


Abstract
For over 10 years, Binding MOAD (Mother of All Databases; http://www.BindingMOAD.org) has been one of the largest resources for high-quality protein–ligand complexes and associated binding affinity data. Binding MOAD has grown at the rate of 1994 complexes per year, on average. Currently, it contains 23 269 complexes and 8156 binding affinities. Our annual updates curate the data using a semi-automated literature search of the references cited within the PDB file, and we have recently upgraded our website and added new features and functionalities to better serve Binding MOAD users. In order to eliminate the legacy application server of the old platform and to accommodate new changes, the website has been completely rewritten in the LAMP (Linux, Apache, MySQL and PHP) environment. The improved user interface incorporates current third-party plugins for better visualization of protein and ligand molecules, and it provides features like sorting, filtering and filtered downloads. In addition to the field-based searching, Binding MOAD now can be searched by structural queries based on the ligand. In order to remove redundancy, Binding MOAD records are clustered in different families based on 90% sequence identity. The new Binding MOAD, with the upgraded platform, features and functionalities, is now equipped to better serve its users.
INTRODUCTION
Advancements in structure-based drug design require high-quality datasets of ligand–protein complexes for training and testing methods. The last decade has seen the development of many data resources derived from the Protein Data Bank (PDB) (1) with different focuses toward ligand binding and binding-site ‘druggability’ (2–11). Binding MOAD (Mother of All Database) is one of the largest databases, which contains high resolution structures from the PDB with ligand annotation (biologically valid/invalid) and protein classifications (enzyme/non-enzyme, homologous families). This annotation and classification makes it unique compared to other databases of its kind. Binding MOAD also links protein–ligand structures with their corresponding experimental binding affinities (2,3). To incorporate new data, Binding MOAD is updated every year, using a semi-automated literature curation of the crystallography references cited in each PDB file. This large database has also been successfully used in many data-mining studies, which explore ligand binding from different perspectives. These studies include insights in protein–ligand recognition (12), differences in binding affinities between enzymes and non-enzymes (13), molecular basis of ligand promiscuity (14), validation of methods for protein functional-site prediction (15) and composition of protein–ligand binding sites (16). Files for docking and scoring studies have been created for a subset of its protein–ligand complexes. The first generation was the CSAR-NRC High Quality (HiQ) set used in the 2010 CSAR Benchmark Exercise (17). After our most recent update, the HiQ set contains 467 complexes. This set has pristine electron density for the ligands and the binding-site residues, which is essential for use in docking studies. The set is comprehensive and not limited to only drug-like molecules.
There are other resources based on the PDB, like Binding MOAD. The one that is most comparable is PDBbind (4), which is also a comprehensive set. PDBbind aims to provide data on all complexes, even those with a resolution poorer than our 2.5 Å limit, and its definition of protein–ligand binding includes protein–protein interactions. The PDBbind ‘core’ set focuses on 195 complexes of drug-like ligands, intended for docking and scoring studies. sc-PDB (5) also focuses on complexes of drug-like ligands, and it has augmented the data with additional information from the literature and calculated physical properties. A unique focus of sc-PDB is the assessment of druggability of binding sites and a pharmacophore-based comparison across the data. The Pocketome (6) also focuses on >2200 binding sites of drug-like ligands. Relibase (7) is comprehensive like Binding MOAD, and its website has unique tools for comparing across binding sites and/or ligands. Each data resource has its own definition of an appropriate binding site or drug-like complex, which can include restrictions on the physicochemical properties of the ligands, the function of the proteins, the number of unique PDB structures for the protein or the availability of additional information from other database resources. Each resource also provides different searching and mining tools on their websites.
