


Abstract
Helminth.net (http://www.helminth.net) is the new moniker for a collection of databases: Nematode.net and Trematode.net. Within this collection we provide services and resources for parasitic roundworms (nematodes) and flatworms (trematodes), collectively known as helminths. For over a decade we have provided resources for studying nematodes via our veteran site Nematode.net (http://nematode.net). In this article, (i) we provide an update on the expansions of Nematode.net that hosts omics data from 84 species and provides advanced search tools to the broad scientific community so that data can be mined in a useful and user-friendly manner and (ii) we introduce Trematode.net, a site dedicated to the dissemination of data from flukes, flatworm parasites of the class Trematoda, phylum Platyhelminthes. Trematode.net is an independent component of Helminth.net and currently hosts data from 16 species, with information ranging from genomic, functional genomic data, enzymatic pathway utilization to microbiome changes associated with helminth infections. The databases’ interface, with a sophisticated query engine as a backbone, is intended to allow users to search for multi-factorial combinations of species’ omics properties. This report describes updates to Nematode.net since its last description in NAR, 2012, and also introduces and presents its new sibling site, Trematode.net.
INTRODUCTION
Parasitic helminth infections are considered ‘the great neglected tropical diseases (NTDs)’ (1), accounting for 8 of the 17 most important NTDs, resulting in a collective burden rivaling that of the major high-mortality conditionsm such as HIV/AIDS or malaria (according to the WHO Factsheet on NTDS; http://www.who.int/neglected_diseases/2010report/en/). The symptoms of diseases caused by helminth parasites range from the dramatic sequelae of elephantiasis, blindness, seizures from neurocysticercosis and bladder and liver cancers from urogenital schistosomiasis and opisthorchiasis, respectively, to the more subtle but widespread effects on child development, pregnancy, productivity and maintenance of poverty and predisposition toward other diseases (1–3).
Helminth.net (www.helminth.net) is the new name for an evolving collection of databases hosting resources for helminths, which includes roundworms (Nematoda; Nematode.net, which has had significant updates since 2012 (4)) and flatworms (Platyhelminthes; Trematode.net, a new addition to the website, and Cestode.net, planned in future updates). Genomes of the major parasitic helminths of medical (hookworm, whipworm, ascaris, filarial species), agricultural (e.g. root-knot and cyst nematodes) and veterinary (e.g. gastrointestinal parasites of small ruminants) significance are now the subject of genome sequencing, annotation and other omics approaches (e.g. (5–10)). Helminth.net complements and expands the functionality of related databases, such as WormBase (11) and its sister site WormBase-Parasite, which provide high quality reference genomes and curated gene models for many of these species. Helminth.net, in addition, provides comprehensive functional gene/protein annotation, stage and tissue-specific expression information, population-based variant annotation, ChEMBL drug target association and interactive tools for performing complex multi-factor searches and analyzes in a user-friendly manner.
With Trematode.net, we will provide the research community with these data and tools for schistosomes and food-borne trematodes (FBTs), as we already provide for Nematoda. Genome sequences of the three major species of human-parasitic schistosomes have been reported over the past 5 years (12). The FBTs represent a major group of NTDs, infecting more than 50 million people, and putting 750 million others worldwide (>10% of the world's population) at risk (1,13). Over 100 species of FBTs are known to infect humans, 10 or so of which are responsible for much of the disease burden caused by infection with FBTs (14). Due to their importance the National Institutes of Health (NIH) is supporting sequencing the genomes of 14 FBT genomes (www.trematode.net/FBT_proposal.html), which will be hosted, along with comprehensive annotations and analysis tools, on Trematode.net as they become available.
IMPROVEMENT AND EXPANSION OF Nematode.net
Hosted data
The amount of data hosted on Nematode.net has grown dramatically over the last few years (Table 1). NemaGene now hosts annotation for almost 1.1 million genes and transcripts spanning 67 nematode species, including 998 226 from the genomes of 54 species, 62 385 Roche/454 cDNA isotigs (49 908 transcripts) from 2 species and 44 475 Sanger EST contigs (40 917 transcripts) from 11 species. These species (plus an additional 17 in other data portals) include 16 human parasites, 36 animal parasites, 20 plant parasites, 2 insect parasites and 10 non-parasitic species. We have also added 14 billion nematode Illumina RNAseq reads, spanning numerous stages and tissues across 16 nematodes (Figure 1), providing accurate genome annotation and normalized expression profiles per gene.
