


Abstract
The rapid advancement of next-generation sequencing technology has generated a deluge of genomic data from domesticated dogs and their wild ancestor, grey wolves, which have simultaneously broadened our understanding of domestication and diseases that are shared by humans and dogs. To address the scarcity of single nucleotide polymorphism (SNP) data provided by authorized databases and to make SNP data more easily/friendly usable and available, we propose DoGSD (http://dogsd.big.ac.cn), the first canidae-specific database which focuses on whole genome SNP data from domesticated dogs and grey wolves. The DoGSD is a web-based, open-access resource comprising ∼19 million high-quality whole-genome SNPs. In addition to the dbSNP data set (build 139), DoGSD incorporates a comprehensive collection of SNPs from two newly sequenced samples (1 wolf and 1 dog) and collected SNPs from three latest dog/wolf genetic studies (7 wolves and 68 dogs), which were taken together for analysis with the population genetic statistics, Fst. In addition, DoGSD integrates some closely related information including SNP annotation, summary lists of SNPs located in genes, synonymous and non-synonymous SNPs, sampling location and breed information. All these features make DoGSD a useful resource for in-depth analysis in dog-/wolf-related studies.
INTRODUCTION
Dogs have been the dearest friends of humans as guardians, companions and working partners for thousands of years (1,2). Our natural curiosity, together with practical and aesthetical needs propelled the exploration of the genetic basis of the remarkable diversity of dog phenotypes (3–5). Furthermore, being one of the most thoroughly domesticated animals has made them a long-term focus and perfect model for domestication genetics studies (6–10). Researches into their parallel evolution with humans has facilitated our understanding of human evolution itself and, intriguingly, genes that cause diseases that are mutually shared, especially those related to cancer and neurological disorders (11,12). As a powerful genetic marker, single nucleotide polymorphism (SNP) plays a pivotal role in dog genetic researches referring to human selection (3,4,10), demographic studies (6), linkage and segregation analysis (5) and genome-wide association studies (13).
To date, the latest and most widely used dog SNP data set is from dbSNP (build139:ftp://ftp.ncbi.nlm.nih.gov/snp/), where ∼80% of the ∼2.7M SNPs were called from two dogs, one Boxer (14) and one Standard Poodle (15). It provides the SNP list from all samples without individual lists, which limits its usability in population genetic analysis. The rapid development of next-generation sequencing (NGS) technology has facilitated the generation of massive dog/wolf genome data sets (7,16). However, SNP calling from the massive data generated by NGS is very laborious and requires a large amount of computational resources.
Here, we develop DoGSD, the first web-based public database for accessing highly dense, broadly representative dog/wolf whole-genome SNP data. We collected SNPs from two unpublished dog/wolf genomes, three recently published works (8,9,11) and the latest dog SNP data set (dbSNP139). A total of 34 our samples are Chinese indigenous dogs which represent a key phase in dog domestication and are not included in any existing SNP databases. DoGSD provides a powerful SNP retrieving interface for each individual samples and a non-redundant data set. We made annotation to integrate information, such as SNP-related genes, transcripts, proteins and calculate allele frequencies. DoGSD has a functionality to search gene-related synonymous and non-synonymous SNPs. In addition, we incorporate the essential genetic statistics, viz. F- statistics (Fst) into DoGSD.
DATABASE CONSTRUCTION
Figure 1 shows the database construction pipeline. Details are described in Data sources, Data processing and Database implementation.
Figure 1.
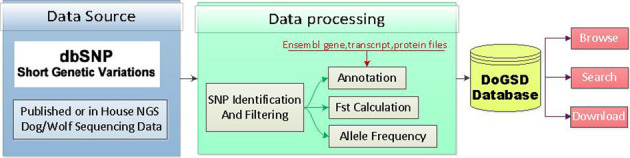
The data sources and pipeline to construct DoGSD.
