

Abstract
Type material is the taxonomic device that ties formal names to the physical specimens that serve as exemplars for the species. For the prokaryotes these are strains submitted to the culture collections; for the eukaryotes they are specimens submitted to museums or herbaria. The NCBI Taxonomy Database (http://www.ncbi.nlm.nih.gov/taxonomy) now includes annotation of type material that we use to flag sequences from type in GenBank and in Genomes. This has important implications for many NCBI resources, some of which are outlined below.
INTRODUCTION
The NCBI Taxonomy Database (1) serves as the standard nomenclature and classification for the International Sequence Database (INSD) comprised of GenBank at the NCBI, ENA at the EBI/EMBL and the DDBJ at the NIG in Japan. Sequence from type is an important subset of GenBank for which we can have a very high level of confidence in the taxonomic identifications, and which we can expect to keep current by following the taxonomic literature. This is particularly relevant for the microbes (the prokaryotes and the cultured fungi, algae and protists) where type material is readily available to the research community from the culture collections.
GenBank is an archive of sequence data, particularly important for sequences associated with publications in the scientific literature. In this case the GenBank entries serve as supplementary material to the publication, and are ‘owned’ by the submitter/author. In this role, it is vital to preserve the original data so that the analysis from the paper can be evaluated and replicated. GenBank also serves as the general reference set of sequence data for current research in biology. There is a tension between these two goals. We try to keep sequences and metadata current, and archive versions of every update to the sequence entries to support the archival requirement. We update the taxonomy with respect to synonymy, but rely on the submitters for the correct original taxonomic identification of their specimens and for the appropriate annotation of their source material (in isolate, strain, culture-collection and specimen-voucher qualifiers). This means that there are misidentified sequences in GenBank, and entries with other annotation problems. It is important to have these corrected (or flagged as problematic) to support the reference database requirement. Third-party updates are passed along to the original submitters but are not implemented in GenBank without the submitters’ permission; entries with egregious errors may be suppressed or flagged as ‘unverified’. Sequence from type can help to alleviate these problems by providing a backbone of reliably identified sequence data.
DISCUSSION
Codes of nomenclature
Carl Linnaeus established the standard for scientific nomenclature in taxonomy with the successive editions of Species Plantarum (2) and Systema Naturae (3). Botanists subsequently selected the first edition of Species Plantarum (1753), and zoologists the 10th edition of Systema Naturae (1758), as the official starting point for the publication of taxonomic names. The animal (in 1843) and plant (in 1867) codes of nomenclature grew out of these two works and have developed independently ever since (4,5). Currently active codes of nomenclature are listed in Table 1. The codes formalized the protocols and requirements for the publication and revision of scientific names, orthographic rules for the proper formation of names and the order of priority when names collide. Each Code is associated with a Judicial Commission that rules on disputes, and occasionally overrides the Code in cases of broader scientific merit. Neither the botanical nor the zoological code maintain an approved list of species names, though there have been attempts to establish official registries for new names—currently, ZooBank for the animals and the combination of MycoBank and Index Fungorum and for the fungi.
Table 1. The Codes of nomenclature.
| ICZN | International Code of Zoological Nomenclature http://iczn.org/ |
| ICN | International Code of Nomenclature for algae, fungi, and plants http://www.iapt-taxon.org/nomen/main.php |
| ICNB | International Code of Nomenclature of Bacteria http://www.ncbi.nlm.nih.gov/books/NBK8808/ |
| ICTV | International Committee on Taxonomy of Viruses http://www.ictvonline.org/ |
| ICNCP | International Code of Nomenclature for Cultivated Plants http://www.ishs.org/sci/icracpco.htm |
| CTPPB | Committee on the Taxonomy of Plant Pathogenic Bacteria http://www.isppweb.org/about_tppb.asp |
| BioCode | an alternative universal code http://www.bionomenclature.net/ |
| PhyloCode | a newer alternative universal code http://www.ohio.edu/phylocode/ |
There are several differences between the plant and animal codes. The zoological code only recognizes one infra-specific rank, the subspecies; the botanical code also recognizes varietas and forma beneath the subspecies level. The zoological code does not regulate names above the family level. Until recently the botanical code supported a dual nomenclatural system for the fungi—sexual and asexual forms of the same species could have different formal scientific names. This was eliminated at the last botanical congress, but it will take many years to resolve the existing dual nomenclature. There are also differences in the ways that names can be formed and revised, and differences in the types of type material that are recognized by the codes.
The bacteriological code (6) separated from the botanical code, and picked 1980 as the starting date for modern bacterial nomenclature. An Approved List of bacterial names (including synonyms and subspecie) was established (7), and has been maintained ever since. The International Journal of Systematic and Evolutionary Microbiology (IJSEM) is the official journal for bacterial nomenclature—a new name is not ‘validly published’ unless it has been described in the IJSEM or until it appears on the list of additions to the Approved Lists that is published monthly in the IJSEM and collects the names of bacterial taxa published elsewhere. Names that have published outside of the IJSEM and have not yet appeared on the Approved Lists are said to be ‘effectively published’ but not ‘validly published’—often this is because the description fails to meet some requirements of the code. The lists discussed below include species with both validly and effectively published names.
The central requirement for the description of a new species in the bacteriological code is the designation of a type strain, which must be deposited in at least two different culture collections. After that the culture collections typically swap types around between themselves so a given type strain might end up in dozens of different collections around the world. These are called co-identical strains. Of course, when hundreds of collections exchange thousands of strains there is potential for error. Simply passaging a strain can lead to contamination, so quality control is an important issue for the culture collections. This is a significant difference from the plant and animal codes, where it is actually not permissible to designate a living specimen as type. Due to the ready availability of type stains from culture collections it is easy to get sequence from type in the microbes.
The requirement of a pure (axenic) culture for the formal description of a new species sets the bar pretty high in the prokaryotes where the vast majority of diversity Is uncultured, much of it unculturable with current techniques. To address this need the category of Candidatus names was created, allowing the introduction of formal names for uncultured species of bacteria into the literature (8,9). These names currently have no standing in the nomenclature under the Bacteriological Code, but the Judicial Commission has approved of their use (10). These names are written in the literature with the category ‘Candidatus’ in italics and the Latin binomial in Roman font, e.g.: Candidatus Phytoplasma trifolii, Candidatus Endobugula sertula. Candidatus taxa will generally not have type material, but some of them may have genomes in GenBank from single-cell sequencing or assembly from metagenomes. We can use the ‘reference material’ name-type in taxonomy to designate reference sequences for these taxa.
The cyanobacteria (aka blue-green algae) have traditionally been treated under the botanical code, which is not a good fit. They do not appear on the Approved Lists of bacterial species names and the research community tends to use genus names with culture collections (e.g. Anabaena PCC7122) in place of binomial species names.
The viruses do not have a code of nomenclature like the other three groups; they maintain an approved list of species names, currently updated annually. New species names are not published in the literature but are proposed to one of 160 committees that rule on changes to the nomenclature for different groups of viruses. Virus species names are not associated with type material, so most of the discussion here will not be of direct relevance to this group.
There are also specialty codes of nomenclature, for the cultivated plants and for the plant pathogenic bacteria, and several modern attempts to develop a universal code—the BioCode and the phylogenetically based Phylocode.
There is no real attempt to keep names unique between the different codes of nomenclature—Bacillus and Bacteria are genera of stick insects and Archaea is a genus of spiders. The zoological code regulates three independent sets of names (species-group names, genus-group names and family-group names) so it is legal to list the damselfly genus Lestoidea in a superfamily with the same name. As many as 10% of the genus names in current use may be duplicated between the three codes, so many species with common epithets are duplicated as well—Agathis montana (both a wasp and a conifer) was the first to be represented with sequence from both species. Another problem of this sort is unique to the viruses, where the code makes a distinction between ‘virus names’ and ‘species names’. In Roman font Enterobacteria phage T4 is a virus name; in italic font Enterobacteria phage T4 is a species name that includes all of the T-even phages and more. Resolving these duplicated names between the several codes of nomenclature will be a major chore for taxonomy in the 21st century.
