


Abstract
Following the discovery of serious errors in the structure of biomacromolecules, structure validation has become a key topic of research, especially for ligands and non-standard residues. ValidatorDB (freely available at http://ncbr.muni.cz/ValidatorDB) offers a new step in this direction, in the form of a database of validation results for all ligands and non-standard residues from the Protein Data Bank (all molecules with seven or more heavy atoms). Model molecules from the wwPDB Chemical Component Dictionary are used as reference during validation. ValidatorDB covers the main aspects of validation of annotation, and additionally introduces several useful validation analyses. The most significant is the classification of chirality errors, allowing the user to distinguish between serious issues and minor inconsistencies. Other such analyses are able to report, for example, completely erroneous ligands, alternate conformations or complete identity with the model molecules. All results are systematically classified into categories, and statistical evaluations are performed. In addition to detailed validation reports for each molecule, ValidatorDB provides summaries of the validation results for the entire PDB, for sets of molecules sharing the same annotation (three-letter code) or the same PDB entry, and for user-defined selections of annotations or PDB entries.
INTRODUCTION
Validation of biomacromolecular structures has become a very important topic, because some published structures have been found to contain serious errors (1–4). The first step in the validation of biomacromolecules and their complexes is checking the standard building blocks, namely, standard amino acids and nucleotides. The usual procedure is to evaluate specific properties of each residue (e.g. electron density, atom clashes, bond lengths, bond angles, torsion angles, etc.). Various software tools have been developed to perform such analyses, e.g. WHAT_CHECK (5), PROCHECK (6), MolProbity (7) and OOPS (8).
The next key step is the validation of ligands and non-standard residues in biomacromolecular structures, which can be performed in a similar manner as for standard residues (focus on electron density, atom clashes, etc.). An example of software specialized on this type of validation is ValLigURL (9). This approach was also added to several software tools focused on the validation of standard residues (Mogul (10), Coot (11), PHENIX (12)).
A different ligand validation approach, which can be denoted as validation of annotation, was developed later. The goal of this approach is to evaluate if the ligand or non-standard residue is annotated correctly (i.e. if its structure corresponds to the three-letter code it was assigned in the Protein Data Bank (PDB) file format). Specifically, the topology and stereochemistry of the validated molecule are compared to those of a reference molecule (model), and any differences found are reported. The first software tool implementing this methodology has been pdb-care (13), a tool specialized on carbohydrates. The next step has been MotiveValidator (14), which allows validation of all ligands and residues, performs basic validation analyses and reports basic warnings (substitutions, foreign atoms, different naming). Because this approach is relatively young, the available tools cover only some of its key topics, leaving many aspects to be explored or improved.
At the same time, with the exponential increase in the size of structural databases, the concept of storing precomputed validation results is becoming increasingly attractive. The first step in this direction was achieved by the PDBREPORT database (5), which is a collection of the outputs from the WHAT_CHECK program. Afterward, the PDB_REDO database (15) of validation results for existing PDB entries was published. Recently, wwPDB included validation reports (16) providing detailed validation results for individual PDB entries directly into their pages.
In our work, we address all challenges described above. We first developed and implemented an improved approach for the validation of annotation, which we subsequently applied to validate all ligands and non-standard residues in the PDB. We then collected all results and built the database ValidatorDB, which offers several advantages over currently available tools (ValLigURL, pdb-care, MotiveValidator):
ValidatorDB is a database of precomputed validation results for all ligands and non-standard residues in the PDB (except small molecules having fewer than seven heavy atoms).
ValidatorDB provides summaries of the validation results for the entire PDB, for sets of molecules sharing the same annotation or the same PDB entry, and for user-defined selections of annotations or PDB entries.
ValidatorDB provides a systematic insight into validation results. The validation analyses are classified into three main categories (Completeness, Chirality and Advanced), each containing several related analyses.
ValidatorDB classifies the types of chirality errors, enabling the user to distinguish between serious chirality issues and minor inconsistencies.
ValidatorDB performs novel analyses and can report completely erroneous ligands, alternate conformations, identity with the model molecules, etc. Such analyses can provide information valuable for further data processing.