Data sources
Our data set integrates SNPs called from two newly sequenced wolf/dog samples and three published dog genetic works (8,9,11) with the newly released dog SNP data set (dbSNP139) (Table 1). The two newly sequenced samples include one grey wolf (solidGW1) sampled from Harbin, China and a Chinese indigenous dog (CID1) sampled from Ya'an, China. The 75 published canines include 7 grey wolves and 68 dogs. The 7 grey wolves are sampled across the Eurasian continent and America, with 33 indigenous dogs from China, another 34 from 5 breeds and 1 Dingo. Dogs from the 5 breeds include 11 German Shepherd Dogs (Germany), 10 Kunming Dogs (China), 11 Tibetan Mastiffs (China), 1 Belgian Malinois (Belgium) and 1 Basenji (Congo). We only used Solexa data of the 5 individuals from Freedman et al. In total, DoGSD includes 8 grey wolves, 34 Chinese indigenous dogs, 13 ancient breed dogs, 22 modern breed dogs and the dbSNP139.
Table 1. Data sets in DoGSD.
| Species/Breed | Number | Average coverage | Group ID | Data source |
|---|---|---|---|---|
| Grey Wolf | 1 | 17.9 | 1 | SOLiD platform |
| Indigenous Dog | 1 | 14 | ||
| Grey Wolf | 4 | 18.17 | ||
| German Shepherd | 1 | 9.53 | ||
| Tibetan Mastiff | 1 | 10.99 | Wang et al. | |
| Belgian Malinois | 1 | 9.97 | ||
| Indigenous Dog | 3 | 14.38 | ||
| Grey Wolf | 3 | 10.45 | 2 | |
| Basenji | 1 | 4.25 | Freedman et al. | |
| Dingo | 1 | 3.67 | ||
| German Shepherd | 10 | 16.75 | ||
| Indigenous Dog | 30 | 15.54 | Gou et al. | |
| Tibetan Mastiff | 10 | 15.42 | ||
| Kunming Dog | 10 | 16.02 | ||
| dbSNP139 | / | / | / | NCBI |
Data processing
Except for dbSNP139, we divided our samples into two groups. Group 1 is the two unpublished dog/wolf genomes (solidGW1 and CID1) generated by the SOLiD system and group 2 is from three recently published works (8,9,11), which were sequenced with the Illumina Solexa technology. All of the individual genomes were sequenced to an average of 15× coverage.
For the two samples sequenced by the SOLiD system, two DNA mate-paired libraries were prepared for solidGW1 with 5 and 1.5 kb insert size, which generated 72.93G bp of raw data. A single mate-paired library with 500 bp insert size was prepared for CID1, producing 45.96G bp raw data. Using the Bioscope pipeline, 42.01 Gb of solidGW1 was aligned to the Boxer genome assembly (canFam2) with 17.9× coverage, while 32.99 Gb of the CID1 was aligned to the canFam2 with 14× coverage.
We used two sets of methods and the camFam2/canFam3 reference genomes download from UCSC (http://hgdownload.soe.ucsc.edu/goldenPath/canFam2 or 3/bigZips/canFam2 or 3.fa.gz) to identify SNPs from these two groups, and integrated the SNPs from all 77 samples and the dbSNP139 data set to obtain a non-redundant SNP set. For group 1, SNPs were detected with the BioScope diBayes tool where valid adjacent two-base mismatches occurred between a read and the reference (canFam2). Depending on whether there was high or low read coverage on the reference, either a Frequentist or Bayesian algorithm was applied, respectively. The Frequentist algorithm is based on the null hypothesis that a given position is homozygous and any other valid adjacent mismatches are errors subject to a Poisson distribution. The Bayesian algorithm calculates the posterior probability of each site according to the expected polymorphism rate in the genome, GC content, coverage, position in a read and the quality value of the colour call and prior errors derived from the 6mer probe annealing error. SNP calling stringency was set as medium. Finally, we converted genome positions of all resulting SNPs from Canfam2.0 to Canfam3.0.
For group 2, we used Burrows–Wheeler Aligner (version: 0.6.2-r126) (17,18) to map Illumina short reads to the dog reference sequence (Canfam3) in the first step. Picard (version: 1.87) (downloaded from http://picard.sourceforge.net) was then used to eliminate duplicated reads generated in the library polymerase chain reaction construction. After that, we used the tools in GATK (version: 2.5–2-gf57256b) (19,20) to realign reads around known indels (downloaded from Ensembl ftp://ftp.ensembl.org/pub/release-73/variation/vcf/canis_familiaris/), and recalibrate base quality score to obtain more accurate quality score for each base. The refined data from all individuals were jointly used to call a raw SNPs set by GATK UnifiedGenotyper, and finally, we identified a high quality set of SNPs, using the variant quality score to recalibrate procedure in GATK.
