


Abstract
The Gene Ontology (GO; http://www.geneontology.org) is a community-based bioinformatics resource that supplies information about gene product function using ontologies to represent biological knowledge. Here we describe improvements and expansions to several branches of the ontology, as well as updates that have allowed us to more efficiently disseminate the GO and capture feedback from the research community. The Gene Ontology Consortium (GOC) has expanded areas of the ontology such as cilia-related terms, cell-cycle terms and multicellular organism processes. We have also implemented new tools for generating ontology terms based on a set of logical rules making use of templates, and we have made efforts to increase our use of logical definitions. The GOC has a new and improved web site summarizing new developments and documentation, serving as a portal to GO data. Users can perform GO enrichment analysis, and search the GO for terms, annotations to gene products, and associated metadata across multiple species using the all-new AmiGO 2 browser. We encourage and welcome the input of the research community in all biological areas in our continued effort to improve the Gene Ontology.
INTRODUCTION
The Gene Ontology (GO) project provides a comprehensive source for functional genomics. The project is a collaborative effort that creates evidence-supported annotations to describe the biological roles of individual genomic products (e.g. genes, proteins, ncRNAs, complexes) by classifying them using our ontologies (1). That is, graph structures comprised of classes for molecular functions, the biological processes these contribute to, the cellular locations where these occur (cellular components), and the relationships connecting these, in a species-independent manner. A ‘GO annotation’ describes the association between a class from the ontology and a gene product, as well as references to the evidence supporting the association. Nearly two decades of efforts make the GO an integrated resource of functional information for genes from over 460 000 species (including strains) covering plants, animals, and the microbial world. The work of the Gene Ontology Consortium (GOC) addresses the need for consistent descriptions of gene products across biological databases, providing not only comprehensive coverage of biological concepts but also community-wide agreement on how those should be used to describe gene functions across all organisms. There are three separate aspects to this effort: (i) the development and maintenance of the ontology, (ii) the annotation of gene products, and (iii) the development and continuous improvement of tools and training that facilitate the creation, maintenance, and use of the ontologies. Here we describe the latest improvements to the tools and resources of the GOC. Ontologies, annotations, and tools are freely available via the Internet at http://www.geneontology.org.
NEW FEATURES AND IMPROVEMENTS
Shared vocabularies are an important step toward unifying biological databases, yet as knowledge changes, the vocabularies and their use necessarily change, resulting in individual curators evaluating data differently. To address the concern of inconsistent data representation, the GOC continuously provides enhancements to its tools, resources, and policies, improving the annotation consistency and ensuring that annotations reflect the current state of biological knowledge. This section discusses our latest advances.
Ontology development
The GOC has engaged in various projects and collaborations with the goal of expanding and improving the representation of biology. The total number of GO terms has been steadily increasing from around 18 000 to more than 40 000 between 2004 and 2014; over 5300 new terms were added to the GO since our last report ((2); Table 1). Compared to the number of GO terms added to describe molecular functions and cellular components, the number of terms to describe biological processes (BP) has increased at a higher rate, averaging 4000 new BP terms every two years since 2011 (2,3). The GOC has also seen a steady increase in the number of manual annotations made by curators (2), and the number of manually annotated gene products has grown to almost 400 000 (Table 1).
Table 1. Annotation production statusa.
| Total number of GO terms | 41 775 | |
| Biological process terms | 27 284 | |
| Molecular function terms | 10 733 | |
| Cellular component terms | 3758 | |
| Species with annotations | 461 573 | |
| Total annotated gene products b | 53 042 843 | |
| Manually annotated (experimental) gene products | 311 335 | |
| Manually annotated (phylogenetic) gene products | 79 839 | |
| Total annotations | 4 185 487 |
aAs of September 2014.
bIncludes isoforms.