The Binding MOAD website was developed in 2004 based on Java 2 Platform, Enterprise Edition (J2EE) using JBoss Application Server (version 4.0). Binding MOAD is updated yearly and has grown from 5331 to 23 269 protein–ligand complexes, with 1375 to 8156 binding affinities, respectively. Since 2010, the affinity data from Binding MOAD have been cross-linked with the PDB website (http://www.rcsb.org) for a quick and direct access. The website receives ∼2000 hits/month. Although the data remained up-to-date, the website itself had not been upgraded in terms of both the underlying platform and additional features since its creation. The legacy of the old JBoss platform had not only restricted Binding MOAD from timely upgrades but also created security holes. To update JBoss required a major rewrite of the code, so we chose to rewrite the platform on the more straightforward ‘LAMP’ environment. In order to bring in the latest technology and meet expectations of users, we have developed a new Binding MOAD website from scratch and added additional features required for improving functionality and security, while handling growth in Binding MOAD.
RESULTS AND DISCUSSION
Binding MOAD statistics
We have seen an increase in Binding MOAD at the rate of ∼2000 complexes and ∼750 affinity measurements per year, on average. Table 1 lists the updates, detailing the increase in data over time. The current version (v2013) of Binding MOAD contains 23 269 high-resolution (2.5 Å or better) protein–ligand complexes. This paper will appear in January 2015, the same time we will initiate our next update. We estimate that v2014 will contain more than 25 000 complexes with binding affinities for ∼9000 protein–ligand pairs. The complexes are grouped into 6960 different families based on 90% sequence identity. It contains 11 173 unique biologically relevant (valid) ligands with invalid ligands additives annotated separately. The experimental binding data—Kd, Ki and IC50 extracted from crystallography literature—are available for 8156 (35%) protein–ligand pairs. The percentage of structures with affinity data grows steady each year as more and more affinity data are reported in the literature. For example, our last update (v2013) added binding constants for 872 (40.4%) complexes out of the 2160 total complexes.
Table 1. Binding MOAD update statistics.
| Release (version, PDB Download date) | Protein–ligand complexes | Protein families (90% identity) | Unique ligands | Binding affinities (coveragea) |
|---|---|---|---|---|
| Initial release in 2004 (2) | 5331 | 1780 | 2630 | 1375 (25.8%) |
| Prior to the website in 2005 | 8250 | 2732 | 3932 | 2374 (28.8%) |
| 1st (v2006, 12/31/2006) (3) | 9836 | 3151 | 4665 | 2950 (30.0%) |
| 2nd (v2007, 12/31/2007) | 11 366 | 3583 | 5348 | 3452 (30.4%) |
| 3rd (v2008, 12/31/2008) | 13 138 | 4078 | 6210 | 4146 (31.6%) |
| 4th (v2009, 12/31/2009) | 14 720 | 4624 | 7064 | 4782 (32.5%) |
| 5th (v2010, 12/31/2010) | 16 948 | 5198 | 8140 | 5630 (33.2%) |
| 6th (v2011, 12/31/2011) | 18 764 | 5772 | 9048 | 6311 (33.6%) |
| 7th (v2012, 12/31/2012) | 21 109 | 6443 | 10 156 | 7284 (34.5%) |
| 8th (v2013, 12/31/2013) | 23 269 | 6960 | 11 173 | 8156 (35.0%) |
aCoverage is the percentage of protein–ligand complexes with binding constants available.
Improvements in the Binding MOAD website
Application platform upgrade
The old Binding MOAD website used the Java-based platform (JBoss 4.0) that could not be updated to the latest JBoss version 7 (or WildFly 8) without a complete rewrite. We used this opportunity to restructure our approach. We selected the LAMP (Linux, Apache, MySQL and PHP) software bundle for our application, which is an open-source, straightforward and well-established platform. Binding MOAD already used a MySQL database in the backend. The Apache server on a Linux platform is used to host the new implementation written in PHP for the new Binding MOAD website. In addition to the LAMP software bundle, we have used CakePHP 2.4 (http://cakephp.org), the rapid development of PHP framework that is based on a Model–View–Controller (MVC) software architecture pattern, which is well suited for websites.