Table 1. The expansion of data sets available on Nematode.net since 2011, and the newly available data sets on Trematode.net.
| Database | Data type | 2011 | 2014 |
|---|---|---|---|
| Nematode.net | ESTs and 454/Roche cDNA sequences | 11 880 572 | 11 880 572 |
| Illumina RNAseq sequences | 0 | 14 046 331 058 | |
| No. species in NemaGene | 34 | 84 | |
| NemaGene entries | 233 125 | 1 089 051 | |
| No. splice isoforms | 208 418 | 349 565 | |
| Codon Usage table codon counts | 17 463 274 | 17 463 274 | |
| No. of species with proteomics data | 0 | 7 | |
| No. microbiome samples | 0 | 219 | |
| Trematode.net | No. species represented | 0 | 16 |
| No. species in TremaGene | 0 | 12 | |
| TremaGene entries | 0 | 221 003 | |
| Illumina RNAseq sequences | 0 | 1 138 918 031 | |
| No. of species with proteomics data | 0 | 1 | |
| No. microbiome samples | 0 | 12 |
Figure 1.
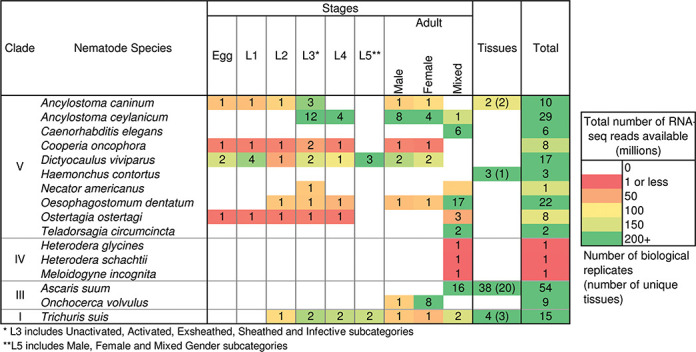
The availability of RNA-Seq data sets newly added to Nematode.net since the last update. A total of just over 14.0 billion reads across 16 species and 187 biological replicates (collected at a particular life cycle stage or from a particular tissue) are currently used for the hosted analysis. Clade phylogeny based on (52).
The NemaBrowse portal has been updated to feature tracks displaying SnpEff-annotated variants (15) from isolates with different phenotypes, which are viewable through GBrowse. This portal will be populated with more species data as more genome-wide single nucleotide polymorphism data based on high-throughput sequencing becomes available, providing an accessible way to explore variants with regard to the acquisition of drug resistance in helminths or other phenotypes.
Alternative splicing (AS) of mRNA is a vital mechanism for enhancing evolutionary complexity, enabling single genes to have diverse molecular and biological functions across organs, tissues, developmental stage and environmental conditions. Predictions of 349 565 isoforms across 10 parasitic nematode species (16) are now hosted on Nematode.net, facilitating deeper investigation of AS and its implications, and AS information based on RNAseq data will be hosted soon.
Many nematodes and trematodes reside in the gastrointestinal tract, directly modulating the immune system, and indirectly influencing the immune response through their effect on the microbiome of the alimentary tract of the host. We have built a ‘Microbiome Interaction’ section of the database, where we host research summaries, highlights of important results and available data sets from publications examining microbiome structure and changes as a result of helminth infections. At present we host microbial communities profiled using targeted 16S rRNA gene sequencing during hookworm infections (17), whipworm infection (18) and polyparasitism (19). In addition, we host currently unpublished metagenome shotgun sequencing data examining microbial communities during nematode infections.
Our Data Download section now hosts additional resources and supplemental data related to the publications (published and in progress) of several dozen nematode pathogens, including RNAseq gene expression data and mass spectrometry proteomic data for available species.
Expansion of analysis and data-mining portals
A number of new tools for exploring data and performing analyses have been introduced to Nematode.net (Figure 2). The NemaGene interface was redesigned to be more user-friendly, and now allows users to define queries using multiple species of interest, InterPro IDs (20,21), Gene Ontology (GO) terms (22), Kegg Orthology (KO) IDs (23) and transcript presence in a given stage and/or tissue according to Sanger EST contigs or 454/Roche cDNA isotigs (where available). NemaGene search results now provide protein or nucleotide sequence FASTA files for all results, and links to individual gene/transcript home pages, which provide: (i) available functional annotations for InterPro (21), GO (22) and KO (23), with links to parent annotation repositories; (ii) a link to view the gene model within NemaBrowse (if available); (iii) sequences, and links to forward sequences directly to NemaBlast; (iv) links from KEGG annotations to our own NemaPath resource (24), allowing users to further explore gene functionality; (v) where available, stage and/or tissue-specific normalized expression data (FPKM) for the genes (Table 1, Figure 1), with new expression values being added as they are produced (Supplementary Information); (vi) where applicable, indication of stage-specific transcript detection according to Sanger-based EST or 454/Roche-based sequences; (vii) links to ChEMBL (25), drug target annotations; (viii) annotations of putative chokepoint enzymes.
Figure 2.