Significant work was recently undertaken in the cellular component branch. The Subcellular Anatomy Ontology (SAO), part of the Neuroscience Information Framework Standard (NIFSTD) suite of ontologies, was merged into the GO cellular component representation. The SAO also describes cell components, but in the domain of neuroscience. The major effort to merging the SAO into GO was the manual examination of terms to determine which terms were already in GO with or without the same name, the addition of terms to GO that were not already in GO, and whether some terms in SAO were out of scope for GO. This resulted in a single, unified ontology designed to serve the needs of both the neuroscience community (4) as well as the broader biomedical research community already served by the GO. The GOC is also currently working with the SYSCILIA Consortium ((5); http://syscilia.org/) to improve the representation of ciliary substructures in the GO cellular component branch, with plans to also improve the biological process branch. Fifty cilia-related terms have been added or modified thus far. Curators at The Mouse Genome Informatics (MGI) resource have already started using the new terms, with a focused effort to annotate ciliary proteins. The new cilia terms will also be used by the SYSCILIA Consortium to annotate human proteins with a focus on ciliopathies. The GOC is also working with researchers from the parasitic flagellates (Diplomonads) community to extend GO coverage of biological concepts that are specific to species in this taxonomic group, such as Giardia and related. Approximately 30 new terms have been added to the cellular component branch to describe substructures that are specific to the Diplomonads.
Ontology editors also carried out an effort to update and refine other areas of the ontology. We have commenced an effort to temporally delimit cellular processes using logical definitions such as the starts_with and ends_with relationships from the OBO Relations Ontology (http://obo-relations.googlecode.com). For example, ‘apoptotic process’ starts_with ‘apoptotic signaling pathway’ and ends_with ‘execution phase of apoptosis’. Our goal is to apply this pattern throughout the cellular process branch of the GO, in order to better enforce annotator consistency across different GO annotation sources, and to allow for a limited form of temporal reasoning over the ontology, all of which results in greater interpretability of GO-based analyses for all users. We have also made a number of enhancements in the OWL version of the GO to better support automated quality control and classification as part of the ontology development cycle, these are described in a separate publication (6). Box 1 describes an example of an OWL stanza for a term that is defined by a logical definition.
Box 1. The ‘L-glutamate import across plasma membrane’ stanza.
name: L-glutamate import across plasma membrane
equivalentTo:
transport that (‘has target start location’ some ‘extracellular region’)
and (‘has target end location’ some cytosol)
and (imports some ‘L-glutamate’)
and (‘results in transport across’ some ‘plasma membrane’)
inferred classifications:
‘import across plasma membrane’
‘L-alpha-amino acid transmembrane transport’
‘L-glutamate import into cell’
In this example, ‘L-glutamate import across plasma membrane’ has a logical definition (OWL equivalentTo) that specifies necessary and sufficient conditions for membership of the class. These conditions include the substance imported, what it is transported across, and its ‘start’ and ‘end’ locations. As shown, this information is sufficient for automated classification under a number of classes including one based on classification of the chemical transported. This automated classification relieves the editors of the unsustainable task of manually finding appropriate classifications for each term they add, and of keeping these classifications up to date as the ontology changes.
It is not necessarily desirable to add logical definitions to all classes. In some cases it may not be possible to come up with necessary and sufficient conditions for class membership that sufficiently reflect the way biologists classify a process. In other cases, we do not yet have the required formalizations. As a result, most complex root processes are not defined using logical definitions. For those processes, we limit ourselves to recording necessary conditions for class membership (relationships), for example apoptosis has relationships defining its beginning and end. Box 2 shows a snipped version of the apoptosis stanza.
Box 2. The ‘apoptotic process’ stanza.
‘apoptotic process’
def: 'A programmed cell death process which begins when a cell receives an internal (e.g. DNA damage) or external signal (e.g. an extracellular death ligand), and proceeds through a series of biochemical events (signaling pathway phase) which trigger an execution phase. The execution phase is the last step of an apoptotic process, and is typically characterized by rounding-up of the cell, retraction of pseudopodes, reduction of cellular volume (pyknosis), chromatin condensation, nuclear fragmentation (karyorrhexis), plasma membrane blebbing and fragmentation of the cell into apoptotic bodies. When the execution phase is completed, the cell has died.'