The new Binding MOAD now incorporates many features that not only increase the accessibility to the data but also improve the user interface. We have embedded commonly used third-party plugins into the website to increase the functionality: Jmol (an open-source Java viewer for chemical structures in 3D; http://www.jmol.org) is used to view the protein–ligand complex; MarvinView (Marvin 5.12.3, 2013, ChemAxon; http://www.chemaxon.com) is used to view the ligand's 2D structure and JChem Base (JChem 6.0.0, 2013, ChemAxon; http://www.chemaxon.com) with MarvinSketch is used for structure-based searching of ligands.
Website flow scheme
There are two main menus for retrieving the Binding MOAD database record: ‘Browse’ and ‘Search’. The old scheme had different and unrelated pages for the two menus. ‘Browse’ was a family-based (leader only) listing and ‘Search’ was a simple listing of the matched PDB IDs with associated ligands. In our new scheme, a single common ‘View Listing’ page is used for both menus to bring uniformity by combining common functionalities (Figure 1). In order to improve the usability, the results in the listings can be further filtered and sorted without changing the underlying search query. Some fields in the ‘View Listing’ page are cross-linked to a new quick search that lists all associated records (e.g. clicking a particular Source or Ligand will search all records associated with that Source or Ligand). The records after searching/filtering/sorting can also be downloaded (maximum 1000) as CSV files (binding affinities) and structural PDB files (biounits). The ‘Search’ or ‘Browse’ menus are used to access detailed individual Binding MOAD records via the ‘View Listing’ page.
Figure 1.
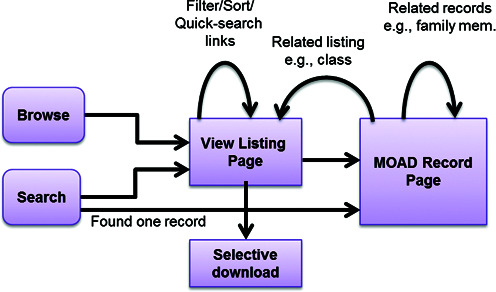
The new Binding MOAD scheme showing page flow and related functionalities in terms of retrieving data from the web interface. An integrated approach allowed the addition of selective downloads based on queries.
Improved user interface and tooltips
We have completely revamped the website and improved the front-end to increase accessibility of the features. Shown below are key improvements in the new website:
- A contextually dynamic sidebar has been added that remains fixed in the page while scrolling. This provides quick links, menus, references, downloads and other information. The sidebar is dynamic as it changes to serve the specific needs of a given page. For example, in a Binding MOAD record page (Figure 2), it provides navigation links, external links and download options related to the current record being displayed. Whereas, in ‘View Listing’ page, it serves as a filtering menu for search refinement.
- A ‘Collapse/Expand’ feature for data tables has been added to provide a summarized view (table headings only) of the page and allow quick access to the relevant data table directly (the navigation menu on the sidebar can also be used; Figure 2). This improves accessibility to the data, especially for large records.
- 3D visualization of the crystal structure is now provided with Jmol using a pre-defined visualization. The backbone is displayed as cartoon and the side chains as lines. A quick menu for Jmol is provided on the right side, which can be used to customize the visualization properties. The binding site cavity and the ligand molecular surface are also generated in the default visualization.
- Infotip (small hover box showing information about the link being hovered) has been added to every important link in order to provide quick help to users of the site. In most of the cases, the information indicates what to expect after clicking.
Figure 2.
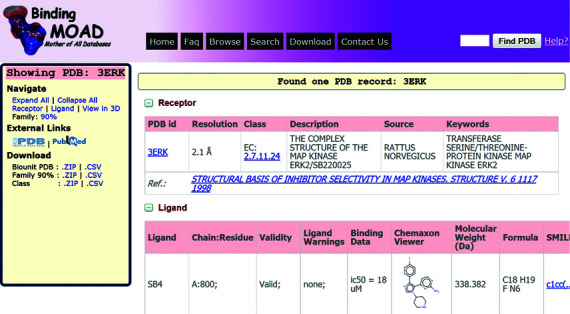
A screenshot of Binding MOAD's new record page for PDB ID: 3ERK. The sidebar provides the related links for navigation, external websites and downloads. The main page contains the data organized in different tables/sections. Receptor and Ligand tables are expanded by default, whereas Jmol molecular viewer and 90% family table are collapsed by default.