ValidatorDB obtains correct structures of ligands and non-standard residues from the wwPDB Chemical Components Dictionary (wwPDB CCD) (17), which it uses as reference molecules (models) during validation. ValidatorDB is updated weekly, and is freely available via the Internet at: http://ncbr.muni.cz/ValidatorDB.
VALIDATION ANALYSES
As ValidatorDB implements the approach of validation of annotation, each validated molecule is compared against a model with the same annotation from wwPDB CCD. The validation analyses performed by ValidatorDB cover the main issues which have been observed in the topology (2D structure) and geometry (3D structure) of ligands and non-standard residues, and which are important for their correct annotation. These validation analyses, along with their respective results, can be classified into three categories, namely, Completeness, Chirality and Advanced analyses (Figure 1). If no issues are found during these analyses, the molecule is marked as having complete structure and correct chirality (Figure 1a).
Figure 1.
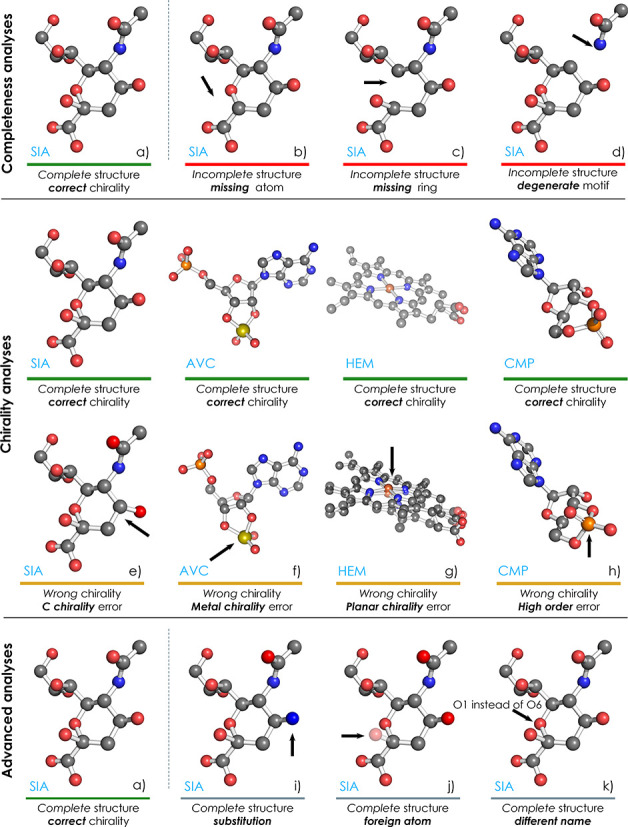
Examples of results provided by different validation analyses. ValidatorDB classifies results into three main categories (Completeness, Chirality, Advanced), each referring to several related analyses. Information about the source of the particular molecules displayed here is given in Supplemental Table S3.
The Completeness analyses attempt to find which atoms are missing (Figure 1b), whether these atoms are part of rings (Figure 1c) or the structure is degenerate, i.e. the molecule contains very severe errors (Figure 1d). These severe errors may refer to residues overlapping in the 3D space, or atoms which are disconnected from the rest of the structure. Validated molecules exhibiting an error in at least one of the Completeness analyses are denoted as incomplete, whereas the remaining molecules are reported as complete.
The Chirality analyses are performed only on complete structures, and aim to evaluate the chirality of each atom in the validated molecule. We distinguish between several types of chirality errors: on carbon atoms (C chirality, Figure 1e), on metal atoms (Metal chirality, Figure 1f), on atoms with four substituents in one plane (Planar chirality, Figure 1g), on atoms connected to at least one substituent by a bond of higher order (High-order chirality, Figure 1h) and the remaining chirality issues (Other chirality). If no issues are detected during the chirality analyses, the validated molecule is marked as having Correct chirality, whereas the remaining molecules are marked as having Wrong chirality. Some types of chirality errors do not constitute real issues, but are artifacts of the automated chirality-determination procedure (i.e. planar chirality and high-order chirality). Therefore, if the validated molecule is found to have these chirality errors, but no other type of chirality issues, the molecule is marked as having ‘Correct chirality (tolerant)’.