SubClassOf: ’programmed cell death’
start_with some ‘apoptotic signaling pathway’
ends_with some ‘execution phase of apoptosis’
A version of the ontology containing all relationships, including information from the Uberon anatomy (or stage) ontology (7), the Chemical Entities of Biological Interest ontology (ChEBI; (8)), the Plant Ontology for plant structure/stage (PO; (9)), the Phenotypic Quality Ontology (PATO; (10)) and the Sequence Ontology (SO; (11)), is called go-plus and is available at http://geneontology.org/page/download-ontology. The GOC also makes other versions of the ontology available at this site.
Cell cycle processes
We have begun extensive improvements to the ontology terms describing the cell cycle, and the revision of annotations using these terms. For two days curators, ontology developers and invited cell cycle experts (Takashi Toda and Jacqueline Hayles CRUK London UK and Rob De'Bruin UCL) met at the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI) in Cambridge, UK. The subsequent cell cycle overhaul has addressed longstanding issues including making the cell cycle node prokaryote compliant and enabling the positioning of cytokinesis, DNA replication and spindle organization annotations under their respective mitotic or meiotic cell cycle nodes. The terms that represent the regulation of eukaryotic cell cycle progression were revised to provide a better representation of known biological events. This was achieved by creating a grouping term for cell cycle transitions and repositioning the checkpoint terms to represent them as negative regulators of the specific transitions which they block (for example the spindle assembly checkpoint negatively regulates the metaphase/anaphase transition). A new node, disjoint from checkpoint signaling was created for checkpoint responses to annotate processes that correct the problems that activate the checkpoints. An additional outcome of the cell cycle work was that cell cycle phases are no longer subclasses or parts of the cell cycle. Instead, they are classified under a new term ‘biological phase’. Biological phases are intervals in which biological processes can occur, and as such are is_a disjoint—i.e. they share no parent terms via the is_a relation—with other biological processes. Biological processes and biological phases are instead related by the happens_during relation. The majority of proposed ontology changes have been implemented; further refinements and improvements are ongoing. Currently, existing annotations are being re-assessed and annotation guidelines are being created to improve the specificity of the existing annotations.
Multi-organism processes
We are now nearing completion of a long-running project to better model multi-organism processes and cellular components in GO. Multi-organism processes include all processes involving hosts and their parasites, including viruses. In collaboration with the Swiss Institute of Bioinformatics (SIB) ViralZone project, we have developed a comprehensive set of GO terms to describe viral processes and cellular components (12) These terms are being used to annotate viral and host gene products from a range of species. We have also extended the GO annotation system to allow annotators to record the relationships between interacting organisms involved in multi-organism processes. To do this we have defined a set of relations that hold between interacting organisms, which include symbiont_of, host_of, parasite_of and vector_for. GO annotations can now record in which of the two organisms process occurs.
TermGenie
One of the ongoing challenges in the GO is to streamline the process of generating new terms in response to requests from curators. TermGenie is a web-based tool for requesting new GO classes ((13); http://geneontology.org/page/termgenie). It exploits the fact that many ontology terms conform to documented design patterns and uses a template-based system, and logical reasoning to facilitate the expansion of GO, enabling curators to rapidly generate new terms while ensuring validity, uniqueness, and proper relationships to other classes. TermGenie also allows for an ontology developer to review all generated terms before they are committed to the ontology. The system makes extensive use of OWL axioms (logical definitions), but can be easily used without understanding these axioms. Using TermGenie helps replace traditional trackers and tools, shifting from an inherently inefficient, entirely manual process to a semi-automatic and scalable approach to adding new terms. Between July 2010 and June 2014 the GO TermGenie instance has been used to generate 4715 terms; this represents more than half (51.4%) of all new terms created during that period. TermGenie relies heavily on reasoning for automatic classification and validation. This requires the GO to be sufficiently axiomatized with equivalent class axioms (a.k.a. logical definitions or cross-products). This formalization effort is still an ongoing task, which includes creating intra-ontology definitions (14), and using other domain-specific ontologies, such as PATO (10), ChEBI (8), PO (9), Uberon (7), Cell Ontology (CL) (15), Sequence Ontology (SO) (11), Ontology of Biological Attributes (OBA; (16)) and the Protein Ontology (PRO) (17) for cross-products definitions. At present, there are over 40 available template forms for requesting new terms, and this number continues to grow. Examples include templates for creating new terms to describe relationships such as ‘regulation of biological process’ or ‘chemical response to’. TermGenie can be found at http://go.termgenie.org/, the source code is available from Google code at https://code.google.com/p/termgenie and all changes to the repository are listed at https://code.google.com/p/termgenie/source/list.