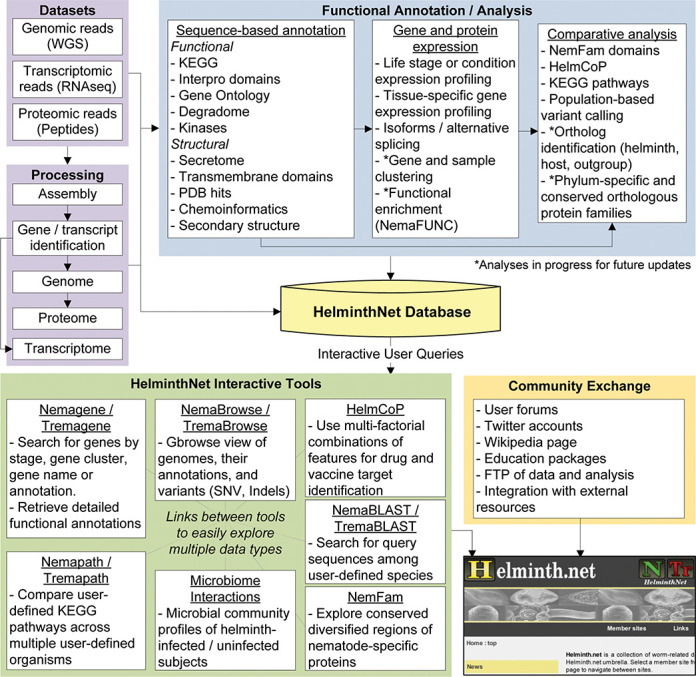
An overview of the input data sets, analyses, tools and community exchange interactions within Helminth.net.
The ChEMBL drug target annotations for our hosted genes contribute to our goal of becoming a central chemogenomic resource for helminths and facilitating systematic identification of anthelminthic drug target(s) and compound(s) targeting them which has already produced promising results for nematode proteins (26,27). We screened all the NemaGene protein products against the ChEMBL database (based on similar sequences and functional annotations) for possible homology to drug targets, annotating targets and the compounds targeting them. The ChEMBL database contains detailed information on the bioactivity, chemical information and structures of more than one million small molecules, providing abundant resources for pursuing nematode proteins as drug candidates.
The NemaPath tool (24) was expanded to host the genesets of 53 nematodes (and transcript pathway annotation for 9 other species) and updated to release 68 of the KEGG genes database (db). Chokepoint enzymes, which catalyze chokepoint reactions (defined as a reaction that produces a unique compound or consumes a unique substrate (28)), were also annotated using a previously published approach (26) since they are potential drug targets due to the lethality resulting from the accumulation of a unique substrate or the organism being starved of a unique substrate (26,29).
The NemaBLAST service has been updated to include the nucleotide sequence (transcript and/or coding DNA sequence (CDS)) for the genesets of 45 nematode species published since the last update. The WU-BLAST–based search engine has also been migrated to a powerful compute cluster to better support queries from concurrent users.
Finally, the NemaBrowse viewer now hosts gene annotations for nine genomes (Ancylostoma caninum, Ancylostoma ceylanicum, Ancylostoma duodenale, Dictyocaulus viviparus, Necator americanus, Oesophagostomum dentatum, Teladorsagia circumcincta, Trichuris suis and Trichinella spiralis), and will soon be expanded further with addition of the upcoming genomes.
Data integration
We have made an ongoing effort to link all annotations we provide to their repositories of origin. NemaGene functional annotations and ChEMBL (25) annotations link back to the parent database entries for every reported ID. HelmCoP's (30) output provides links into the Protein Data Bank (PDB) (31) and DrugBank (32) and our species hub pages provide links to the Sanger Pathogens unit (http://www.sanger.ac.uk), NemBase4 (33) and the appropriate NCBI BioProject ID summary page (34), where available. The hubs also provide links to the species-specific pages available in WormBase (11) and WormBase-Parasite (parasite.wormbase.org) for organisms hosted in those complementary resources.
Education
Nematode.net's ‘Education’ section now features the ‘Introduction to Nematodes’ teaching package presentation, a comprehensive introduction to the field of nematology created by E.C. McGawley, C. Overstreet, M.J. Pontif and A.M. Skantar (Society of Nematologists http://www.nematologists.org). Our team also participated the NIH-funded filarial resource FR3 (35), an annual course organized, among others, to train parasitologists to use parasitic nematode websites/databases. The ‘Education’ section features this tutorial outlining the use of Nematode.net, and we also detailed the use of each portal of our new site expansion (Trematode.net) as Supplementary Information (Supplementary Information SI1).
Site navigation
URL redirection has been provided for jumping directly to species pages as well as to the major analytical tools. Species pages can be accessed directly using the URL nematode.net/<Species_name>.html (e.g. nematode.net/Necator_americanus.html), and the various analysis portals can be accessed similarly (e.g. nematode.net/nemagene.html). This feature is also available for Trematode.net pages.
INTRODUCTION TO Trematode.net
Trematode.net was recently developed to provide omics data dissemination, from an initiative to study the genomes of the etiological agents of FBTs, with a primary goal of studying trematode (fluke) genome-wide gene and protein annotations online via GBrowse (36). However, Trematode.net now also provides numerous additional services and tools to serve the Trematode research community (Supplementary Information S1, Figure 2), and currently houses information for 16 trematode species. Our overall design priority was to make Trematode.net mirror Nematode.net as much as possible (both in terms of layout and functionality) to create a seamless user experience across Helminth.net. The navigation menu interfaces for Nematode.net (Figure 3A) and Trematode.net (Figure 3B) link to each other, and are organized in a similar fashion, providing one-click access to interactive tools used to mine hosted data (Figure 3C).