The Advanced analyses are focused on issues which are not real chemical problems, but which can complicate further processing and exploration of data, and thus should be noted. When issues are found during an advanced analysis, a warning is reported: Substitution, Foreign atom, Different naming, Zero root mean square deviation (RMSD) or Alternate conformations. The Substitution analysis (Figure 1i) reports the replacement of some atom by an atom of a different chemical element. The Foreign atom analysis (Figure 1j) detects atoms which originate from the neighborhood of the validated molecule (i.e. having different PDB residue ID than the majority of the validated molecule), and generally marks sites of intermolecular linkage. The Different naming analysis (Figure 1k) identifies atoms whose name in PDB format are different than the standard convention for the validated molecule. The Zero RMSD analysis reports molecules whose structure is identical (RMSD = 0 Å) to the model from wwPDB CCD. The Alternate conformation analysis informs about the occurrence of alternate conformations in the validated PDB entry.
DATA PREPARATION
Validation procedure for a single molecule
The starting information characterizing the investigated molecule consists of a PDB residue ID, annotation and PDB ID. According to this information, the input motif is extracted from the PDB entry under investigation. The input motif contains all atoms with the given PDB residue ID, along with their surroundings (atoms within two bonds from any atom of the investigated molecule). The annotation of the molecule is used to identify a suitable model from wwPDB CCD, which then serves as the correct reference structure. The validation proceeds by identifying the maximum common subgraph between the input motif and the model. The atoms of the input motif which belong to this common subgraph make up the validated molecule, which can thus be reliably identified in the PDB entry under investigation. The validated molecule and the model are then superimposed (18) in such a way that their RMSD is minimal. The superimposition provides a pairing (bijection) between atoms in the validated molecule and the corresponding (chemically equivalent) atoms in the model. This bijection allows comparing various properties of each atom in the validated molecule with those of the chemically equivalent atom from the model. All the validation analyses are based on this comparison of atom properties (presence, chirality, element symbol, PDB name, etc.). Other unusual aspects encountered during validation are reported as processing warnings (e.g. which conformer was validated if several conformers were present). A scheme of the validation procedure is depicted in Supplemental Figure S1.
Generation of validation data for all ligands and non-standard residues in the entire Protein Data Bank
The latest versions of the PDB and wwPDB CCD are downloaded once a week, and the following steps ensue.
Obtaining a set of models for validation
Select all models from wwPDB CCD which contain at least seven heavy atoms, excluding the five standard nucleotides and their common deoxy- forms, the 20 standard amino acids and selenomethionine (MSE). ValidatorDB does not focus on the standard building blocks of biomacromolecules because many tools already cover these. Additionally, MSE is also excluded from validation due to its extremely high occurrence in the PDB (markedly higher than other ligands and non-standard residues) and high incidence of circumstantial inclusion in biomacromolecules (to aid X-ray crystallography experiments).
Obtaining validation results for all ligands and non-standard residues in a single PDB entry
For a PDB entry with a given PDB ID, identify the PDB residue IDs of all molecules sharing the annotation with any model obtained in the previous step. Using the procedure described in the first step, detect all validated molecules (via PDB residue ID and corresponding annotation) and compare them to the appropriate models. Collect the validation results for all molecules validated in this PDB entry, and summarize the results of each validation analysis.
Obtaining PDB-wide validation results for each ligand or non-standard residue
For each set of molecules sharing the same annotation, collect validation results from all PDB entries and summarize the results of each validation analysis.
Obtaining a validation overview for the entire PDB
Collect and summarize the results of all types of validation analyses for all validated molecules, irrespective of annotation or PDB entry.
While the algorithm we use for data preparation is generally applicable, highly automated and produces results with straightforward interpretation, it does have limitations. These limitations are described in detail in the Supplementary Material.
DATABASE ORGANIZATION
ValidatorDB provides the user with direct access to a wide range of validation reports, where the results of the validation analyses are organized on several levels. Specifically:
Validation report for a particular molecule or a set of molecules (accessible via Search → Molecule Identifier), depicted in Figure 2.
Validation report for a particular PDB entry or a set of PDB entries (accessible via Search → PDB Entry).
Validation report for a particular annotation or a set of annotations (accessible via Search → Molecule Annotation).
Table with validation results for all PDB entries (accessible via Details by PDB Entry).
Table with validation results for all annotations (accessible via Details by Molecule).
Graph with results of all validation analyses for the entire PDB (accessible via Overview).
Figure 2.
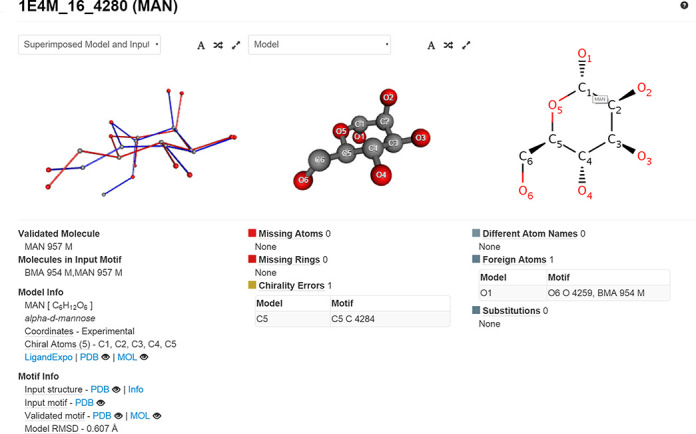
Detailed validation report for the saccharide MAN 957 from PDB entry 1E4M. The structure is complete, but exhibits a chirality error on atom C5. Additionally, the warning of foreign atom at position O1 indicates that this molecule is part of an oligosaccharide chain.
A description of the ValidatorDB user interface is provided in the ValidatorDB Wiki Manual.
RESULTS AND DISCUSSION
Validation results for the entire PDB
One of the advantages of ValidatorDB is that it can provide a straightforward overview of the quality of ligands and non-standard residues in the entire Protein Data Bank. The results in Supplemental Table S1 show that currently the PDB (10 August 2014) contains about 9% incomplete ligands and non-standard residues, out of which about 6% miss at least one atom and 2.6% miss rings. Chirality problems occur in less than 8% of the validated molecules. The frequency of basic chirality errors is even lower—only 2.4% of molecules exhibit chirality errors on a carbon atom, and 1.4% on a metal atom. Other chirality issues are generally reported more frequently—i.e. 4.3% of molecules have wrong High-order chirality plus 1.1% wrong Planar chirality, but the majority of these are very probably artifacts (as mentioned in the section Validation Analyses). Therefore, about 83% of validated molecules are complete and have correct chirality. This statement is slightly more optimistic than previous estimations, which are based on the fit to electron density and 3D structure of the ligands and place the expected percentage of erroneous molecules between 20 and 30% (19,20). The situation appears even better if we exclude the chirality errors reported during the Planar and High-order chirality analyses. Specifically, about 88% of molecules are complete and have correct chirality for all carbon and metal atoms.
On the other hand, the issues found by the Advanced analyses occur more frequently than completeness and chirality errors. More than 20% of the validated molecules contain substitutions, and about 35% have at least one atom formally located in the neighbor residue. Additionally, 38% contain atoms which are not named in agreement with the standard PDB atom naming convention. Overall, the validation was carried out uneventfully for about 30% of the molecules. While the results of the Advanced analyses have no bearing over the chemical soundness of the validated molecules, they indicate that further, especially automated processing of these structures can be very problematic. Therefore, it is indeed useful to validate the structure of ligands or non-standard residues of interest before performing further investigations, especially where a high degree of automation is involved.
Samples
To show the functionality of ValidatorDB and also the importance of such validation analyses, we selected a few interesting samples and included them in the ValidatorDB web page.
Case studies
One important question is how the quality of the structures varies for different classes of molecules. We have thus designed and conducted several case studies to show how ValidatorDB can answer such questions. We selected the molecules according to a combination of features related to chemical structure, biological function, area of application, availability, etc. The following classes were defined as subsets of models from wwPDB CCD:
Polycyclic molecules: contain three or more conjugated rings. The molecules containing metals were excluded, as their quality is influenced more by the presence of the metal than by their polycyclic structure.