Continuous integration
As reported before, the GOC uses an open-source continuous integration system (Jenkins; http://jenkins-ci.org/) to validate the ontology. The same approach is in use for many other ontology related tasks, such as generating the custom ontology subsets for each external ontology in the GO, and generating derivative files (such as the external mapping files). Furthermore, we use the same approach for validating the annotations submitted to the GOC from 26 different contributing groups around the world. There are also Jenkins jobs for integrating the annotations generated using with the Phylogenetic Annotation Inference Tool (PAINT (18)), and to generate summary statistics for the current annotations. At the same time we use Jenkins to continuously test and build the software tools required for these task.
Annotation
Over the last year, the GOC has introduced additional metadata to better describe the biological context of an annotation (14,19). These ‘annotation extensions’ represent relationships such as localization dependencies, substrates of protein modifiers and regulation targets of signaling pathways, and transcription factors as well as spatial and temporal aspects of processes such as cell or tissue type or developmental stage. The information expressed by these extensions refines the functional annotations by representing relationships between a basic annotation and contextual information from either within the GO or from external ontologies. Extended annotations can enable complex queries and reasoning such as inquiring about the type of cell or anatomical structures in which an annotated biological process occurs.
The GOC phylogenetic annotation project (18) has been expanded since our last published update. This project continues to produce expert human-reviewed inferred annotations, by integrating experimental annotations from ‘model organisms’ into detailed gene family-specific models of gene function evolution and conservation. The project now provides inferred annotations for 85 organisms (http://pantherdb.org/panther/summaryStats.jsp). Currently, inferred annotations are available for over 500 gene families, totaling about 370 000 annotations for about 80 000 genes. These annotations can be identified using the ‘IBA’ (inferred from biological ancestor) evidence code, and downloaded from the GO web site using AmiGO 2.
Model Organism Databases (MOD) and the Gene Ontology Annotation group (GOA) at UniProt provide the bulk of the annotations that the GOC distributes. As is the case with MODs, the GOA group incorporates manual literature-based annotations and is responsible for providing annotations for human, cow, dog, and chicken. The manually annotated gene products with experimental evidence are distributed across the MODs. These annotations are created by experienced biocurators using both published experimental results and tools developed for their own projects. Many contributing groups are transitioning to using Protein2GO. Developed by the GOA group, Protein2GO processes only protein sequences but is currently being expanded to include RNA gene products and macromolecular complexes. GOA provides access to roughly 98% of the total number of species with annotations available from InterPro, Ensembl, and UniProt. GOA produces these using a sophisticated computational pipeline that implements several rules and methods to assert annotations including shared protein domains and sequence homology (20).
The GOC encourages and welcomes experts to provide input in various biological areas. For example, a recent collaboration with the Transcription Factor Checkpoint database (http://www.tfcheckpoint.org/) has expanded annotation to human, mouse, and rat transcription factors (21), and the Developmental Functional Annotation at Tufts (DFLAT) project improved the quality of annotations of genes involved in fetal development curating human fetal gene functions using both manual and semi-automated annotation (22). In a joint collaboration between Gramene (www.gramene.org) and Ensembl Plants (http://plants.ensembl.org) initial GO annotations are now provided for ∼37 sequenced plant genomes as of the current release (23).
Summary of gene ontology annotations
All GO annotations here described are summarized in Table 1.