For both groups and the dbSNP139 data set, we filtered SNPs with two-thirds derived alleles. SNPs were then annotated to genes, transcripts and proteins downloaded from the Ensembl FTP site (ftp://ftp.ensembl.org/pub/release-75/fasta/canis_familiaris/). Allele frequency was calculated for wolf/dog populations for each SNP from 77 sequenced samples. Also the Fst was calculated using vcftools (21) for the batch two data.
Database implementation
Several tables containing all resulting high-quality SNPs and their annotations were processed with Perl scripts and put into MySQL database. We use JSP/JAVA to implement data visualization, searching and downloading. GBrowse was also integrated for chromosome-based data visualization.
Data characteristics
In total, we obtained 19,333,098 non-redundant SNPs, approximately seven times greater than in dbSNP139. The average length of SNP intervals of dbSNP139 is 862.0 bp, with a median value of 241.0 bp, while the same statistics for DoGSD are 120.4 bp and 68.0 bp, respectively, showing that the SNP distribution of our database is denser and more uniform than that of dbSNP139. Ancestral LD/cross-breed LD blocks of the dog have an average size of 10 Kb (14). Thus, we compared the amount of intervals  10 Kb with no SNP of both data sets. Our results show that there were 333.4 Mb (covering 13.8% of the genome) without SNPs in dbSNP139 while only 7.4 Mb (0.3%) in DoGSD, clearly demonstrating that our database has fewer blocks without SNPs than dbSNP139 data set. This pattern is still true when calculating SNP intervals
10 Kb with no SNP of both data sets. Our results show that there were 333.4 Mb (covering 13.8% of the genome) without SNPs in dbSNP139 while only 7.4 Mb (0.3%) in DoGSD, clearly demonstrating that our database has fewer blocks without SNPs than dbSNP139 data set. This pattern is still true when calculating SNP intervals  10 Kb with only one (191.1 Mb/7.93% versus 1 Mb/0.04%) or two (148.6 Mb/6.17% versus 1.1 Mb/0.05%) SNPs. These superior characteristics are primarily due to the wider geographical sampling locations and the unbiased resequencing strategy. Together, higher density, more uniform distribution and fewer SNP rare blocks make our database more effective and accurate in specifying genome locations closely related to certain phenotypes and sequence sites under selection (22).
10 Kb with only one (191.1 Mb/7.93% versus 1 Mb/0.04%) or two (148.6 Mb/6.17% versus 1.1 Mb/0.05%) SNPs. These superior characteristics are primarily due to the wider geographical sampling locations and the unbiased resequencing strategy. Together, higher density, more uniform distribution and fewer SNP rare blocks make our database more effective and accurate in specifying genome locations closely related to certain phenotypes and sequence sites under selection (22).
In the 77 samples collected in DoGSD, 34 are Chinese indigenous dogs, which have never been provided by any other databases. The Chinese indigenous dog has been demonstrated as the first stage in dog domestication (11) and its SNP data set should undoubtedly help researchers to profoundly understand the evolutionary gap between wolf and breed dog.
In addition to SNPs and their annotations, we also provide Fst for almost all SNP, which is an important and widely used population genetic statistic for selection detection. Users may browse and locate SNPs with potential selection signals. The Weir and Cocker-ham method (23) was used to calculate Fst between the wolf and dog populations using the inferred genotypes. Fst values provided in DoGSD are based on all 75 samples from the group 2 data set.
USAGE AND ACCESS
The DoGSD database can be accessed through a simple user interface. Online documentation is provided to help users to access the database. DoGSD has been designed with two main functionalities for data retrieval, Browse and Search. Users may browse the no-redundant and individual sample SNPs either with text format in tables (Figure 2A), or with a chromosome-based graphical GBrowse (24) interface. In the text format tables, SNP ID, sample and sequencing platform information, chromosome location, reference and derived alleles, three-fifths flank sequences are given for each SNP and annotated gene, transcript and protein, derived residual and counterparts from other samples are given if available (Figure 2B). In GBrowse interface, users can obtain Fst values, a pie chart showing the allele frequency of the dog and wolf populations, SNP density in 300 kb windows size, related gene and transcript information (Figure 2C).