Type material
The idea of type material dates to the codes of nomenclature, a century after Linnaeus. Type specimens serve as the exemplars of the species. There are many types of type—Table 2 lists the types that are mentioned by one or more of the codes of nomenclature. The situation is simplest in the bacteria—the only complication is ‘neotype strain’, which is designated whenever the original type strain is lost or contaminated. The eukaryotes are much more complex, but in practice most of the types we see will be holotypes or paratypes. Holotype is the important one; this is the single specimen that is designated as the ‘name-bearing’ type of the new species. The other less common name-bearing types are the neotype (designated when the type specimen is lost), syntypes, lectotype and hapanotype. Any number of paratypes may also be designated by the original author. There are two main reasons to designate paratypes—to show the range of variation in the new species, but also to deposit them in a variety of museums around the world making them more accessible to the taxonomic community. We also list three name-types not covered by code—the generic ‘type material’ for types of unknown type, ‘reference material’ which we use to designate source material that is not from type for reference purposes and a series of ‘culture from type’ name-types. These are for use with eukaryotic microbes, where living cultures derived from the type specimen (or cultures from which the type specimen was prepared) are not themselves formally considered to be type material in either the botanical or zoological codes—though these will be the source of most of the sequence we see in GenBank.
Table 2. Types of type. Parenthesized letters indicate the code of nomenclature for which the type is valid (B) botanical code, (Z) zoological code and (P) bacteriological code.
| type strain | (P) | deposited in at least two different culture collections |
| neotype strain | (P) | replacement culture for a type that has been lost |
| holotype | (BZ) | single name-bearing type specimen |
| paratype | (BZ) | other specimens designated in the original type series |
| neotype | (BZ) | single name-bearing specimen designated when holotype is lost |
| allotype | (Z) | designated type specimen of opposite sex to the holotype |
| epitype | (B) | other specimen designated in subsequent type series |
| isotype | (B) | duplicate specimen of the holotype |
| syntype | (BZ) | one of a name-bearing series of type specimens |
| isosyntype | (B) | duplicate specimen of a syntype |
| lectotype | (BZ) | syntype subsequently designated as the single name-bearing type |
| paralectotype | (B) | syntype not subsequently designated as lectotypes |
| hapanotype | (B) | protist assemblage collectively designated as holotype |
| type material | (-) | type material of unknown type |
| reference material | (-) | non-type material designated for reference purposes |
| culture from type | (-) | living eukaryotic culture derived from type material |
If a species is revised, and broken into several new species, the name-bearing type (type strain or holotype) determines which of the successor species will retain the old name. On the other hand, it is possible that a paratype specimen of an existing species could be designated as the holotype specimen of a new species.
There are any number of other types of types that can be found in the literature (11), from phylotypes (a molecular sequence chosen to represent an operational taxonomic unit (OTU) to splatotypes (samples recovered from windshields, or jet engines). One of the most useful is the topotype, a specimen collected from the ‘type locality’ where the holotype was originally collected. We have chosen not to represent topotypes in the taxonomy database, in part because they are not mentioned in any of the codes, but also because there is no rigorous definition of what constitutes ‘from the type locality’.
Type specimens should not be confused with type species, which function at a higher level in the taxonomic hierarchy. When a new genus is described, one of the requirements of the codes is to designate a type species. The subgenus that includes the type species (the ‘nominal subgenus’) uses the same name as the corresponding genus, and the subspecies that includes the type specimen (the ‘nominal subspecies’) uses the same name as the corresponding species—as in the name Gorilla (Gorilla) gorilla gorilla. If the genus is later broken apart, the original genus name remains with the type species. If the genus Drosophila is revised by promoting the subgenera, the name Drosophila melanogaster will change to Sophophora melanogaster, since it is not in the same subgenus as the type species Drosophila funebris. A proposal to change the type species of Drosophila to melanogaster was sent to the judicial commission of the zoological code, but that was rejected (Case 3407, Opinion 2245).
Two types of synonyms are recognized by the codes—objective synonyms (also known as homotypic synonyms) and subjective synonyms (also known as heterotypic synonyms). When a species is first described it is placed in a particular genus, reflected in the first token of the scientific name. If a subsequent revision transfers this species to a different genus, or demotes it to a subspecies, it gets a new name, e.g.: Cobitis longicauda > Nemacheilus longicaudus > Nemacheilus malapterus longicaudus > Paracobitis longicauda. The species epithet (the last token in the scientific name) usually stays the same during this process (though the ending may be tweaked to agree with the gender of the genus)—unless it is transferred into a genus that already contains a species with the same epithet (in which case one of the species will get a replacement name). All of these are different names for the same thing—taxonomists can disagree on which of them is the best name to use, but they would not use two of them to mean different things. In particular, type material associated with the first name in the chain (called the basionym, in this case Cobitis longicauda) applies equally to every name in the chain.
Subjective synonyms are a different story. In this case two species are first described independently, each with its own type material. If another taxonomist decides that they actually represent the same species, one is sunk into synonymy with the other (priority by first publication date determines which name remains). The types remain tied to their respective names even in synonymy, and if the name is later rescued from synonymy it emerges with its original types intact. Another important difference is that subjective synonymies are a matter of taxonomic opinion—taxonomists can (and often do) disagree on whether or not the two names represent the same species.
Curators at most natural history collections will be reluctant to allow destructive sampling of any type material (particularly for the holotypes), and depending on the age and the manner of preservation the specimen may not be a productive source of sequence data—so we may not see a lot of submissions from existing type specimens. New sequencing techniques and less destructive sampling methods may change this situation in the future. As a counter example, see the stunning photo (Figure 5.3, p. 74) of the arm bone from the type specimen of Homo sapiens neanderthalensis (12), which was the source of the first published Neanderthal mitochondrial genome sequence (13).
Species in GenBank
Sequence entries in GenBank are identified with varying degrees of certainty. Some are taken from specimens (or cultures) that can be independently identified by a specialist—some of these come with species-level identifications (formal names), the others get informal names of several sorts. Species with a formal name in the appropriate code of nomenclature are indexed in Taxonomy Entrez with the specified [property]. Sequences from specimens that have not been identified to the species level are annotated with an informal name of one form or another—these represent 66% of the names in Taxonomy (but only 20% of the entries in GenBank). The rest of this discussion will apply only to species (and sequences) with formal scientific names.
Taxonomy was first indexed in Entrez in 1993—at the time there were just over 5000 species with formal scientific names represented in GenBank. Table 3 charts the growth of these numbers for several taxonomic groups. It took just over 10 years to reach the first 100 000 species, 5 years to reach 200 000, but then another 5 years to reach 300 000. No one knows how many species have actually been described in the taxonomic literature, but common estimates run around 2 million—if so, the subset with sequence in GenBank represents 15% of the total. Many of the remaining species are rare, some only represented by a single specimen, and the slowdown may reflect this. Several initiatives (e.g. Barcode of Life) are explicitly focused on extending sequence coverage to all species of life.