New features and functionalities
Search refinement and selective download
A new feature in Binding MOAD allows users to further refine (filter) any current search/browse results. This filtering not only lets users select a customized subset of the records but also provides a mechanism allowing better understanding of the data by trying different quick-filtering. The filtered results can be downloaded as CSV files (binding affinities) or structural PDB files (biounits). The filtering can be performed on enzyme/non-enzyme class and subclass, redundancy (all/leaders), range of binding affinities, PDB resolutions and molecular weight of the ligand (Figure 3). These filterable columns are also sortable, which provides further control on the data visualization. Also, improved pagination allows quick access to all pages of the data.
Figure 3.
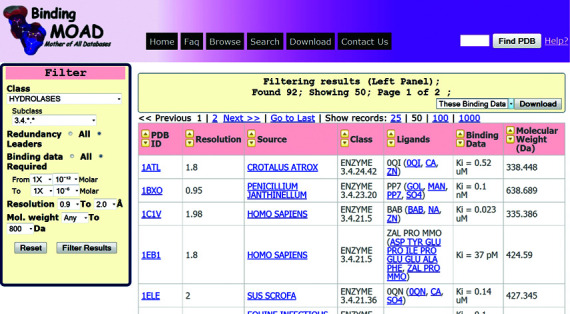
A screenshot of the browse/search listing page showing an example of filtered results based on class/subclass (Hydrolases; EC: 3.4.*.*), redundancy (leaders only), binding data range (pico to micro Molar), pdb resolutions (0.9–2 Å) and molecular weights for ligand (<800 Da).
Ligand structure-based searching
In addition to the previous field-based search, we have now incorporated structure-based searching of ligands using ChemAxon tools. This allows users to search ligands very efficiently by structure (e.g. ‘Substructure’, ‘Superstructure’, ‘Similarity’ and ‘Full structure’) in Binding MOAD. Users can draw chemical structures into the embedded applet MarvinSketch that is used to search the stored Binding MOAD ligands via the JChem Base tool. The matched ligand structures resulting from the search query can be seen in MarvinView and the associated Binding MOAD records for each result can be accessed by the ‘Search MOAD records’ button, as shown in Figure 4.
Figure 4.

A screenshot of the search page that has the additional feature for ligand-structure searching along with the previous field-based searching. Ligand structure/substructure searching is provided using JChem (17), which allows drawing the ligand structure (not shown), searching the database and displaying the results (shown). Here is an example of substructure ligand searching for Naphthalene.
Our Binding MOAD is one of the largest databases for high-quality protein–ligand complexes. We have constantly updated and maintained our database for a decade. The improvements in the Binding MOAD website are necessary to embrace new technology and add new functionalists, which will benefit scientific community in their research toward structure-based drug design.
Acknowledgments
We thank the many coworkers who have contributed to the Binding MOAD project, particularly Dr Mark Benson, PhD.
FUNDING
National Science Foundation [MCB 0546073 to H.A.C.]; National Institutes of Health [U01 GM086873 to H.A.C.]; National Center for Advancing Translational Sciences (NCATS) [UL1TR000433 to A.A.]. Funding for open access charge: National Science Foundation [MCB 0546073]; National Institutes of Health [GM086873, TR000433].
Conflict of interest statement. None declared.