Figure 2.
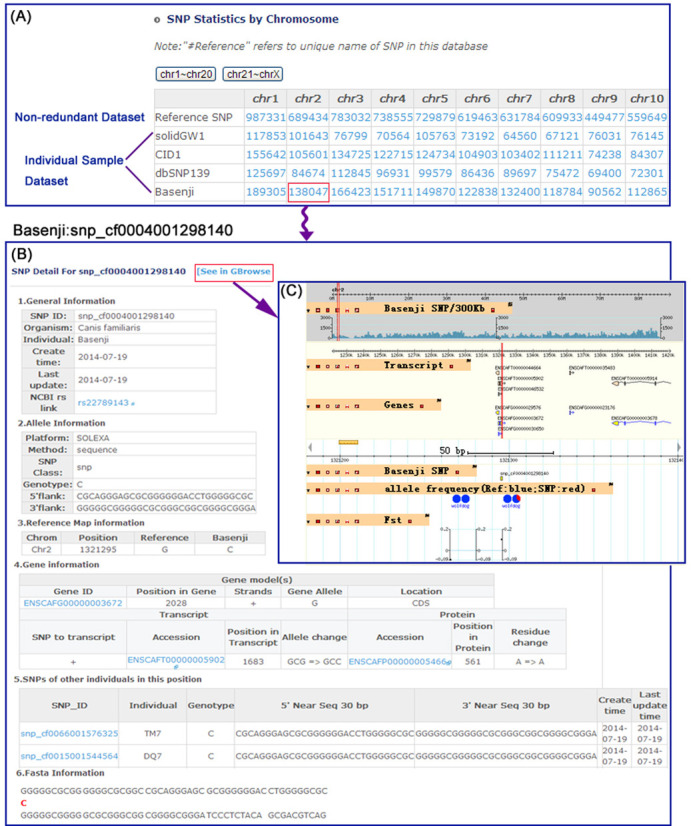
The screen dumps of the SNP lists of DoGSD and typical browse results. The curly arrow indicates two steps are not directly linked and the straight arrow means two steps are directly linked. (A) The non-redundant and individual sample SNP lists. (B) The text format browse result of an SNP. (C) GBrowse visualization of a SNP.
Users may type in chromosome numbers, start and end positions as input to retrieve data for either a single SNP or a group of SNPs. Comparative search of SNPs between two or more individuals were also implemented (Figure 3A). Figure 3B shows a typical comparative search result. Users can compare the SNPs of individuals interested them within a chromosome range.
Figure 3.
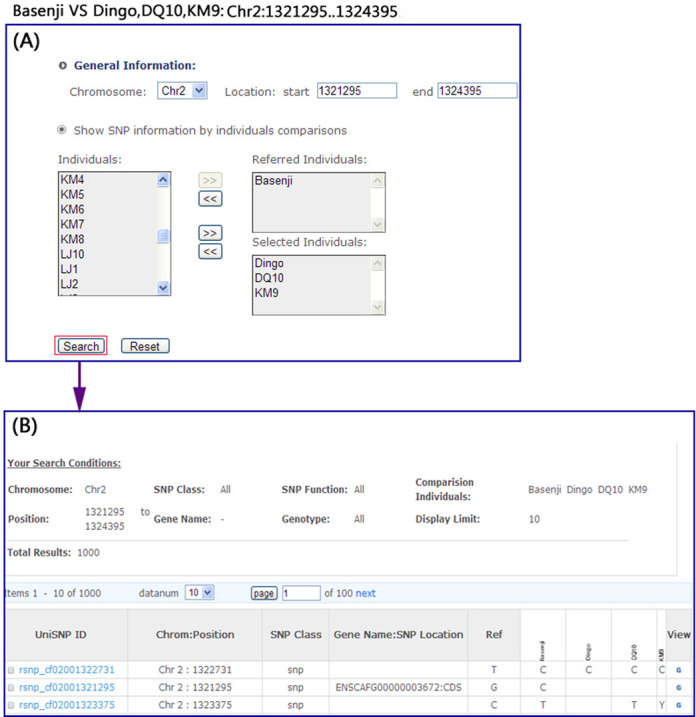
Example of a comparative search. (A) An example of search entry. (B) The comparative search result.
Considering that SNPs locate in genes and coding sequences (CDSs), and especially since non-synonymous SNPs are often of greater interests for further study, we provide several functions within the Browse pull-down menu: SNP Hit On Gene, SNP Coding, SNP Coding Synonymous and SNP Coding NonSynonymous. Users may obtain useful information by summaries of SNPs hit on genes or CDSs and examine whether they are synonymous or non-synonymous.
All the SNP data can be downloaded freely as tab-delimited files and bam/fastq/sra format files of the 75 cited samples are provided.
DISCUSSION
Nowadays, researches towards dog evolution, traits and human co-evolution are still hotspots in biological field. As the first database concentrated on dog/wolf whole genome SNPs, DoGSD stores huge amount of uniformly distributed high-quality SNPs, which compensates for the scarcity of wolf/dog SNPs provided by other databases. With high re-sequencing depth, sampling coverage and calling accuracy from 77 individual samples, our non-redundant SNP data set can be used as a dog SNP reference. Also, users can download individual sample SNP list from our database for population genetic analyses. Especially, SNPs indentified in the Chinese indigenous dog are valuable to researches about early phase of dog evolution. Fst value will also shed light on the selection signals in dog/wolf genomes, guiding researchers to in-depth investigation of these signals on a larger population scale and thoroughly explain the evolutionary driving forces between dog and wolf.
We will keep up with whole-genome SNP data releases and update DoGSD in a timely manner with new released data from population studies of dogs and wolves either by our own research groups or from publicly available resources. We would add uploading functionality to DoGSD by which users could submit whole-genome sequence data or SNP list directly. Once we get the whole-genome sequence, we would identify SNPs in it by group 2 pipeline described in Data processing. Interfaces for calculating Fst and θw will also be developed. We will add additional genetic statistical parameters, such as Tajima D, EHH and recombination maps to the database. Since detection of large chromosomal events promotes our understanding of complex traits and evolution from a perspective that complements SNP, we will further integrate structural variations of dog/wolf genomes into our database in the future.
Footnotes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.
FUNDING
973 program [2013CB835200 and 2013CB835202 to G.D.W.]; The Strategic Priority Research Program of the Chinese Academy of Sciences,Grant No.XDB13040500; Chinese Academy of Sciences [1731200000001 to W.Z.]; The Youth Innovation Promotion Association, Chinese Academy of Sciences [to G.D.W.]. Funding for open access charge: 973 program.
Conflict of interest statement. None declared.
REFERENCES
- 1.Savolainen P., Zhang Y.P., Luo J., Lundeberg J., Leitner T. Genetic evidence for an East Asian origin of domestic dogs. Science (New York, N.Y.) 2002;298:1610–1613. doi: 10.1126/science.1073906. [DOI] [PubMed] [Google Scholar]
- 2.Pang J.F., Kluetsch C., Zou X.J., Zhang A.B., Luo L.Y., Angleby H., Ardalan A., Ekstrom C., Skollermo A., Lundeberg J., et al. mtDNA data indicate a single origin for dogs south of Yangtze River, less than 16,300 years ago, from numerous wolves. Mol. Biol. Evol. 2009;26:2849–2864. doi: 10.1093/molbev/msp195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoenebeck J.J., Hutchinson S.A., Byers A., Beale H.C., Carrington B., Faden D.L., Rimbault M., Decker B., Kidd J.M., Sood R., et al. Variation of BMP3 contributes to dog breed skull diversity. PLoS Genet. 