Figure 3.
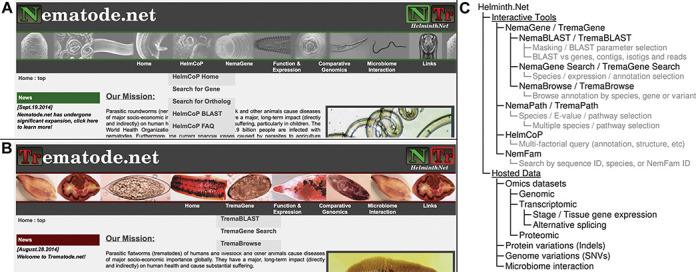
Accessing and interacting with data on Helminth.net. The navigation menu interface for the major tools and datasets available on (A) Nematode.net and (B) Trematode.net is easily accessible from all areas of the site. (C) A site map outlining the interactive tools, the major searchable components of each tool (in grey), and the hosted data on Helminth.net.
TremaGene
TremaGene (Supplementary Figure S1) is the central repository of trematode data hosted within Trematode.net, and currently houses 221 003 annotated genes from 12 trematode species (Table 2). Genes are annotated with InterPro IDs and GO terms (using InterProScan) (20–22), KO IDs (KEGG version 68.0, using WU-BLAST 2.0) (23), and we also have stage and/or tissue-specific expression data for Fasciola hepatica and Schistosoma mansoni, which are displayed in the gene details pages. The TremaGene search interface operates similarly to NemaGene, where users can search based on any combination of species, with filters based on combinations of InterPro, GO and KO IDs, or specific genes (Supplementary Figure S1). Compared to NemaGene, TremaGene only lacks the stage-based expression filter (which was based on identifying stage-specific sanger EST sequences or 454/Roche cDNA sequences in nematodes), because our TremaGene data is entirely based upon analysis of draft genome assemblies. Search results can be downloaded in their entirety, or each gene can be accessed for a view of the detailed annotation, with links to TremaPath from annotated KOs, TremaBrowse to view gene models (if available), and TremaBlast to search for putative orthologs (Supplementary Figures S2 and S3).
Table 2. The number of genes (and deduced proteins) hosted for all organisms available in TremaGene and the names and status of the species with genomes in progress.
| Status | Species | Annotated gene count or project status |
|---|---|---|
| Published or annotated | ||
| Clonorchis sinensis | 13 634 | |
| Echinostoma caproni | 18 607 | |
| Fasciola hepatica | 15 739 | |
| Opisthorchis viverrini | 16 356 | |
| Schistosoma curassoni | 23 546 | |
| Schistosoma haematobium | 13 073 | |
| Schistosoma japonicum | 12 743 | |
| Schistosoma mansoni | 11 828 | |
| Schistosoma margrebowiei | 26 189 | |
| Schistosoma mattheei | 22 997 | |
| Schistosoma rodhaini | 24 089 | |
| Trichobilharzia regenti | 22 202 | |
| Genome sequencing project in progress | ||
| Fasciola gigantica | assembly | |
| Fasciola buski | material acquisition | |
| Haplorchis taichui | material acquisition | |
| Opisthorchis felineus | material acquisition | |
| Opisthorchis viverrini | annotation | |
| Paragonimus kellicotti | data production | |
| Paragonimus miyazaki | assembly | |
| Paragonimus westermani | annotation | |
| Paragonimus spp. (3x) | material acquisition | |
TremaBlast
TremaBlast allows users to search custom sequence(s) directly against deduced protein sets from TremaGene (Supplementary Figure S4). Our currently available database covers 12 trematodes (Table 2), which can be selected in any combination and used as the subject for mapping using WU-BLAST 2.0 (ran in either BLASTx or BLASTp mode). SEG (ftp://ftp.ncbi.nih.gov/pub/seg/seg/) and RepeatMasker (http://www.repeatmasker.org) filters are available if the user wishes to screen out low-complexity sequence or mask repeats in their query. Jobs are submitted to a backend compute farm and results are mailed directly to the user (Supplementary Figure S5).
TremaBrowse
TremaBrowse provides a window into gene annotations of finished and/or draft genomic assemblies using the GBrowse viewer (36). Currently, we host the current draft build of Fasciola hepatica (Supplementary Figure S6) as our first annotated FBT, with an aim to provide at least five more novel genomes within the next 6 months. Displayed information can include Maker (37) gene predictions, RNA genes predicted by RNAmmer (38), tRNAs predicted by tRNAscan (39) and Single Nucleotide Polymorphism (SNP) loci annotated using SnpEff (15) (Supplementary Figure S7). One goal of the TremaBrowse resource is to provide the research community with a view of in-progress trematode genomes, representing our current best draft, in advance of final genome submissions.