Carbohydrates: contain the pyran or furan ring. Molecules containing P (e.g. ATP) were excluded, as their quality is influenced more by the occurrence of phosphate derivatives than by the sugar part.
Mannose derivatives: subclass of carbohydrates.
Organometals: contain a metal atom.
Experimental drugs: described in DrugBank (21) as experimental drugs, i.e. have been shown to bind specific proteins in mammals, bacteria, viruses, fungi or parasites.
Approved drugs: described in DrugBank as approved drugs, i.e. have received approval in at least one country.
A list of the annotations of the molecules from each class can be found in the Supplementary Material. Summaries of the validation results for each class are given in Supplemental Table S2.
Compared to the PDB-wide statistics for all ligands and non-standard residues (see above), polycyclic molecules have overall higher quality (higher percentage of molecules with complete structure and correct chirality). Nonetheless, they exhibit more errors in C chirality, probably due to their more complicated, carbon-based scaffolds. Carbohydrate molecules show similar trends as polycyclic molecules, since their structure is also ring-based. However, they exhibit a higher rate of errors in C chirality, a consequence of the fact that they generally contain more chiral atoms. Mannose derivatives play an important role in cell–cell recognition, a biological function which relies heavily on chirality. Therefore, they must have a characteristic structure (determined by chirality) and are also strongly predisposed to have C chirality errors. We found that the percentage of errors in C chirality is over three times higher for mannose derivatives than the PDB-wide evaluation for all ligands and non-standard residues.
Organometals seem to have overall lower quality. Part of the errors is artifacts of our validation algorithm, as such molecules can have very complicated scaffolds (see algorithm limitations in the Supplementary Material). However, the majority of the reported errors are significant, proving that many challenges remain in the field of structure determination for organometals.
On the other hand, the overall quality of the structure of experimental drugs is clearly much higher than the PDB-wide statistics for all ligands and non-standard residues. For approved drugs, i.e. drugs already on the market, the situation is even better. About 95% of these molecules are complete and have correct chirality, a consequence of the fact that markedly more effort is expended in the determination of their structure in biomacromolecular complexes.
CONCLUSIONS
In this article we introduced ValidatorDB, a database of up-to-date validation results for all ligands and non-standard residues from the Protein Data Bank (all molecules with seven or more heavy atoms). The validation of annotation approach implemented here employs correct reference molecules in the form of models from the wwPDB CCD. ValidatorDB offers analyses which cover the main aspects of validation of annotation, by systematically evaluating the completeness, chirality and other features of the validated molecules. ValidatorDB is the only validation tool able to report several types of chirality errors, which allows distinguishing between serious chirality issues and formal inconsistencies. ValidatorDB can further report completely erroneous ligands, alternate conformations, identity with the model, etc. The validation results are organized systematically, from detailed reports for single molecules, to a PDB-wide general summary, and fully customized reports. All results are available in interactive graphical and tabular form via the web interface, and can be readily downloaded in convenient formats.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
This work was funded by the Ministry of Education, Youth and Sports of the Czech Republic [LH13055], the CEITEC - Central European Institute of Technology [CZ.1.05/1.1.00/02.0068] from the European Regional Development Fund, the “Capacities” specific program [286154] and by INBIOR [CZ.1.07/2.3.00/20.0042] from the European Social Fund and the state budget of the Czech Republic. Additional support was provided by the project “Employment of Newly Graduated Doctors of Science for Scientific Excellence” [CZ.1.07/2.3.00/30.0009] co-financed from the European Social Fund and the state budget of the Czech Republic. Funding for open access charge: European Social Fund and the State Budget of the Czech Republic [CZ.1.07/2.3.00/20.0042].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kleywegt G.J. On vital aid: the why, what and how of validation. Acta Crystallogr. D. Biol. Crystallogr. 2009;65:134–139. doi: 10.1107/S090744490900081X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matthews B.W. Five retracted structure reports: inverted or incorrect. Protein Sci. 2007;16:1013–1016. doi: 10.1110/ps.072888607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rupp B. Detection and analysis of unusual features in the structural model and structure-factor data of a birch pollen allergen. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 2012;68:366–376. doi: 10.1107/S1744309112008421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnston C.A., Kimple A.J., Giguère P.M., Siderovski D.P. Structure of the parathyroid hormone receptor C terminus bound to the G-protein dimer Gbeta1gamma2. Structure. 2008;16:1086–1094. doi: 10.1016/j.str.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Hooft R.W., Vriend G., Sander C., Abola E.E. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 6.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 7.Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleywegt G.J., Jones T.A. Efficient rebuilding of protein structures. Acta Crystallogr. D. Biol. Crystallogr. 1996;52:829–832. doi: 10.1107/S0907444996001783. [DOI] [PubMed] [Google Scholar]
- 9.Kleywegt G.J., Harris M.R. ValLigURL: a server for ligand-structure comparison and validation. Acta Crystallogr. D. Biol. Crystallogr. 2007;63:935–938. doi: 10.1107/S090744490703315X. [DOI] [PubMed] [Google Scholar]
- 10.Bruno I.J., Cole J.C., Kessler M., Luo J., Motherwell W.D.S., Purkis L.H., Smith B.R., Taylor R., Cooper R.I., Harris S.E., et al. Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci. 44:2133–2144. doi: 10.1021/ci049780b. [DOI] [PubMed] [Google Scholar]
- 11.Debreczeni J.É., Emsley P. Handling ligands with Coot. Acta Crystallogr. D. Biol. Crystallogr. 2012;68:425–430. doi: 10.1107/S0907444912000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lütteke T., von der Lieth C.-W. pdb-care (PDB carbohydrate residue check): a program to support annotation of complex carbohydrate structures in PDB files. BMC Bioinformatics. 2004;5:69–74. doi: 10.1186/1471-2105-5-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vařeková R.S., Jaiswal D., Sehnal D., Ionescu C.-M., Geidl S., Pravda L., Horský V., Wimmerová M., Koča J. MotiveValidator: interactive web-based validation of ligand and residue structure in biomolecular complexes. Nucleic Acids Res. 2014;42:W227–W233. doi: 10.1093/nar/gku426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joosten R.P., te Beek T.A.H., Krieger E., Hekkelman M.L., Hooft R.W.W., Schneider R., Sander C., Vriend G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011;39:D411–D419. doi: 10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berman H.M., Kleywegt G.J., Nakamura H., Markley J.L. The Protein Data Bank archive as an open data resource. J. Comput. Aided Mol. Des. 2014;28:1009–1014. doi: 10.1007/s10822-014-9770-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng Z., Chen L., Maddula H., Akcan O., Oughtred R., Berman H.M., Westbrook J. Ligand Depot: a data warehouse for ligands bound to macromolecules. Bioinformatics. 2004;20:2153–2155. doi: 10.1093/bioinformatics/bth214. [DOI] [PubMed] [Google Scholar]
- 18.Sehnal D., Vařeková R.S., Huber H.J., Geidl S., Ionescu C.-M., Wimmerová M., Koča J. SiteBinder: an improved approach for comparing multiple protein structural motifs. J. Chem. Inf. Model. 2012;52:343–359. doi: 10.1021/ci200444d. [DOI] [PubMed] [Google Scholar]
- 19.Lütteke T., Frank M., von der Lieth C.-W. Data mining the protein data bank: automatic detection and assignment of carbohydrate structures. Carbohydr. Res. 2004;339:1015–1020. doi: 10.1016/j.carres.2003.09.038. [DOI] [PubMed] [Google Scholar]
- 20.Liebeschuetz J., Hennemann J., Olsson T., Groom C.R. The good, the bad and the twisted: a survey of ligand geometry in protein crystal structures. J. Comput. Aided Mol. Des. 2012;26:169–183. doi: 10.1007/s10822-011-9538-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y., Maciejewski A., Arndt D., Wilson M., Neveu V., et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]