Table 3. Species with formal scientific names represented with sequence entries in GenBank.
| 1993 | 2004 | 2009 | 2014 | |
|---|---|---|---|---|
| animals | 1682 | 41 668 | 82 706 | 139 469 |
| plants | 942 | 42 934 | 78 149 | 110 303 |
| fungi | 439 | 11 915 | 19 887 | 28 340 |
| protists | 305 | 4080 | 6544 | 8997 |
| bacteria | 1284 | 6897 | 11 024 | 13 029 |
| archaea | 64 | 299 | 439 | 511 |
| viruses | 442 | 1504 | 1953 | 2051 |
| 5158 | 108 297 | 200 702 | 302 702 |
There are some interesting asymmetries in this table. The greatest growth has been in the eukaryotes, but this still represents a small fraction of the total number of described species. For the viruses and bacteria we have at least some sequence (typically a 16S for the bacteria and a genome for the viruses) from virtually all of the formally described species, so these numbers grow only as new species are described in the literature. For the bacteria this is approaching a thousand species a year, for the viruses something closer to a hundred. Estimates of the total number of species remaining to be described are even hazier, but here the asymmetry clearly goes in the other direction. A common estimate for the total number of eukaryotic species is 10 million (though some run to a 100 million), a growth of one or two orders of magnitude. On the other hand, it is clear from environmental sample surveys that the vast majority of bacterial diversity is not represented by the subset that has been cultured and described to date, and that the total number of prokaryotic species could easily grow by four or five orders of magnitude or more.
Adding to the problem, many of these new species are ‘cryptic species’—not species that are easily recognizable as new, but existing species that turn out to be an assortment of different species when you look at them closely (particularly with sequence data). When this happens some of the existing sequence entries in GenBank may be left with inappropriate names.
Unpublished names (a.k.a. manuscript names) are a special name class in the taxonomy database. We often see proposed new species names prior to publication as authors submit sequence to get GenBank accession numbers to use in their papers. These species descriptions are sometimes never published at all—scientific names that appear in the literature without ever having been formally described are called nomen nudum (literally ‘naked name’). Those that do eventually appear are often published with a slightly (or entirely) different name. When this happens the submitters rarely bother to update the name in their sequence entries in spite of the fact that this is a vital piece of supplementary data in the species descriptions. To avoid these problems we list manuscript names in the ‘unpublished name’ name-type in taxonomy and index the entries with an informal name until the description of the species has actually been published. Unpublished names can be used as search and retrieval terms in the taxonomy, but will not themselves appear on any of our public web pages. We do register type material for unpublished names, but it will not be processed as detailed below until the name has been published.
It is also important to realize that just because a name is valid and an entry is correctly identified that does not necessarily mean that the classification makes sense (Figure 1). Bacteroides pectinophilus is a validly published name, and ATCC 43243 is the type strain (from which we have a whole-genome shotgun (WGS) genome)—but it clearly does not belong with the rest of the genus Bacteroides (which we list in a different bacterial division, the equivalent of a eukaryotic phylum). That is the significance of the square brackets around the genus name in the species binomial. This species and others like it are waiting for someone to publish a revision that updates the nomenclature in an appropriate fashion.
Figure 1.

Correctly identified but taxonomically misplaced species, in need of a formal taxonomic revision.
Type material in taxonomy
The importance of identifying sequence from type material in GenBank has been previously recognized (14–16). To support this we have embarked on a project to curate type material in the taxonomy, and use it to flag sequence from type in GenBank. We added ‘type material’ as a name type in the Taxonomy database, and we keep track of all of the types of type listed in Table 2. Type material is annotated at the species (or subspecies, varietas or forma) nodes in taxonomy, and inherited throughout the corresponding subtrees (though type material at a subspecies overrides type material at the species level). In particular, type material is not annotated at strain-level nodes if they have been created for genome sequencing projects in the past—type is an attribute of the species (or subspecies) level names themselves. Type data for nominal subspecies are stored at the corresponding species-level node.
We have obtained type data from several sources (17–27) (Table 4)—first of all from the literature. The prokaryotes are particularly well endowed, with the IJSEM acting as the gatekeeper for the nomenclature, and Bergey's Manuals as comprehensive references. Unfortunately, these data are not freely available in computer-readable form, but given the current pace of new species description it is possible to stay current manually. There are several resources that collect type data. StrainInfo has the largest collection of type data (gathered from the culture collections) and some very useful tools (including strain history trees that track the passage of strains between culture collections)—but any errors in the culture collections are reflected at StrainInfo. The List of Prokaryotes with Standing in Nomenclature (LPSN) has a smaller but more carefully curated list of types; we use this as our standard reference. The LPSN also collects data on names that are effectively but not validly published. NamesforLife has a smaller but even more reliable list of types (only the ones associated with nomenclatural acts published in the taxonomic literature). This is the database used by the IJSEM, Bergey's Manuals and by GOLD/JGI, but the licensing cost to redistribute the data was prohibitive for us. And finally, the DSM maintains ‘Prokaryotic Nomenclature Up-to-date’, the most comprehensive freely available machine-readable data set of prokaryotic type strains that we have found. We have also obtained type data from the official registries of fungal names (MycoBank and Index Fungorum), from several culture collections (in particular, the DSM for the bacteria, the CBS for the fungi and the SAG for the algae) and from several of the museums that are collaborating with us in providing hotlinks for our structured voucher qualifiers (MVZ, MCZ, YPM, CAS). In addition, we have compared our type material with that collected in GOLD, the primary outside resource for genome metadata, although we have not used this as a primary source of type data. We have also started to get type data directly from some taxonomic authors, as they realize that it is possible to flag their sequences in GenBank and in Entrez. We have sequences from type for 36 new species of reptiles and amphibian described by S. Blair Hedges, and for 27 new species of red algae described by Gary Saunders. Pedro Crous holds the record, having submitted sequences from type for more than 746 species of fungi, most of his own description.
Table 4. Sources of type material data.
| NamesforLife (17) | proprietary subscription database of prokaryotic nomenclature |
| http://services.namesforlife.com/home | |
| LPSN/Euzeby (18,19) | List of prokaryotic names with standing in nomenclature |
| http://www.bacterio.net/ | |
| StrainInfo (20) | type data collected from culture collections |
| http://www.straininfo.net/ | |
| DSM | DSMZ culture collection and Prokaryotic Nomenclature Up-to-date |
| http://www.dsmz.de/ | |
| http://www.dsmz.de/bacterial-diversity/prokaryotic-nomenclature-up-to-date.html | |
| CBS/MycoBank (21) | fungal culture collection and nomenclature site |
| http://www.cbs.knaw.nl/ | |
| http://www.mycobank.org/ | |
| Index Fungorum (22) | fungal nomenclature site |
| http://www.indexfungorum.org/ | |
| SAG | Culture Collection of Algae at Goettingen University |
| http://www.uni-goettingen.de/en/184982.html | |
| MCZ | Museum of Comparative Zoology, Harvard |
| http://www.mcz.harvard.edu/ | |
| MVZ | Museum of Vertebrate Zoology, Berkeley |
| http://mvz.berkeley.edu/ | |
| CAS:HERP | California Academy of Sciences, Herpetology Collection |
| http://research.calacademy.org/herp | |
| RDP (23) | Ribosomal Database Project |
| http://rdp.cme.msu.edu/ | |
| SILVA (24) | another ribosomal RNA database |
| http://www.arb-silva.de/ | |
| LTP (25) | All-Species Living Tree Project |
| http://www.arb-silva.de/projects/living-tree/ | |
| SOS (26) | Sequencing Orphan Species |
| JGI/GOLD (27) | Genomes On-line Database |
| https://gold.jgi-psf.org/index |
As mentioned above, we have reserved ‘reference material’ to designate strains that are not formally type material. For example, Candidatus Pelagibacter ubique is one of the most abundant species on the planet, and is represented by 11 different genome sequences in GenBank—but it has not yet been possible to publish a formal description that satisfies the Bacteriological Code, and therefore has no type strain (28). We have designated HTCC1062 as ‘reference material’ for Candidatus Pelagibacter ubique, as this is the longest studied and best characterized strain (S. Giovannoni, personal communication). We plan to extend this to other Candidatus taxa, and will also use ‘reference material’ to identify strains of particular medical, regulatory or scientific interest that have been identified by outside agencies or standards groups.
There is plenty of potential for expansion—to date we have focused on collecting types from culture collections and from microbial taxonomy databases (since these give the highest yield of sequence from type in GenBank) but there are several sources of type data for the plants and animals as well (ZooBank, IPNI, taxonomic databases). At some point it may even be possible to harvest type material directly from the literature—Plazi/Pensoft et al. have developed TaxPub, an XML extension to the NLM journal archiving DTD (used by PMC) that allows detailed markup of taxonomic descriptions (29,30).