TremaPath
TremaPath provides a visualization of pathway usage for trematodes, based on KO annotations (23) for all genes, which are then ‘painted’ onto predefined KEGG pathway maps. Users are provided a graphical distribution of the number of KO hits with varying e-value confidence scores for their chosen species, and then set a desired threshold stringency to assign KOs (Supplementary Figure S8). Users are then presented with a menu of pathways supported by TremaPath (Supplementary Figure S9). Currently, we support four broad KEGG categories: Metabolism, Genetic Information Processing, Environmental Information Processing and Cellular Processes. After pathway selection, a graphic displaying the compounds and reactions of that pathway for their species of choice is shown, with identified enzymes colored green and darker shading indicating multiple genes annotated (Supplementary Figure S10). The user can then optionally choose a second species for comparison, mapping genes onto the same pathway and highlighting differences in pathway usage between the species. TremaPath is currently populated with 204 647 proteins from 11 trematodes (with Opisthorchis viverrini coming soon).
Microbiome interaction
As with its sister site, Trematode.net also hosts microbial community structure information for trematode-infected subjects, including research summaries, highlights of important results and available data sets related to the interaction of trematodes and their host environment. Currently, we host data from a recent study on infection with Opisthorchis viverrini (40) (Supplementary Figure S11). We will continue to expand this section as research findings emerge.
CONCLUSION AND FUTURE PLANS
The primary goal of these databases is to provide the helminth research community with access to integrated data and tools for helminths undergoing targeted active research studies, as well as those available in the public domain.
The focus of this release was on: (i) the dramatic increase in the number of gene sets and RNAseq data sets providing functional genomics information on these species; (ii) the major improvements made to the NemaGene (and now also TremaGene) interface, enabling a much more user-friendly experience; (iii) providing chemogenomic information, i.e. annotation of helminth genes as putative targets, and the compounds putatively targeting them and (iv) the introduction Trematode.net, providing similar assistance and value to the community as Nematode.net does. We also described the expansion of a number of veteran Nematode.net tools and novel data types, including NemaGene, NemaPath, NemaBlast, NemaBrowse and our Microbiome Interaction data collection.
Future expansions and improvements
With over 15.1 billion reads of RNAseq currently in hand, and much more coming soon, one of our major priorities is to effectively disseminate useful analyses of this data through Helminth.net, by implementing several new data analyses and visualizations. For example, we will implement a dynamic gene expression plot viewer, allowing users to select single or multiple species of interest, life cycle stages of interest and/or genes of interest (from a custom list, or imported from other Helminth.net tools). We also plan to implement a fuzzy c-means clustering tool for gene expression data, to group sets of genes of interest according to expression patterns across stages of development and/or longitudinal sections of tissues of interest. This will include statistical cutoffs clustering, color-coded visualization of clusters and annotation information for gene members within each cluster. Genes within a cluster will also be able to be fed directly to our planned expansion of enrichment testing tools (described below), allowing for a custom de novo analysis of stage and tissue-specific functions with just a few clicks.
NemaBrowse and TremaBrowse represent our central repository for the display of genomic information, and we intend to continue their use for new helminth genomes, and to expand its functionality. As we receive sequence data from clinical/field isolates of the same species, we will annotate isolate-specific variant loci in coding regions by mapping to the latest genome assembly references, and we will annotate population-specific effects of each SNP (15). These annotated SNPs, and the underlying sequence alignments to the reference, will be available as separate tracks within NemaBrowse/TremaBrowse and will be available for download as isolate-specific Variant Call Format (VCF) files. Our data currently hosted in our NemaSNP database will also be merged into NemaBrowse to simplify access to this data, and NemaSNP will be decommissioned. Eventually, we plan to provide a comparative view among user-defined sets of orthologous genes within NemaBrowse/TremaBrowse. Initially, we plan to use GBrowse with views of groups of SNP-annotated genes in individual tabs, scaled equivalently for easy comparison, but later iterations may provide more elegant solutions to view in a single window against a common reference.
The NemaGene/TremaGene resource will be further expanded to allow users to download gene annotations directly to a tab-delimited text file after searching using custom filters (as described above). We will also calculate and annotate gene expression values (in units of FPKM) per available stage and/or tissue for all genes/species, and display this data in search results, with links to view the mapping information in GBrowse. We will provide links to the complete RNAseq read data set(s), either as accession IDs within NCBI's SRA (http://www.ncbi.nlm.nih.gov/sra) or as direct links if the data is pending official release. Additional annotation for all genes, including transmembrane domains and detected signal peptides (41) or predicted non-classical secretion (42), as well as degradome information for peptidases and inhibitors (43) will also be added. We also plan to track and annotate isoforms within NemaGene/TremaGene, initially using the gene and/or transcript as the central database entity, but eventually annotating individual isoforms with the same comprehensive annotations we provide for simple genes and transcripts. Isoform information will also be viewable in GBrowse.