REFERENCES
- 1.Rose P.W., Bi C., Bluhm W.F., Christie C.H., Dimitropoulos D., Dutta S., Green R.K., Goodsell D.S., Prlic A., Quesada M., et al. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 2013;41:D475–D482. doi: 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu L., Benson M.L., Smith R.D., Lerner M.G., Carlson H.A. Binding MOAD (Mother Of All Databases) Proteins Struct. Funct. Bioinform. 2005;60:333–340. doi: 10.1002/prot.20512. [DOI] [PubMed] [Google Scholar]
- 3.Benson M.L., Smith R.D., Khazanov N.A., Dimcheff B., Beaver J., Dresslar P., Nerothin J., Carlson H.A. Binding MOAD, a high-quality protein–ligand database. Nucleic Acids Res. 2008;36:D674–D678. doi: 10.1093/nar/gkm911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y., Liu Z., Li J., Han L., Liu J., Zhao Z., Wang R. Comparative assessment of scoring functions on an updated benchmark: 1. Compilation of the test set. J. Chem. Inf. Model. 2014;54:1700–1716. doi: 10.1021/ci500080q. [DOI] [PubMed] [Google Scholar]
- 5.Desaphy J., Azdimousa K., Kellenberger E., Rognan D. Comparison and druggability prediction of protein–ligand binding sites from pharmacophore-annotated cavity shapes. J. Chem. Inf. Model. 2012;52:2287–2299. doi: 10.1021/ci300184x. [DOI] [PubMed] [Google Scholar]
- 6.Kufareva I., Ilatovskiy A.V., Abagyan R. Pocketome: an encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012;40:D535–D540. doi: 10.1093/nar/gkr825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hendlich M., Bergner A., Günther J., Klebe G. Relibase: design and development of a database for comprehensive analysis of protein-ligand interactions. J. Mol. Biol. 2003;326:607–620. doi: 10.1016/s0022-2836(02)01408-0. [DOI] [PubMed] [Google Scholar]
- 8.Liu T., Lin Y., Wen X., Jorissen R.N., Gilson M.K. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007;35:D198–D201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golovin A., Dimitropoulos D., Oldfield T., Rachedi A., Henrick K. MSDsite: a database search and retrieval system for the analysis and viewing of bound ligands and active sites. Proteins Struct. Funct. Bioinform. 2005;58:190–199. doi: 10.1002/prot.20288. [DOI] [PubMed] [Google Scholar]
- 10.Michalsky E., Dunkel M., Goede A., Preissner R. SuperLigands—a database of ligand structures derived from the Protein Data Bank. BMC Bioinformatics. 2005;6:122. doi: 10.1186/1471-2105-6-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puvanendrampillai D., Mitchell J.B.O. Protein Ligand Database (PLD): additional understanding of the nature and specificity of protein–ligand complexes. Bioinformatics. 2003;19:1856–1857. doi: 10.1093/bioinformatics/btg243. [DOI] [PubMed] [Google Scholar]
- 12.Smith R.D., Hu L., Falkner J.A., Benson M.L., Nerothin J.P., Carlson H.A. Exploring protein-ligand recognition with Binding MOAD. J. Mol. Graph. Model. 2006;24:414–425. doi: 10.1016/j.jmgm.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Carlson H.A., Smith R.D., Khazanov N.A., Kirchhoff P.D., Dunbar J.B., Benson M.L. Differences between high- and low-affinity complexes of enzymes and non-enzymes. J. Med. Chem. 2008;51:6432–6441. doi: 10.1021/jm8006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sturm N., Desaphy J., Quinn R.J., Rognan D., Kellenberger E. Structural insights into the molecular basis of the ligand promiscuity. J. Chem. Inf. Model. 2012;52:2410–2421. doi: 10.1021/ci300196g. [DOI] [PubMed] [Google Scholar]
- 15.Verspoor K.M., Cohn J.D., Ravikumar K.E., Wall M.E. Text mining improves prediction of protein functional sites. PLoS One. 2012;7:e32171. doi: 10.1371/journal.pone.0032171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khazanov N.A., Carlson H.A. Exploring the composition of protein-ligand binding sites on a large scale. PLoS Comput. Biol. 2013;9:e1003321. doi: 10.1371/journal.pcbi.1003321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunbar J.B., Jr, Smith R.D., Yang C.Y., Ung P.M., Lexa K.W., Khazanov N.A., Stuckey J.A., Wang S., Carlson H.A. CSAR benchmark exercise of 2010: selection of the protein-ligand complexes. J. Chem. Inf. Model. 2011;51:2036–2046. doi: 10.1021/ci200082t. [DOI] [PMC free article] [PubMed] [Google Scholar]