Implementation and public access
The new GO website
In the summer of 2014, we publicly released a new website for the GOC. It is a major reimplementation with a fresh look, user-friendly features to facilitate reading and navigation, and the latest data and documentation about the project. This reorganization aims to keep all content consistent and up to date, as well as clarifying use policies and licensing, which were standardized on CC-BY 4.0 (https://creativecommons.org/licenses/by/4.0/) for GO data and content. This new site (Figure 1) is also the cornerstone in an effort to centralize community messaging and outreach, bringing together news, current documentation, social media, sub-project information, integration (both stylistically and functionally) with AmiGO 2, and more. Some highlights include: direct access to GO Term Enrichment tools and annotation statistics organized by species, source database, and supporting evidence; highlights on recent news and publications from the members of the consortium and integrated interactions with the wider research community through social media outlets such as Facebook and Twitter. To accomplish this, the new GO website is based on the latest stable version of Drupal (Drupal 7; http://drupal.org), a robust open-source content management system that offers a flexible and extensible way to design and organize content. In the case of social media data, the Facebook feed is pulled into our aggregator, and the Twitter feed (including the widget currently available on the front page), is updated through an automatic ‘publish’ action that feeds the GO Twitter profile when new ‘article’ pages are created on the GO website. This database-backed environment also allows for better control of revisions through time as well as enforced workflows that enable the GOC to allow the consortium members to keep information up to date independent of developer availability. We encourage the public to access and contribute to the efforts of the GOC at http://geneontology.org.
Figure 1.
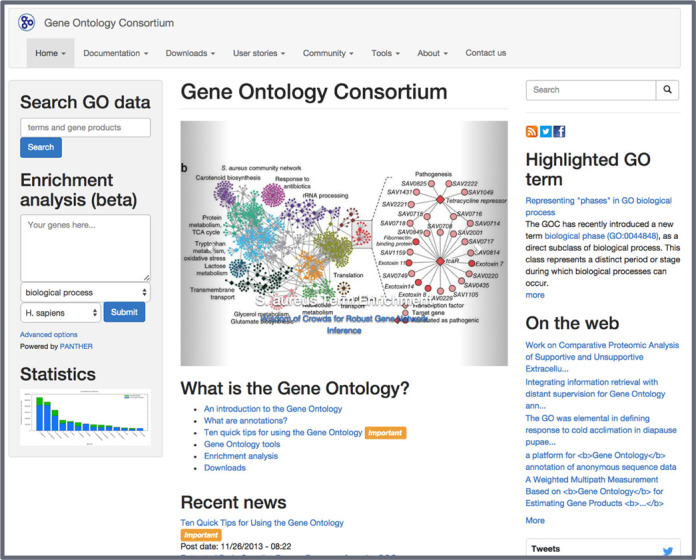
The new Gene Ontology web page. In addition to access to documentation, ontologies, and annotation sets (drop-down menus on top), users can immediately search on GO data (terms and annotations) using the search box, and even perform gene enrichment analysis.
Browsing GO annotations
AmiGO 2 is the new official web-based open-source set of tools for querying, browsing, and visualizing the GO data (Figure 2). Publicly released in the spring of 2014, this new framework provides many architectural changes and improvements to help keep pace with the needs of the community. There are huge improvements in speed and in the variety of search modes, as well as the availability of additional data types, such as the display of annotation extensions and display of protein forms (splice variants and proteins with post translational modifications) (Figure 3). The AmiGO 2 set of tools also provides a JavaScript API for better access and integration with other tools, and both provides and consumes REST APIs to help better integrate resources, such as the PANTHER database (http://pantherdb.org, (24)) for enrichment analysis. There has also been a complete re-skinning of most of the tools with modern methods and styles to improve usability and access across diverse platforms. Under the hood, AmiGO 2 is broken into two distinct layers: (i) the various client software tools and (ii) the data backend. This client/server-oriented architecture allows for greater flexibility and more efficient addition of new functionality. The client software is now mostly written in JavaScript, allowing search-as-you-type interfaces and greater user interactivity, and some Perl for handling synchronous operations. For the backend, the data store for AmiGO 2 has moved away from a MySQL relational database backend and instead uses a specially generated schema for the Solr search platform (http://lucene.apache.org/solr/; dubbed GOlr) to allow for complex and efficient ontology and data queries.
Figure 2.