NemaPath/TremaPath metabolic pathway reconstruction will be expanded by improving both enzyme predictions and pathway mapping. This will be accomplished by undertaking alternate and independent methods for functional annotation including (i) analyzing enzyme class sequence diversity to refine the likelihood estimation in protein annotation (44); (ii) performing Functionally Discriminating Residue recognition (45); (iii) discriminating between the module characteristics of discrete enzyme activities (46) and (iv) comparing pathways across diverse taxa to detect similar topologies (47–49) and translate pathway information into adjacency matrices amenable to topological alignments (50).
Other planned updates include: (i) NemaFUNC/TremaFUNC, using the FUNC tool (51) to allow users to statistically analyze GO functional enrichment of a custom set of genes against a custom background set of genes; (ii) NemaIPR/TremaIPR, to perform a similar enrichment analysis on InterPro domains using internally developed tools; (iii) a tool for exploring pathway enrichment, utilizing KO ID annotations; (iv) a drug-target prioritization approach based on numerical weights assigned to annotation criteria used for querying; (v) NemaGroup/TremaGroup, to view a gene of interest in the context of the global orthologous group collection with filters to restrict the view to specific phylogenetic levels (e.g. clade-specific analyses (52)); (vi) a database hosting microbial community structure (bacterial taxa and their abundance) on a per sample basis, and related results including alpha and beta diversity, and/or metabolic capability of the community (for shotgun metagenomic data). Users will be able to perform advanced parsing and compare microbiomes among infected or non-infected individuals, as well as across infected and non-infected individuals and (vii) more expanded integration with other community resources, particularly WormBase (11) and WormBase Parasite due to the high quality of reference genomes and curated gene models that they provide. By adding information such as comprehensive functional annotation, stage and tissue-specific expression, genome-wide detection and variant annotation, ChEMBL drug target association and more, Helminth.net is an excellent complement to Wormbase.
Overall, these planned expansions will ease user accessibility to more data, and to more types of emerging data, to better disseminate information to the community in a way that is intuitive and that provides extremely useful analysis tools to the end user.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We sincerely thank the numerous collaborators in the helminth community (nematode.net/collaborators.html and trematode.net/collaborators.html), for providing invaluable worm material and being involved in data generation/analysis activities, and the dedicated members of the production group at The Genome Institute (http://genome.wustl.edu/) for the library construction and sequencing.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
National Institutes of Health (NIH) [AI081803 and GM097435 to M.M.]; NIFA [2013-01109 to M.M.]; OPP [GH 1083853]. NIH-NHGRI [U54HG003079]. NIH [AI098639, CA164719 and CA155297 to P.J.B.]. Funding for open access charge: NIH [AI081803].
Conflict of interest statement. None declared.
REFERENCES
- 1.Hotez P.J., Brindley P.J., Bethony J.M., King C.H., Pearce E.J., Jacobson J. Helminth infections: the great neglected tropical diseases. J. Clin. Invest. 2008;118:1311–1321. doi: 10.1172/JCI34261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brindley P.J., Mitreva M., Ghedin E., Lustigman S. Helminth genomics: the implications for human health. PLoS Negl. Trop. Dis. 2009;3:e538. doi: 10.1371/journal.pntd.0000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brooker S., Akhwale W., Pullan R., Estambale B., Clarke S.E., Snow R.W., Hotez P.J. Epidemiology of plasmodium-helminth co-infection in Africa: populations at risk, potential impact on anemia, and prospects for combining control. Am. J. Trop. Med. Hygiene. 2007;77:88–98. [PMC free article] [PubMed] [Google Scholar]
- 4.Martin J., Abubucker S., Heizer E., Taylor C.M., Mitreva M. Nematode.net update 2011: addition of data sets and tools featuring next-generation sequencing data. Nucleic Acids Res. 2012;40:D720–D728. doi: 10.1093/nar/gkr1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Protasio A.V., Tsai I.J., Babbage A., Nichol S., Hunt M., Aslett M.A., De Silva N., Velarde G.S., Anderson T.J., Clark R.C., et al. A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni. PLoS Negl. Trop. Dis. 2012;6:e1455. doi: 10.1371/journal.pntd.0001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young N.D., Nagarajan N., Lin S.J., Korhonen P.K., Jex A.R., Hall R.S., Safavi-Hemami H., Kaewkong W., Bertrand D., Gao S., et al. The Opisthorchis viverrini genome provides insights into life in the bile duct. Nat. Commun. 2014;5:4378. doi: 10.1038/ncomms5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai I.J., Zarowiecki M., Holroyd N., Garciarrubio A., Sanchez-Flores A., Brooks K.L., Tracey A., Bobes R.J., Fragoso G., Sciutto E., et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature. 2013;496:57–63. doi: 10.1038/nature12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young N.D., Jex A.R., Li B., Liu S., Yang L., Xiong Z., Li Y., Cantacessi C., Hall R.S., Xu X., et al. Whole-genome sequence of Schistosoma haematobium. Nat. Genet. 2012;44:221–225. doi: 10.1038/ng.1065. [DOI] [PubMed] [Google Scholar]