2012;8:e1002849. doi: 10.1371/journal.pgen.1002849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akey J.M., Ruhe A.L., Akey D.T., Wong A.K., Connelly C.F., Madeoy J., Nicholas T.J., Neff M.W. Tracking footprints of artificial selection in the dog genome. Proc. Natl. Acad. Sci. U.S.A. 2010;107:1160–1165. doi: 10.1073/pnas.0909918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kerns J.A., Cargill E.J., Clark L.A., Candille S.I., Berryere T.G., Olivier M., Lust G., Todhunter R.J., Schmutz S.M., Murphy K.E., et al. Linkage and segregation analysis of black and brindle coat color in domestic dogs. Genetics. 2007;176:1679–1689. doi: 10.1534/genetics.107.074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vonholdt B.M., Pollinger J.P., Lohmueller K.E., Han E., Parker H.G., Quignon P., Degenhardt J.D., Boyko A.R., Earl D.A., Auton A., et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature. 2010;464:898–902. doi: 10.1038/nature08837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Axelsson E., Ratnakumar A., Arendt M.L., Maqbool K., Webster M.T., Perloski M., Liberg O., Arnemo J.M., Hedhammar A., Lindblad-Toh K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495:360–364. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- 8.Gou X., Wang Z., Li N., Qiu F., Xu Z., Yan D., Yang S., Jia J., Kong X., Wei Z. Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Res. 2014;24:1315–1308. doi: 10.1101/gr.171876.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freedman A.H., Gronau I., Schweizer R.M., Vecchyo D., Han E., Silva P.M., Galaverni M., Fan Z., Marx P., Lorente-Galdos B., et al. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014;10:e1004016. doi: 10.1371/journal.pgen.1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larson G., Karlsson E.K., Perri A., Webster M.T., Ho S.Y., Peters J., Stahl P.W., Piper P.J., Lingaas F., Fredholm M., et al. Rethinking dog domestication by integrating genetics, archeology, and biogeography. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8878–8883. doi: 10.1073/pnas.1203005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G.D., Zhai W., Yang H.C., Fan R.X., Cao X., Zhong L., Wang L., Liu F., Wu H., Cheng L.G. The genomics of selection in dogs and the parallel evolution between dogs and humans. Nat. Commun. 2013;4 doi: 10.1038/ncomms2814. 1860. [DOI] [PubMed] [Google Scholar]
- 12.Wang G.D., Fan R.X., Zhai W., Liu F., Wang L., Zhong L., Wu H., Yang H.C., Wu S.F., Zhu C.L. Genetic convergence in the adaptation of dogs and humans to the high altitude environment of the Tibetan plateau. Genome Biol. Evol. 2014;6:2122–2128. doi: 10.1093/gbe/evu162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cadieu E., Neff M.W., Quignon P., Walsh K., Chase K., Parker H.G., Vonholdt B.M., Rhue A., Boyko A., Byers A., et al. Coat variation in the domestic dog is governed by variants in three genes. Science (New York, N.Y.) 2009;326:150–153. doi: 10.1126/science.1177808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindblad-Toh K., Wade C.M., Mikkelsen T.S., Karlsson E.K., Jaffe D.B., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., 3rd, Zody M.C., et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 15.Kirkness E.F., Bafna V., Halpern A.L., Levy S., Remington K., Rusch D.B., Delcher A.L., Pop M., Wang W., Fraser C.M., et al. The dog genome: survey sequencing and comparative analysis. Science (New York, N.Y.) 2003;301:1898–1903. doi: 10.1126/science.1086432. [DOI] [PubMed] [Google Scholar]
- 16.Kim R.N., Kim D.S., Choi S.H., Yoon B.H., Kang A., Nam S.H., Kim D.W., Kim J.J., Ha J.H., Toyoda A., et al. Genome analysis of the domestic dog (Korean Jindo) by massively parallel sequencing. DNA Res. 2012;19:275–287. doi: 10.1093/dnares/dss011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang G.-D., Xie H.-B., Peng M.-S., Irwin D., Zhang Y.-P. Domestication genomics evidence with animals. Annu. Rev. Animal Biosci. 2014;2:1–24. doi: 10.1146/annurev-animal-022513-114129. [DOI] [PubMed] [Google Scholar]
- 23.Weir B.C.C. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 24.Donlin M.J. Using the Generic Genome Browser (GBrowse) Curr. Protoc. Bioinformatics. 2009;28:9.9.1–9.9.25. doi: 10.1002/0471250953.bi0909s28. [DOI] [PubMed] [Google Scholar]