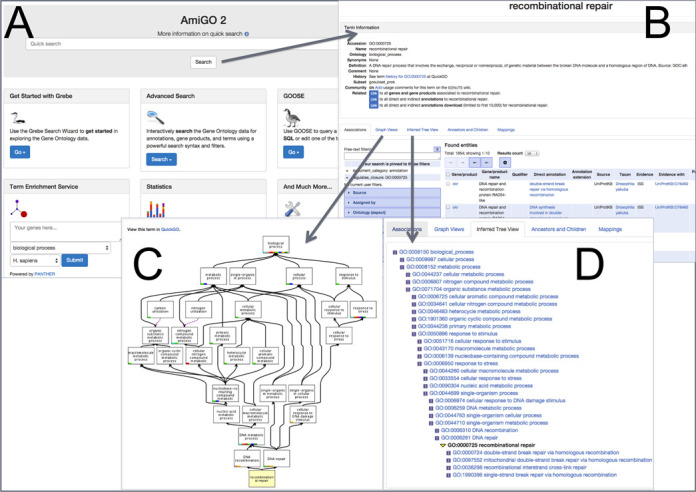
The new Gene Ontology browser AmiGO 2. (A) The entry portal page, where simple or complex queries can be performed, as well as term enrichment analysis. (B) Sample output from a ‘Quick Search’ on a GO term (recombinational repair). (C) Graphical Display and (D) Tree View of this GO term and its placement within the ontology.
Figure 3.
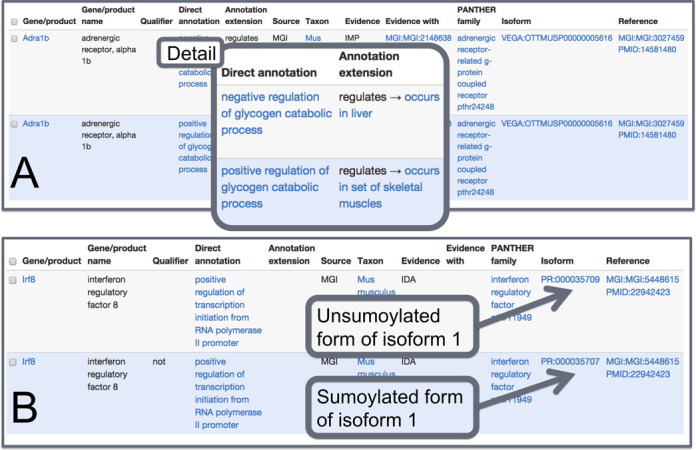
Sample search results from AmiGO2. (A) Shows two annotations of the same gene to both ‘negative’ and ‘positive’ regulation of glycogen catabolic process; the difference between the two lies in data entry on the ‘Annotation Extension’ column, showing that the experiments were performed in different tissues, i.e. liver and skeletal muscle. (B) Shows two annotations that only differ on whether the gene product ‘does’ or ‘does not’ positively regulate transcription initiation from an RNA polymerase II promoter. The data in the ‘Isoform’ column represents that the unsumoylated form ‘does’, whereas the sumoylated from ‘does not’.
Community outreach and user support
The GOC provides platforms of interaction and welcomes participation from the community through our Helpdesk (http://geneontology.org/form/contact-go), to address general inquiries, and the Sourceforge tracker (http://sourceforge.net/p/geneontology/ontology-requests/) to address specific requests for the ontology.
FUTURE DIRECTIONS
Our plans to the future include the consolidation of an Education and Outreach Portal on the GO website, which will include instructional materials via slide presentations, web content, and video tutorials to facilitate understanding and usage of GO resources. Additionally, as we wish to continuously expand the scope of GO, we are extending an invitation to research groups interested in conducting and submitting annotations using data from microbiome experiments to please contact us with ideas and proposed approaches. These and all efforts allow us to work toward developing a coordinated set of web-based tools to streamline and semi-automate annotation and help curators become more efficient, as well as to lower the barrier for others in the broader research community to participate in GO annotation.
Acknowledgments
We gratefully acknowledge the many other contributors to GO annotation efforts: Gramene, Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR, USA; The J. Craig Venter Institute, Rockville, MD, USA; PAMGO, Wells College, Aurora, NY, USA and PAMGO, Virginia Bioinformatics Institute, VA, USA; AspGD, Stanford, CA, USA; CGD, Stanford, CA, USA; Sanger GeneDB, Hinxton, UK; InterPro European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI), Wellcome Trust Genome Campus, Hinxton, UK; IntAct, (EMBL-EBI), Hinxton, UK; seudoCAP, British Columbia, Canada; SGN, Ithaca, NY, USA.