- 9.Tang Y.T., Gao X., Rosa B.A., Abubucker S., Hallsworth-Pepin K., Martin J., Tyagi R., Heizer E., Zhang X., Bhonagiri-Palsikar V., et al. Genome of the human hookworm Necator americanus. Nat. Genet. 2014;46:261–269. doi: 10.1038/ng.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foth B.J., Tsai I.J., Reid A.J., Bancroft A.J., Nichol S., Tracey A., Holroyd N., Cotton J.A., Stanley E.J., Zarowiecki M., et al. Whipworm genome and dual-species transcriptome analyses provide molecular insights into an intimate host-parasite interaction. Nat. Genet. 2014;46:693–700. doi: 10.1038/ng.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris T.W., Baran J., Bieri T., Cabunoc A., Chan J., Chen W.J., Davis P., Done J., Grove C., Howe K., et al. WormBase 2014: new views of curated biology. Nucleic Acids Res. 2014;42:D789–D793. doi: 10.1093/nar/gkt1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zerlotini A., Aguiar E.R.G.R., Yu F., Xu H., Li Y., Young N.D., Gasser R.B., Protasio A.V., Berriman M., Roos D.S., et al. SchistoDB: an updated genome resource for the three key schistosomes of humans. Nucleic Acids Res. 2012;41:D728–D731. doi: 10.1093/nar/gks1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keiser J., Utzinger J. Food-borne trematodiases. Clin. Microbiol. Rev. 2009;22:466–483. doi: 10.1128/CMR.00012-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sripa B., Kaewkes S., Intapan P.M., Maleewong W., Brindley P.J. In: Advances in Parasitology. Xiao-Nong Zhou RBRO, Jürg U, editors. Vol. 72. Waltham, MA: Academic Press; 2010. pp. 305–350. [DOI] [PubMed] [Google Scholar]
- 15.Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abubucker S., McNulty S.N., Rosa B.A., Mitreva M. Identification and characterization of alternative splicing in parasitic nematode transcriptomes. Parasit Vectors. 2014;7:1756–3305. doi: 10.1186/1756-3305-7-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cantacessi C., Giacomin P., Croese J., Zakrzewski M., Sotillo J., McCann L., Nolan M.J., Mitreva M., Krause L., Loukas A. Impact of experimental hookworm infection on the human gut microbiota. J. Infect. Dis. 2014;210:1431–1434. doi: 10.1093/infdis/jiu256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper P., Walker A.W., Reyes J., Chico M., Salter S.J., Vaca M., Parkhill J. Patent human infections with the whipworm, Trichuris trichiura, are not associated with alterations in the faecal microbiota. PLoS ONE. 2013;8:e76573. doi: 10.1371/journal.pone.0076573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S.C., Tang M.S., Lim Y.A.L., Choy S.H., Kurtz Z.D., Cox L.M., Gundra U.M., Cho I., Bonneau R., Blaser M.J., et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl. Trop. Dis. 2014;8:e2880. doi: 10.1371/journal.pntd.0002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones P., Binns D., Chang H.Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter S., Jones P., Mitchell A., Apweiler R., Attwood T.K., Bateman A., Bernard T., Binns D., Bork P., Burge S., et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Consortium T.G.O. Gene ontology annotations and resources. Nucleic Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wylie T., Martin J., Abubucker S., Yin Y., Messina D., Wang Z., McCarter J.P., Mitreva M. NemaPath: online exploration of KEGG-based metabolic pathways for nematodes. BMC Genom. 2008;9:1471–2164. doi: 10.1186/1471-2164-9-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bento A.P., Gaulton A., Hersey A., Bellis L.J., Chambers J., Davies M., Kruger F.A., Light Y., Mak L., McGlinchey S., et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42:D1083–D1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor C.M., Martin J., Rao R.U., Powell K., Abubucker S., Mitreva M. Using existing drugs as leads for broad spectrum anthelmintics targeting protein kinases. PLoS Pathog. 2013;9:e1003149. doi: 10.1371/journal.ppat.1003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor C.M., Wang Q., Rosa B.A., Huang S.C., Powell K., Schedl T., Pearce E.J., Abubucker S., Mitreva M. Discovery of anthelmintic drug targets and drugs using chokepoints in nematode metabolic pathways. PLoS Pathog. 2013;9:e1003505. doi: 10.1371/journal.ppat.1003505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeh I., Hanekamp T., Tsoka S., Karp P.D., Altman R.B. Computational analysis of Plasmodium falciparum metabolism: organizing genomic information to facilitate drug discovery. Genome Res. 2004;14:917–924. doi: 10.1101/gr.2050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palumbo M.C., Colosimo A., Giuliani A., Farina L. Essentiality is an emergent property of metabolic network wiring. FEBS Lett. 2007;581:2485–2489. doi: 10.1016/j.febslet.2007.04.067. [DOI] [PubMed] [Google Scholar]
- 30.Abubucker S., Martin J., Taylor C.M., Mitreva M. HelmCoP: an online resource for helminth functional genomics and drug and vaccine targets prioritization. PLoS One. 2011;6:e21832. doi: 10.1371/journal.pone.0021832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rose P.W., Bi C., Bluhm W.F., Christie C.H., Dimitropoulos D., Dutta S., Green R.K., Goodsell D.S., Prlić A., Quesada M., et al. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 2013;41:D475–D482. doi: 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y., Maciejewski A., Arndt D., Wilson M., Neveu V., et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elsworth B., Wasmuth J., Blaxter M. NEMBASE4: the nematode transcriptome resource. Int. J. Parasitol. 2011;41:881–894. doi: 10.1016/j.ijpara.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Clark K., Pruitt K., Tatusova T., Mizrachi I. Bioproject: The NCBI Handbook. Bethesda, MD: National Center for Biotechnology Information; 2013. [Google Scholar]
- 35.Michalski M.L., Griffiths K.G., Williams S.A., Kaplan R.M., Moorhead A.R. The NIH-NIAID Filariasis Research Reagent Resource Center. PLoS Negl. Trop. Dis. 2011;5:e1261. doi: 10.1371/journal.pntd.0001261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein L.D. Using GBrowse 2.0 to visualize and share next-generation sequence data. Brief Bioinform. 2013;14:162–171. doi: 10.1093/bib/bbt001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cantarel B.L., Korf I., Robb S.M., Parra G., Ross E., Moore B., Holt C., Sanchez Alvarado A., Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lagesen K., Hallin P., Rodland E.A., Staerfeldt H.H., Rognes T., Ussery D.W. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plieskatt J.L., Deenonpoe R., Mulvenna J.P., Krause L., Sripa B., Bethony J.M., Brindley P.J. Infection with the carcinogenic liver fluke Opisthorchis viverrini modifies intestinal and biliary microbiome. FASEB J. 2013;27:4572–4584. doi: 10.1096/fj.13-232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kall L., Krogh A., Sonnhammer E.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 42.Bendtsen J.D., Jensen L.J., Blom N., Heijne G., Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Design Selection. 2004;17:349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 43.Rawlings N.D., Waller M., Barrett A.J., Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014;42:D503–D509. doi: 10.1093/nar/gkt953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hung S.S., Wasmuth J., Sanford C., Parkinson J. DETECT–a density estimation tool for enzyme classification and its application to Plasmodium falciparum. Bioinformatics. 2010;26:1690–1698. doi: 10.1093/bioinformatics/btq266. [DOI] [PubMed] [Google Scholar]
- 45.Kumar N., Skolnick J. EFICAz2.5: application of a high-precision enzyme function predictor to 396 proteomes. Bioinformatics. 2012;28:2687–2688. doi: 10.1093/bioinformatics/bts510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Claudel-Renard C., Chevalet C., Faraut T., Kahn D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res. 2003;31:6633–6639. doi: 10.1093/nar/gkg847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ay F., Dang M., Kahveci T. Metabolic network alignment in large scale by network compression. BMC Bioinformat. 2012;13:S2. doi: 10.1186/1471-2105-13-S3-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ay F., Kellis M., Kahveci T. SubMAP: aligning metabolic pathways with subnetwork mappings. J. Comput. Biol. 2011;18:219–235. doi: 10.1089/cmb.2010.0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graca G., Goodfellow B.J., Barros A.S., Diaz S., Duarte I.F., Spagou K., Veselkov K., Want E.J., Lindon J.C., Carreira I.M., et al. UPLC-MS metabolic profiling of second trimester amniotic fluid and maternal urine and comparison with NMR spectral profiling for the identification of pregnancy disorder biomarkers. Mol. bioSyst. 2012;8:1243–1254. doi: 10.1039/c2mb05424h. [DOI] [PubMed] [Google Scholar]
- 50.Zhang J.D., Wiemann S. KEGGgraph: a graph approach to KEGG PATHWAY in R and bioconductor. Bioinformatics. 2009;25:1470–1471. doi: 10.1093/bioinformatics/btp167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prufer K., Muetzel B., Do H.H., Weiss G., Khaitovich P., Rahm E., Paabo S., Lachmann M., Enard W. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformat. 2007;8:41. doi: 10.1186/1471-2105-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blaxter M.L., De Ley P., Garey J.R., Liu L.X., Scheldeman P., Vierstraete A., Vanfleteren J.R., Mackey L.Y., Dorris M., Frisse L.M., et al. A molecular evolutionary framework for the phylum Nematoda. Nature. 1998;392:71–75. doi: 10.1038/32160. [DOI] [PubMed] [Google Scholar]