There is a small set of keywords and a little syntax to the structure of type data in the taxonomy database. ‘strain’ indicates that the type is a strain name, and we should not expect to find the next token in our collections database—also that the name might not be unique (10 different species have type strains named ‘S1’). ‘host’ is used for endosymbionts, where the type is determined by the host species and the relevant metadata is in the /host source feature qualifier. ‘accession’ is used to directly identify the sequence from type when the source feature annotation is not sufficient for this purpose. For example, many museums accession fish in ‘lots’ and the museum catalog number is often not sufficient to identify a type specimen. ‘not’ is used to identify strains that should not be recognized as type for a particular species—for example, a particular culture may have become contaminated, or a paratype specimen may have been designated as the holotype of a different species. And finally, we use double square brackets to indicate that a strain is type of a subjective synonym, e.g. ‘DSM 30147 [[Rhizobium radiobacter]]’. Whenever species are synonymized or rescued from synonymy (a.k.a. resurrected) in Taxonomy (the corresponding nodes merged or unmerged, respectively) care must be taken to ensure that any associated type material is properly apportioned with respect to the relevant names.
We currently list 72 750 items of type data, from 18 847 different species. These are available in the taxonomy dump files on our FTP site, and are searchable in Taxonomy Entrez and in the taxonomy browser.
Sequence from type in GenBank
We have indexed the Nucleotide domain of Entrez with ‘sequence from type [filter]’ which retrieves sequence entries that are derived from type material. This process involves comparing the source feature annotation in the sequence entries with the type material annotation in the taxonomy database. One important aspect of this approach is that type material is added to the taxonomy database independently of source feature annotation in the sequence entries—entries will be flagged as sequence-from-type only if organism and specimen data from both sources agree. This is currently implemented by executing a long list of Entrez queries constructed from the type material annotation in Taxonomy. These queries look for matching source feature annotation in GenBank entries in several places (isolate, strain, culture_collection and specimen_voucher qualifiers) in several different formats (with or without internal spaces, with or without trailing ‘T’ or ‘(T)’, in semicolon-separated lists, &c.). The sequence from type [filter] first appeared in Entrez on November 2013 with just over 400K entries—but is approaching a million entries as of this writing. Table 5 shows several Entrez queries involving type material.
Table 5. Entrez queries with type material.
| Taxonomy | has type material [filter] | 18 847 |
| Taxonomy | has type material [filter] AND Bacteria [subtree] | 11 301 |
| Taxonomy | has type material [filter] AND subspecies [rank] | 247 |
| Nucleotide | sequence from type [filter] | 936 674 |
| Nucleotide | sequence from synonym type [filter] | 9006 |
| Nucleotide | sequence from type [filter] AND Archaea [orgn] | 17 061 |
| Nucleotide | sequence from type [filter] AND Escherichia coli [orgn] | 337 |
| Nucleotide | sequence from type [filter] AND Zootaxa [jour] | 231 |
| Nucleotide | sequence from type [filter] AND Crous P [au] | 3807 |
| Assembly | ‘type material’ [filter] AND latest [prop] | 3365 |
| Assembly | ‘synonym type material’ [filter] AND latest [prop] | 25 |
| Assembly | ‘type material’ [filter] AND Fungi [orgn] | 71 |
| Assembly | ‘type material’ [filter] AND ‘complete genome’ [assembly level] | 813 |
| Assembly | ‘type material’ [filter] AND scaffold [assembly level] | 1193 |
| Assembly | ‘type material’ [filter] AND contig [assembly level] | 1310 |
Type material is currently indexed in three different Entrez databases, all derived from data stored in the taxonomy database. Filter queries in the Assembly database have to be quoted if they contain more than one word. ‘sequence from type’ and ‘type material’ will both work as filter queries in both Nucleotide and Assembly Entrez. Counts reflect entries indexed in Entrez on 9 September 2014.
Figure 2 shows the taxonomic distribution of sequences from type material in GenBank at several different levels. The vast majority are from the microbes—71% of our sequences from type are from the bacteria and 28% from the fungi, with 17K (1.8%) from the archaea and even 9K (0.9%) from the cyanobacteria (in spite of the code of nomenclature issues with this group). These numbers are inflated by WGS contigs, but they reflect the fact that type material is readily accessible for organisms that can be cultured. The middle panel shows the 2013 type sequences from eukaryotes other than fungi, generated by following the ‘more…’ hotlink near the bottom of the first portlet. Even at this scale most of these sequences are from cultures, the exceptions being the animals and the red algae. The final panel shows the distribution of type sequence from the animals. This pitifully small number is overwhelmingly from new species descriptions. While we may never see sequence from many of the existing type specimens in museums and herbaria, it is becoming increasingly common to include at least a little sequence data in new species descriptions (often a COI or matK/rbcL barcode)—often from the holotype specimen. It is also useful to preserve a tissue and/or DNA sample from the types, for future analysis.
Figure 2.
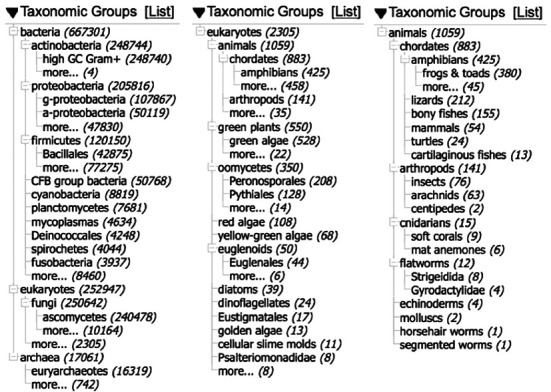
Taxonomic distribution of sequence from type. Taken from the Tree view of the interactive taxonomy portlet on the sidebar of Nucleotide Entrez. (a) All type material. (b) Type material from eukaryotes other than fungi. (c) Type material from the animals.
Figure 3 (taken from the January 2014 NCBI News announcement of sequence-from-type) shows one of the entries associated with the description of Cercopithecus lomamiensis, a new species of primate described in 2012 (31). As is often the case, this was first submitted to GenBank with an unpublished manuscript name, and was originally indexed with the informal name ‘Cercopithecus sp. ASB-2012’, now listed as an ‘equivalent name’. Type material (taken from the paper) is listed explicitly in the entry—YPM MAM 14080 (circled) is the holotype specimen; the rest are paratypes (this information is not surfaced in the taxonomy browser, but it is recorded in the database). The GenBank flatfile entry is annotated with the structured specimen voucher ‘YPM:MAM:14080’—this is a Darwin Core triplet identifier, and indicates that the institution and collection codes can be found in our BioCollection database, where they are registered as the Yale Peabody Museum, Mammal Collection. This is one of the collections that are associated with a URL formula, so the voucher is hotlinked to the corresponding specimen page at the museum web site, shown in the lower left. This is not the flashiest example of a specimen page—some include photos, some keep track of sequence derived from their specimens and link back to GenBank—but this is the first animal sequence entry from a holotype specimen which links to both the full-text description of the species (in PubMed Central and at PLoS ONE) and to the specimen page at the museum. This is common in the prokaryotes, where the full text of the taxonomic literature is generally accessible from PubMed and the culture collections are online, but is rare for the higher eukaryotes, where the taxonomic literature is often not in PubMed at all (though Zootaxa has just started to send abstracts, and ZooKeys sends full-text to PubMed Central), and most natural history collections are just starting to digitize their collections (the NSF is actively funding this effort). We will continue to capture as much sequence from type as possible in the higher eukaryotes, concentrating on sequence submissions from papers that describe new species, but the real type bonanza for GenBank is in the microbes, which will be the focus of the remainder of this paper.
Figure 3.
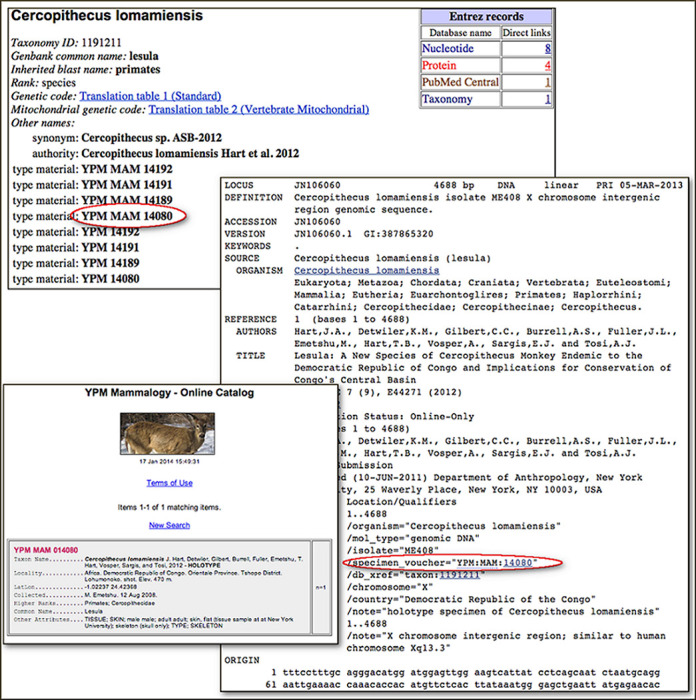
Sequence from type in Cercopithecus lomamiensis. Clockwise from top: (1) taxonomy entry for Cercopithecus lomamiensis. (2) GenBank flatfile format for accession JN106060. (3) Holotype specimen page at the Yale Peabody Museum, Mammal Collection.
The INSD Collaboration (GenBank/ENA/DDBJ) has approved a new source feature qualifier ‘type_material’ which will be populated only by the taxonomy lookup (not supplied directly by submitters). This will replace the /notes seen in some entries (as in Figure 3) and will be used to directly index the sequence-from-type filter without resorting to the Entrez queries that are currently used to create the filter.
RefSeq Targeted Locus reference sets
The RefSeq Targeted Loci project (http://www.ncbi.nlm.nih.gov/refseq/targetedloci/) maintains curated sets of full-length reference sequences from type wherever possible, for ribosomal RNAs. The recently released fungal ITS reference set was developed in collaboration with the fungal community (bioproject PRJNA177353)—2130 of the 2235 reference sequences in this set are from type (32). The 16S reference set is a long-standing collaboration with the RDP, SILVA, GreenGenes and the DSM (bioproject PRJNA33175 for the bacteria and PRJNA33317 for the archaea). When the sequence-from-type filter appeared in Entrez, the RefSeq reference set contained 6800 entries that were identified as type. The sequence-from-type filter recovered 85% of these. We examined each of the 975 entries that were not recovered by the filter. Most were easily corrected either by adding new types to the taxonomy database, or by updating the source feature annotation in the sequence entries—luckily the relevant literature is readily accessible online.
Some entries had no source material annotation (strain, culture collection or isolate) at all, and it was added from the paper. Others had too much—we have not enforced an annotation standard for multiple values in source material qualifiers, so we find them in many formats: ‘HRC/518T (NCTC10147)’ and ‘HD100 = DSM 50701 = ATCC 15356 = ICPB 3268 NCIB 9529’ and ‘type strain: Lam5 = DSM 18033’. Some of these we can resolve by splitting them into strain and culture-collection qualifiers—the rest we are converting to semicolon-separated lists to make them accessible to Entrez queries. Others had simple submission errors—ATCC 12662 instead of IAM 12662, ATCC 49724 instead of ATCC 49725 and NCTC 12370 instead of NCTC 12376. Another common problem is variant spellings of the strain name, particularly cases involving space/no space/hyphen/underscore, or capital I/lowercase l/number 1. There is a correct spelling (published with the original description) but even that can be hard to ascertain, depending on the journal type font. For example, Cryobacterium roopkundense was described with type strain RuGl7 (33), which is correctly represented with an ‘l’ at the LPSN, has a ‘1’ at the DSM and for a while had an ‘I’ at GenBank where I personally replaced one error with a different error while trying to clean up the reference set.
Other problems ran deeper: some were types of subjective synonyms or types of subspecies, not the names they claimed to be types of, and a few were not from type at all. Some reflected a serious problem mentioned above—when as new species is described based on strains that are already represented in the sequence database, there is no mechanism in place to update the earlier entries. Submitters rarely update their own entries, and we do not allow them to update other submitters’ entries. To cope with this problem, we have instituted a new policy. When it comes to our attention that a new species description cites sequence accessions that are already public in GenBank, we update the names in the earlier entries and send a notification to the original submitter (rather than sending a request and waiting for a response). We can maintain this consistently with a feature of the taxonomy lookup known as strain forwarding, which essentially establishes synonymies at the strain level—any combination of organism name and source modifier value can be forwarded to a different name and source modifier value.
For example, AB046995-AB047001 were submitted as part of a study to revise the species Pseudomonas fulva (34). Eventually, a paper was published that split this one species into three, but the earlier entries were not updated with the new names, and the sequences were resubmitted as new entries, AB060131-AB060137. Entries with dated annotation in GenBank can cause considerable confusion. In a particularly egregious example, LPSN listed AB046999 as the reference 16S sequence for Pseudomonas parafulva, the GenBank entry listed the organism name as Pseudomonas fulva, but the strain annotation indicated that it was actually type of the other new species, Pseudomonas cremicolorata. It was straightforward to work this out with the type material explicit in the taxonomy database, and strain forwarding allows us to ensure that other existing (and future) entries will be annotated consistently by transferring everything with Pseudomonas fulva and ‘IAM 1541’ to Pseudomonas cremicolorata.
Throughout this process, we found (and corrected) at least some errors in all of the outside resources that we examined—including the primary literature. There we found cases where the same strain had been designated as type by the same authors for two different species in two different papers (35), where different types had been designated for the same species in different places in the same paper (36), and of course typographical errors in both GenBank accessions and culture collection accessions relating to type strains.
We are using the sequence-from-type filter to recruit new candidate entries for the 16S reference set, which currently includes 16K sequences from 10K species of prokaryotes. We can perform some consistency checks on these data—comparing 16S sequences within the same species (mindful of the fact that some strains show considerable internal variation in their 16S sequences, even though many are homogenized by gene conversion) 6.4% for Thermobispora bispora (37) and 5.7% for Haloarcula marismortui (38). Two large insertions in the 16S sequences of Desulfotomaculum kuznetsovii drive the internal variation to 8.3% (39). We can also compare 16S sequences between different species (mindful of the fact that some closely related species share the same 16S sequence—16S is not really a good barcode marker since it evolves so slowly). The most extreme example of this is between Seliberia stellata and Bradyrhizobium betae (NR_104886 and NR_114198)—these two species share identical 16S sequences, but are listed in different families. In this particular case, Seliberia stellata is a very poorly studied species that was described a long time ago and is probably in need of a taxonomic revision, but we have many examples of real species with identical 16S sequences (40).
The 16S references set is available as a separate Basic Local Alignment Search Tool (BLAST) database, with a pushbutton to restrict the search to type strains (the reference set also includes 16S sequences from a subset of our complete genomes). We are in the process of upgrading the other curated RefSeq sets of prokaryotic ribosomal RNAs with respect to the sequence from type filter.
Genomes from type
Prokaryotic genomes are being sequenced at an increasingly rapid pace, some to completion and some as unfinished partial WGS assemblies. In contrast with the eukaryotes, it is becoming increasingly common to sequence a complete genome to accompany the description of a new prokaryotic species, and there are efforts underway, analogous to the SOS initiative with 16S sequences, to sequence genomes from type for all of species that do not yet have one (41). This flood of data has led to the development of several new methods for building trees based on whole-genome data and will inevitably lead to new models of the species concept in the prokaryotes, which is currently still based on physical DNA reannealing profiles (42–44). The NCBI genomes group currently maintains two such trees, a ‘k-mer tree’ based on shared DNA 28-mers, and a ‘marker tree’ based on length-averaged protein BLAST scores of the ribosomal proteins. Unfortunately, there are misidentified genomes in GenBank just as in the rest of the database—these are easy to spot in the genome trees.
Information about sequenced genomes at NCBI is currently maintained in our Assembly database. We used the sequence from type filter in Nucleotide Entrez to flag the genomes from type in Assembly Entrez. We currently list 3365 genomes from type (3098 bacteria, 204 archaea and 63 fungi) as well as 25 genomes from types of subjective synonyms (e.g. Rhizobium radiobacter, a subjective synonym of Agrobacterium tumefaciens). This represents a slightly smaller number of species because we have several genome sequences from co-identical type strains (e.g. WGS genomes from three different type strains of Escherichia coli—JCM 1649, ATCC 1175 and DSM 30083). One-third of these are complete genome sequences, the rest are unfinished WGS assemblies in various states of completion. We currently have genomes in Assembly for 4019 different prokaryotic species with formal names (including 105 Candidatus species)—in some cases lots of them (4147 genomes from Staphylococcus aureus, 2323 from Escherichia coli and even 11 from Candidatus Pelagibacter ubique). Of these, 2974 have a genome from type in Assembly; 1045 do not.
For species with genomes from type, we can use those as landmarks of correct identification to help resolve misidentified genomes in the k-mer and marker trees (Figure 4a). Several misidentified genomes are immediately apparent once the types are flagged—CP001654 was submitted as Dickeya dadantii, but it is virtually identical to the type genome from Dickeya paradisiaca. CP001655 was submitted as Dickeya zeae, but is virtually identical to the genome of Dickeya chrysanthemi NCPPB 3533—while neither of these are from type, they do fall in the Dickeya chrysanthemi clade. These genomes will be suppressed unless the submitters resolve the discrepancies. There are several other potentially misidentified genomes in this figure that will require further analysis.
Figure 4.
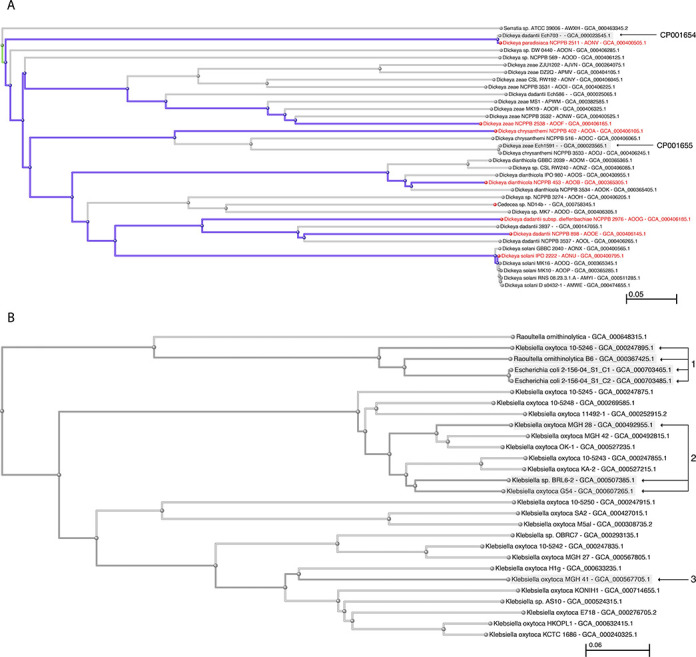
(a) Type genomes in the Dickeya clade of the k-mer tree. (b) Proxytype genomes for Raoultella ornithinolytica and Klebsiella oxytoca in the k-mer tree. (1) Best proxytype candidate genomes for Raoultella ornithinolytica, including one submitted as Klebsiella oxytoca and two misidentified as Escherichia coli. (2) Best proxytype candidate genomes for Klebsiella oxytoca. (3) Best BLAST hit for AB008147, submitted as type sequence from Klebsiella oxytoca.
For the species without genomes from type, we will generally have an assortment of gene sequences from type in GenBank—these can be used to identify the genomes that are closest to type, which can serve as proxies for the type (proxytypes). Proxytypes can be ranked and rated by measuring the similarity with the corresponding sequence-from-type entries in GenBank. Inconsistencies in the sequence similarities that are correlated with a particular co-identical type strain can uncover errors and contamination at the culture collections (without even having any type sequences in common from any of the co-identical strains). Inconsistencies correlated with a particular gene locus can uncover horizontal transfers. For example, two misidentified genomes, submitted as Escherichia coli, were found in the k-mer tree in the neighborhood of Raoultella ornithinolytica and Klebsiella oxytoca (Figure 4b). We have genomes from three different co-identical type strains of Escherichia coli so it is easy to demonstrate that the two misidentified genomes are not from Escherichia coli—but we do not have genomes from type for either Raoultella ornithinolytica or Klebsiella oxytoca. We have two genomes from Raoultella ornithinolytica (which cluster together in a clade with our two misidentified genomes) and 22 genomes from Klebsiella oxytoca (all of which cluster together in a different clade, except for one that is in with Raoultella ornithinolytica).
We do have sequences from type for both of these species—for Raoultella ornithinolytica we have 24 sequences from 10 different loci and 4 different co-identical type strains, and for Klebsiella oxytoca we have 28 sequences from 16 different loci and 6 different co-identical type strains. We blasted these sequences against our genomes to identify the proxytypes, which are summarized in Supplementary Table S1 and highlighted in Figure 4b. rRNA and ITS sequences are excluded from this analysis—for complete genomes the results are complicated by multiple targets and fragmented BLAST hits; for partial (WGS) genomes the rRNA operons are often missing (repeat regions can be difficult to assemble). The Klebsiella oxytoca proxytypes all cluster within a particular subclade—the best candidate genome was submitted as ‘Klebsiella sp. BRL6-2’. The Raoultella ornithinolytica proxytypes also cluster tightly in the k-mer tree, the best include the misidentified Escherichia coli genomes and another submitted as ‘Klebsiella oxytoca 10-5246’. This is easier to understand in light of the taxonomic history of the species Raoultella ornithinolytica—it was originally know as ‘ornithine-positive Klebsiella oxytoca’, subsequently described as Klebsiella ornithinolytica, and later transferred to the new genus Raoultella—many of our Raoultella ornithinolytica sequences were originally submitted as Klebsiella oxytoca (45,46).
Note that one of these strains is not like the others—most of BLAST scores decay consistently, but AB008147 (from JCM 1665, reportedly type of Klebsiella oxytoca) shows a different pattern. This sequence matches best with a different subclade in the k-mer tree, and shares the most similarity with Klebsiella oxytoca MGH 41. There are several possible explanations for this discrepancy—AB008147 may not be from JCM 1665, JCM 1665 may not be from type or the GroES/EL locus may be problematic. As it happens we also have a 16S sequence from JCM 1665 in the RefSeq-targeted locus set (NR_113341 in Supplementary Table S1b)—this tracks with the other co-identical type strains, so it seems likeliest that AB008147 may not be from JCM 1665. Other more complicated explanations are also possible—the type strain of Klebsiella oxytoca could have undergone a horizontal transfer at the GroES/EL locus that changed its affinity in the k-mer tree at this locus. This could be resolved by resequencing this locus in JCM 1665 and in one of the other co-identical type strains. We excluded AB008147 from the proxytype calculation for Klebsiella oxytoca.
The same situation arose in our next proxytype genome calculation (Supplementary Table S1c). AF515643, an rpoB sequence reportedly from a type strain of Enterococcus faecium (DSM 20477), did not appear in any of the BLAST hit lists for the candidate proxytype genomes—instead, it shares 100% identity with two strains of Serratia grimesii. The 16S sequence from DSM 20477 does behave as expected, as does the rpoB sequence from a different type strain of Enterococcus faecium (ATCC 19434), so in this case the problem appears to be with AF515643 itself, and not something more complicated with the locus. The submitter has agreed with this analysis, and AF515643 has been suppressed.
In summary, analysis of sequence-from-type suggests that the two misidentified Escherichia coli genomes are actually Raoultella ornithinolytica, but also that the two genomes that represent the best proxytypes for their respective species should be updated as well, GCA_000507385 from Klebsiella sp. to Klebsiella oxytoca, and GCA_000247895 from Klebsiella oxytoca to Raoultella ornithinolytica. In addition, the expectation of internal consistency among type sequences makes the data set self-correcting—we identified potential problems with putative type sequences from both Klebsiella oxytoca and Enterococcus faecium. We plan to perform this analysis with all of the prokaryotic sequence from type in GenBank.
CONCLUSION
Sequence from type is a high-value subset of GenBank for which we can maintain a very high level of confidence in the taxonomic identification. Nomenclatural acts involving type material are carefully documented in the taxonomic literature, so we can reasonably hope to keep these identifications current. For the microbes, in particular, this gives us a comprehensive set of reliably identified sequence entries that can be used by many other applications (BLAST, pathogen pipeline, RefSeq reference sets, submission triage, &c.). We cannot have complete confidence even in sequences reported to be from type—they may not actually be from the strain they are annotated with, and the strains reported to be from type may not actually be from type—but this is a self-correcting process; 16S and genomic analysis using sequence from type helps to validate the entries themselves, and inconsistencies can uncover errors that reflect on the status of the type material. Diligent curation of sequences from type material in GenBank as outlined above can make this set even more reliable. Species with problematic taxonomy are still problematic, but egregious misidentifications can be found and corrected. This does not solve the more general problem of misidentified entries in GenBank, but does provide a reliable backbone of correctly identified entries that could help support a more general solution.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
NCBI, Jim Ostell and David Lipman. Taxonomy group, esp. Conrad Schoch and Barbara Robbertse for fungal taxonomy and the ITS reference set; Sean Turner, prokaryotic taxonomy and types; Carol Hotton algal taxonomy and types. Taxonomy Software and Database support, Vladimir Soussov and Mikhail Domrachev. GenBank, Ilene Mizrachi and Mark Cavanaugh. WGS, Karen Clark. Genomes, Tatiana Tatusov and Igor Tolstoy. Entrez Indexing, Leonid Khotomiansky. Collections Database, Shobha Sharma. 16S reference set, Rich McVeigh, Kathleen O'Neill and Bill Klimke. Assembly Database, Avi Kimchi and Paul Kitts. Genome Workbench, Mike DiCuccio. k-mer tree, Richa Agarwala. Marker tree, Leonid Zaslavsky. Aiden Parte at the LPSN. Peter Dawyndt at StrainInfo. Raúl Muñoz at SILVA/LTP. Pablo Yarza at SOS. George Garrity at Bergeys/NamesforLife. Vincent Robert and Pedro Crous at CBS/MycoBank. Paul Kirk at Index Fungorum. Hans-Peter Klenk and Brian Tindall at the DSM. Nikos Kyrpides, Tanja Woyke and T.B.K. Reddy at JGI/GOLD. Gordon Jarrell and Dusty McDonald at Arctos. John Wieczorek at the MVZ. Brendan Haley at the MCZ. Lawrence Gall at the YPM. Donat Agosti at Plazi. Richard Pyle at ZooBank. Lyubomir Penev at ZooKeys. Zhi-Qiang Zhang at Zootaxa. Blair Hedges for the herps and Gary Saunders for the red algae.
FUNDING
Intramural Research Program of the National Institutes of Health, National Library of Medicine. Funding for open access charge: Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1.Federhen S. The NCBI Taxonomy Database. Nucleic Acids Res. 2012;40:D13–D25. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linnaeus C. Species Plantarum. 1st edn. Stockholm: 1753. Two volumes. Laurentius Salvius. [Google Scholar]
- 3.Linnaeus C. Systema Naturae. 10th edn. Stockholm: 1758. Two volumes. Holmiae Salvius. [Google Scholar]
- 4.International Commission on Zoological Nomenclature. International Code of Zoological Nomenclature, Fourth Edition. International Trust for Zoological Nomenclature. London: The Natural History Museum; 1999. [Google Scholar]
- 5.McNeill J, Barrie FR, Buck WR, Demoulin V, Greunter W, Hawksworth DL, Herendeen PS, Knapp S, Marhold K, Prado J, et al., editors. International Code of Nomenclature for Algae, Fungi, and Plants (Melbourne Code), Adopted by the Eighteenth International Botanical Congress Melbourne, Australia, July 2011. Bratislava: International Association for Plant Taxonomy; 2012. [Google Scholar]
- 6.Lapage S.P., Sneath P.H.A., Lessel E.F., Skerman V.B.D., Seeliger H.P.R., Clark W.A. International Code of Nomenclature of Bacteria (Bacteriological Code, 1990 Revision) Washington, D.C: American Association for Microbiology; [PubMed] [Google Scholar]
- 7.Skerman V.B.D., McGowan V., Sneath P.H.A. Approved Lists of Bacterial Names, Amended Edition. Washington, D.C: American Society for Microbiology; 1989. [PubMed] [Google Scholar]
- 8.Murray R.G.E., Schleifer K.H. Taxonomic notes: a proposal for recording the properties of putative taxa of procaryotes. Int. J. Syst. Bacteriol. 1994;44:174–176. doi: 10.1099/00207713-44-1-174. [DOI] [PubMed] [Google Scholar]
- 9.Murray R.G.E., Stackebrandt E. Taxonomic note: implementation of the provisional status Candidatus for incompletely described prokaryotes. Int. J. Syst. Bacteriol. 1995;45:186–187. doi: 10.1099/00207713-45-1-186. [DOI] [PubMed] [Google Scholar]
- 10.Judicial Commission of the International Committee on Systematic Bacteriology. Minutes of the meetings, 2 and 6 July 1994, Prague, Czech Republic. Int. J. Syst. Bacteriol. 1995;45:195–196. [Google Scholar]
- 11.Evenhuis N.L. A Compendium of Zoological Type Nomenclature: A Reference Source. Honolulu, Hawaii: Bishop Museum Technical Report 41; 2008. [Google Scholar]
- 12.Pääbo S. Neanderthal Man: In Search of Lost Genomes. NY: Basic Books; 2014. [Google Scholar]
- 13.Krings M., Stone A., Scmnitz R.W., Krainitzki H., Sotoneking M., Pääbo S. Neandertal DNA sequences and the origin of modern humans. Cell. 1997;90:19–30. doi: 10.1016/s0092-8674(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 14.Chakrabarty P. Genetypes: a concept to help integrate molecular systematics and traditional taxonomy. Zootaxa. 2010;2632:67–68. [Google Scholar]
- 15.Harrison I.J., Chakrabarty P., Freyhof J., Craig J.F. Correct nomenclature and recommendations for preserving and cataloguing voucher material and genetic sequences. J. Fish Biol. 2011;78:1283–1290. doi: 10.1111/j.1095-8649.2011.02979.x. [DOI] [PubMed] [Google Scholar]
- 16.Chakrabarty P., Warren M., Page L., Baldwin C. GenSeq: a updated nomenclature and ranking for genetic sequences from type and non-type sources. ZooKeys. 2013;346:29–41. doi: 10.3897/zookeys.346.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garrity G.M., Lyons C. Future-proofing biological nomenclature. OMICS. 2003;7:31–33. doi: 10.1089/153623103322006562. [DOI] [PubMed] [Google Scholar]
- 18.Euzeby J.P. List of bacterial names with standing in nomenclature: a folder available on the internet. Int. J. Syst. Bacteriol. 1997;47:590–592. doi: 10.1099/00207713-47-2-590. [DOI] [PubMed] [Google Scholar]
- 19.Parte A.C. LPSN–list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2014;42:D613–D616. doi: 10.1093/nar/gkt1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verslype B., De Smet W., De Baets B., De Vos P., Dawyndt P. StrainInfo introduces electronic passports for microorganisms. Syst. Appl. Microbiol. 2014;37:42–50. doi: 10.1016/j.syapm.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Crous P.W., Gams W., Stalpers J.A., Robert V., Stegehuis G. MycoBank: an online initiative to launch mycology into the 21st century. Stud. Mycol. 2004;50:19–22. [Google Scholar]
- 22.Kirk P.M. World catalogue of 340 K fungal names. Mycol. Res. 2000;104:516–517. [Google Scholar]
- 23.Cole J.R., Wang Q., Fish J.A., Chai B., McGarrell D.M., Sun Y., Brown C.T., Porras-Alfaro A., Kuske C.R., Tiedje J.M. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quast C., Pruese E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. The SILVA ribosomal RNA database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yilmaz P., Parfrey L.W., Yarza P., Gerken J., Pruesse E., Quast C., Schweer T., Peplies J., Ludwig W., Glöckner F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42:D643–D648. doi: 10.1093/nar/gkt1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yarza P., Sproer C., Swiderski J., Mrotznek N., Spring S., Tindall B.J., Gronow S., Pukall R., Klenk H.P., Lang E., et al. “Sequencing orphan species initiative (SOS): filling the gaps in the 16S rRNA gene sequence database for all species with validly published names. Syst. Appl. Microbiol. 2013;36:69–73. doi: 10.1016/j.syapm.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Pagani I., Liolios K., Jansson J., Chen I.M., Smimova T., Nosrat B., Markowitz V.M., Kyrpides N.C. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2013;40:D571–D579. doi: 10.1093/nar/gkr1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris R.M., Rappe M.S., Connon S.A., Vergin K.L., Siebold W.A., Carlson C.A., Giovanni S.J. SAR11 clade dominates ocean surface bacterioplankton communities. Nature. 2002;420:806–810. doi: 10.1038/nature01240. [DOI] [PubMed] [Google Scholar]
- 29.Catapano T. Bethesda, MD: 2010. TaxPub: AN Extension of the NLM/NCBI Journal Publishing DTD for Taxonomic Descriptions. http://www.ncbi.nlm.nih.gov/books/NBK47086/10.1093/nar/gku1127.html . [Google Scholar]
- 30.Penev L., Lyal C.H., Weitzman A., Morse D.R., King D., Sautter G., Gregoriev T., Morris R.A., Catapano T., Agosti D. XML schemas and mark-up practices of taxonomic literature. Zookeys. 2011;150:89–116. doi: 10.3897/zookeys.150.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hart J.A., Detwiler K.M., Gilbert C.C., Burrell A.S., Emetshu M., Hart T.B., Vosper A., Sargis E.J., Tosi A.J. Lesula: a new species of Cercopithecus monkey endemic to the Democratic Republic of Congo and implications for conservation of Congo's central basin. PLoS ONE. 2012;7:e44271. doi: 10.1371/journal.pone.0044271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoch C.L., Robbertse B., Robert V., Vu D., Cardinali G., Irinyi L, Meyer W., Nilsson R.H., Hughes K., Miller A.N., et al. Finding needles in haystacks: linking scientific names, reference specimens and molecular data for Fungi. Database. 2014;2014:1–21. doi: 10.1093/database/bau061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy G.S., Pradhan S., Manorama R., Shivaji S. Cryobacterium roopkundense sp. nov., a psychrophilic bacterium isolated from glacial soil. Int. J. Syst. Evol. Microbiol. 2010;60:866–870. doi: 10.1099/ijs.0.011775-0. [DOI] [PubMed] [Google Scholar]
- 34.Uchino M., Shiad O., Uchimura T., Komagata K. Recharacterization of Pseudomonas fulva Iizuka and Komagata 1963, and proposals of Pseudomonas parafulva sp. nov. and Pseudomonas cremicolorata sp. nov. J. Gen. Appl. Microbiol. 2001;47:247–261. doi: 10.2323/jgam.47.247. [DOI] [PubMed] [Google Scholar]
- 35.Zhao G.A., Li J., Huang H.Y., Park D.J., Kim C.J., Xu L.H., Li W.J. Pseudonocardia kunmingensis sp. nov., an actinbacterium isolated from surface-sterilized roots of Artemisia annua L. Int. J. Syst. Evol. Microbiol. 2011;61:2292–2297. doi: 10.1099/ijs.0.027607-0. [DOI] [PubMed] [Google Scholar]
- 36.Vandamme P., Goris J., Coenye T., Hoste B., Janssens D., Kersters K., De Vos P., Falsen E. Assignement of Centers for Disease Control group IVc-2 to the genus Ralstonia as Ralstonia paucula sp. nov. Int. J. Syst. Evol. Microbiol. 1999;49:663–669. doi: 10.1099/00207713-49-2-663. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y., Zhang Z., Ramanan N. The actinomycete Thermobispora bispora contains two distinct types of transcriptionally active 16S rRNA genes. J. Bacteriol. 1997;179:3270–3276. doi: 10.1128/jb.179.10.3270-3276.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mylvaganam S., Dennis PP. Sequence heterogeneity between the two genes encoding 16S rRNA from the halophilic archaebacterium Haloarcula marismortui. Genetics. 1992;130:399–410. doi: 10.1093/genetics/130.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tourova T.P., Kuznetov B.B., Novikova E.V., Poltaraus A.B., Nazina T.N. Heterogeneity of the nucleotide sequences of the 16S genes of the type strain of Desulfotomaculum kuznetosovii. Microbioloby. 2001;70:678–684. [PubMed] [Google Scholar]
- 40.Fox G.E., Wisotkey J.D., Jurtshuk P., Jr How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int. J. Syst. Evol. Microbiol. 1992;42:166–170. doi: 10.1099/00207713-42-1-166. [DOI] [PubMed] [Google Scholar]
- 41.Krypides N.C., Hugenholtz P., Eisen J.A., Woyke T., Goker M., Parker C.T., Amann R., Beck B.J., Chain P.S., Chun J., et al. “Genomic encyclopedia of bacteria and archaea: sequencing a myriad of type strains”. PLoS Biol. 2014;12:1–7. doi: 10.1371/journal.pbio.1001920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mende D.R., Sunagawa S., Zeller G., Bork P. Accurate and universal delineation of prokaryotic species. Nat. Methods. 10:881–887. doi: 10.1038/nmeth.2575. [DOI] [PubMed] [Google Scholar]
- 43.Jolley K.A., Bliss C.M., Bennett J.S., Bratcher H.B., Brehony C., Colles F.M., Wimalarathna H., Harrison O.B., Sheppard S.K., Cody A.J., et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology. 2012;158:1005–1015. doi: 10.1099/mic.0.055459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meier-Kolthoff J.P., Auch A.F., Klenk H.-P., Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformat. 2013;14:1–14. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakazaki R., Tamura K., Kosako Y., Yoshizaki E. Klebsiella ornithinolytica sp. nov., formerly known as ornithine-positive Klebsiella oxytoca. Curr. Microbiol. 1989;18:201–206. [Google Scholar]
- 46.Drancourt M., Bollet C., Carta A., Rousselier P. Phylogenetic analyses of Klebsiella species delineate Klebsiella and Raoultella gen. nov., with description of Raoultella ornithinolytica comb. nov., Raoultella terrigena comb. nov. and Raoultella planticola comb. nov. Int. J. Syst. Evol. Microbiol. 2001;51:925–932. doi: 10.1099/00207713-51-3-925. [DOI] [PubMed] [Google Scholar]