APPENDIX
The following is a list of the members of the Gene Ontology Consortium, whom together author this article. Authors marked with a star (*) prepared the bulk of the manuscript. MGI, The Jackson Laboratory (Bar Harbor, ME, USA): J.A. Blake, K.R. Christie, M.E. Dolan, H.J. Drabkin*, D.P. Hill*, L. Ni, D. Sitnikov; AgBase, Mississippi State University (Starkville, MS, USA): S. Burgess, T. Buza, C. Gresham, F. McCarthy, L. Pillai, H. Wang; Berkeley Bioinformatics Open-Source Projects, Genomics Division, Lawrence Berkeley National Laboratory (Berkeley, CA, USA): S. Carbon*, H. Dietze*, S.E. Lewis, C.J. Mungall*, M.C. Munoz-Torres*; CALIPHO group, SIB Swiss Institute of Bioinformatics (Geneva, Switzerland): M. Feuermann, P. Gaudet; dictyBase, Northwestern University (Chicago, IL, USA): S. Basu, R.L. Chisholm, R.J. Dodson, P. Fey; Division of Bioinformatics, Department of Preventive Medicine, University of Southern California (Los Angeles, CA, USA): H. Mi, P.D. Thomas, A. Muruganujan, S. Poudel; EcoliWiki, Departments of Biology and Biochemistry and Biophysics, Texas A&M University (College Station, TX, USA): J.C. Hu, S.A. Aleksander, B.K. McIntosh, D.P. Renfro, D.A. Siegele; FlyBase, Gurdon Institute and Department of Genetics, University of Cambridge (Cambridge, UK): H. Attrill, N.H. Brown, S. Tweedie; GO-EMBL-EBI (Hinxton, UK): J. Lomax*, D. Osumi-Sutherland*, H. Parkinson, P. Roncaglia*; Institute of Cardiovascular Science, University College London (London, UK): R.C. Lovering, P.J. Talmud, S.E. Humphries, P. Denny, N.H. Campbell, R.E. Foulger; Institute for Genome Sciences, University of Maryland School of Medicine (Baltimore, MD, USA): M.C. Chibucos, M. Gwinn Giglio; InterPro, EMBL-EBI (Hinxton, UK): H.Y. Chang, R. Finn, M. Fraser, A. Mitchell, G. Nuka, S. Pesseat, A. Sangrador, M. Scheremetjew, S.Y. Young; Collection and Refinement of Physiological Data on Mycobacterium tuberculosis MTBBASE (Berlin, Germany): R. Stephan; PomBase, University of Cambridge (Cambridge, UK): M.A. Harris, S.G. Oliver, K. Rutherford, V. Wood; PomBase, University College (London UK): J. Bahler, A. Lock; PomBase, EMBL-EBI (Hinxton UK): P.J. Kersey, M.D. McDowall, D.M. Staines; RGD, Medical College of Wisconsin (Milwaukee, WI, USA): M. Dwinell, M. Shimoyama, S. Laulederkind, G.T. Hayman, S.J. Wang, V. Petri; Reactome, Department of Biochemistry & Molecular Pharmacology, NYU School of Medicine (New York, NY, USA): P. D'Eustachio, L. Matthews; SGD, Department of Genetics, Stanford University (Stanford, CA, USA): R. Balakrishnan, G. Binkley, J.M. Cherry, M.C. Costanzo, J. Demeter, S.S. Dwight, S.R. Engel, B.C. Hitz, D.O. Inglis, P. Lloyd, S.R. Miyasato, K. Paskov, G. Roe, M. Simison, R.S. Nash, M.S. Skrzypek, S. Weng, E.D. Wong; TAIR, Phoenix Bioinformatics (Redwood City, CA, USA): T.Z. Berardini, D. Li, E. Huala; UniProt: EMBL-EBI (Hinxton, UK), SIB Swiss Institute of Bioinformatics (SIB) (Geneva, Switzerland), and Protein Information Resource (PIR) (Washington, DC, USA): J. Argasinska, C. Arighi, A. Auchincloss, K. Axelsen, G. Argoud-Puy, A. Bateman, B. Bely, M.C. Blatter, C. Bonilla, L. Bougueleret, E. Boutet, L. Breuza, A. Bridge, R. Britto, C, Casals, E. Cibrian-Uhalte, E. Coudert, I. Cusin, P. Duek-Roggli, A. Estreicher, L. Famiglietti, P. Gane, P. Garmiri, A. Gos, N. Gruaz-Gumowski, E. Hatton-Ellis, U. Hinz, C. Hulo, R. Huntley, F. Jungo, G. Keller, K. Laiho, P. Lemercier, D. Lieberherr, A. MacDougall, M. Magrane, M. Martin, P. Masson, P. Mutowo, C. O'Donovan, I. Pedruzzi, K. Pichler, D. Poggioli, S. Poux, C. Rivoire, B. Roechert, T. Sawford, M. Schneider, A. Shypitsyna, A. Stutz, S. Sundaram, M. Tognolli, C. Wu, I. Xenarios; WormBase, California Institute of Technology (Pasadena, CA, USA): J. Chan, R. Kishore, P.W. Sternberg, K. Van Auken, H.M. Muller, J. Done, Y.Li; ZFIN, University of Oregon (Eugene, OR, USA): D. Howe, M. Westerfield.
FUNDING
National Institutes of Health/National Human Genome Research Institute grant [HG002273] awarded to the PI group formed by Judith A. Blake, J. Michael Cherry, Suzanna E. Lewis, Paul W. Sternberg, and Paul D. Thomas, as well as additional funding awarded to each participating institution. For more details please visit: http://geneontology.org/page/go-consortium-contributors-list. Funding for open access charge: National Institutes of Health/National Human Genome Research Institute [HG002273].
Disclaimer: Portions of this document include an account of work sponsored by the United States Government at Lawrence Berkeley National Laboratory. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California.
Conflict of interest statement. None declared.
REFERENCES
- 1.The Gene Ontology Consortium. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The Gene Ontology Consortium. Gene Ontology annotations and resources. Nucleic Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The Gene Ontology Consortium. The Gene Ontology: enhancements for 2011. Nucleic Acids Res. 2012;40:D559–D564. doi: 10.1093/nar/gkr1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roncaglia P., Martone M., Hill D., Berardini T., Foulger R., Imam F., Drabkin H., Mungall C., Lomax J. The Gene Ontology (GO) Cellular Component Ontology: integration with SAO (Subcellular Anatomy Ontology) and other recent developments. J. Biomed. Semant. 2013;4:20. doi: 10.1186/2041-1480-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Dam T., Wheway G., Slaats G., Group S.S., Huynen M., Giles R. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia. 2013;2:7. doi: 10.1186/2046-2530-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mungall C.J., Dietze H., Osumi-Sutherland D. Proceedings of the 11th International Workshop on OWL: Experiences and Directions. Riva del Garda, Italy: 2014. Use of OWL within the Gene Ontology; pp. 25–36. [Google Scholar]
- 7.Mungall C., Torniai C., Gkoutos G., Lewis S., Haendel M. Uberon, an integrative multi-species anatomy ontology. Genome Biol. 2012;13:R5. doi: 10.1186/gb-2012-13-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hastings J., de Matos P., Dekker A., Ennis M., Harsha B., Kale N., Muthukrishnan V., Owen G., Turner S., Williams M., et al. The ChEBI reference database and ontology for biologically relevant chemistry: enhancements for 2013. Nucleic Acids Res. 2013;41:D456–D463. doi: 10.1093/nar/gks1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper L., Walls R.L., Elser J., Gandolfo M.A., Stevenson D.W., Smith B., Preece J., Athreya B., Mungall C.J., Rensing S., et al. The Plant Ontology as a tool for comparative plant anatomy and genomic analyses. Plant Cell Physiol. 2013;54:e1. doi: 10.1093/pcp/pcs163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gkoutos G., Green E., Mallon A.-M., Hancock J., Davidson D. Using ontologies to describe mouse phenotypes. Genome Biol. 2004;6:R8.1–R8.10. doi: 10.1186/gb-2004-6-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eilbeck K., Lewis S., Mungall C., Yandell M., Stein L., Durbin R., