [PubMed] [Google Scholar]
- 7.Bonilla Guerrero R., Roberts L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005;42:760–777. doi: 10.1016/j.jhep.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Poreba E., Broniarczyk J.K., Gozdzicka-Jozefiak A. Epigenetic mechanisms in virus-induced tumorigenesis. Clin. Epigenetics. 2011;2:233–247. doi: 10.1007/s13148-011-0026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wentzensen N., Vinokurova S., von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 2004;64:3878–3884. doi: 10.1158/0008-5472.CAN-04-0009. [DOI] [PubMed] [Google Scholar]
- 10.Ishikawa T. Clinical features of hepatitis B virus-related hepatocellular carcinoma. World J. Gastroenterol. 2010;16:2463–2467. doi: 10.3748/wjg.v16.i20.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujimoto A., Totoki Y., Abe T., Boroevich K.A., Hosoda F., Nguyen H.H., Aoki M., Hosono N., Kubo M., Miya F., et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 12.Sung W.K., Zheng H., Li S., Chen R., Liu X., Li Y., Lee N.P., Lee W.H., Ariyaratne P.N., Tennakoon C., et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 13.de Jong J., Akhtar W., Badhai J., Rust A.G., Rad R., Hilkens J., Berns A., van Lohuizen M., Wessels L.F., de Ridder J. Chromatin landscapes of retroviral and transposon integration profiles. PLoS Genet. 2014;10:e1004250. doi: 10.1371/journal.pgen.1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ojesina A.I., Lichtenstein L., Freeman S.S., Pedamallu C.S., Imaz-Rosshandler I., Pugh T.J., Cherniack A.D., Ambrogio L., Cibulskis K., Bertelsen B., et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saigo K., Yoshida K., Ikeda R., Sakamoto Y., Murakami Y., Urashima T., Asano T., Kenmochi T., Inoue I. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum. Mutat. 2008;29:703–708. doi: 10.1002/humu.20701. [DOI] [PubMed] [Google Scholar]
- 16.Kan Z., Zheng H., Liu X., Li S., Barber T.D., Gong Z., Gao H., Hao K., Willard M.D., Xu J., et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–1433. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dewey F.E., Grove M.E., Pan C., Goldstein B.A., Bernstein J.A., Chaib H., Merker J.D., Goldfeder R.L., Enns G.M., David S.P., et al. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311:1035–1045. doi: 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao X., Liu Q., Cai Q., Li Y., Xu C., Li Y., Li Z., Zhang X. Dr.VIS: a database of human disease-related viral integration sites. Nucleic Acids Res. 2012;40:D1041–D1046. doi: 10.1093/nar/gkr1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M., et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie C., Yuan J., Li H., Li M., Zhao G., Bu D., Zhu W., Wu W., Chen R., Zhao Y. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2014;42:D98–D103. doi: 10.1093/nar/gkt1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karolchik D., Barber G.P., Casper J., Clawson H., Cline M.S., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W., Zeng X., Lee N.P., Liu X., Liu X., Chen S., Guo B., Yi S., Zhuang X., Chen F., Wang G., et al. HIVID: an efficient method to detect HBV integration using low coverage sequencing. Genomics. 2013;102:338–344. doi: 10.1016/j.ygeno.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Leamon J.H., Link D.R., Egholm M., Rothberg J.M. Overview: methods and applications for droplet compartmentalization of biology. Nat. Methods. 2006;3:541–543. doi: 10.1038/nmeth0706-541. [DOI] [PubMed] [Google Scholar]
- 24.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M., et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X., Zhang J., Yang Z., Kang J., Jiang S., Zhang T., Chen T., Li M., Lv Q., Chen X., et al. The function of targeted host genes determines the oncogenicity of HBV integration in hepatocellular carcinoma. J. Hepatol. 2014;60:975–984. doi: 10.1016/j.jhep.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 26.Rozenblatt-Rosen O., Deo R.C., Padi M., Adelmant G., Calderwood M.A., Rolland T., Grace M., Dricot A., Askenazi M., Tavares M., et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature. 2012;487:491–495. doi: 10.1038/nature11288. [DOI] [PMC free article] [PubMed] [Google Scholar]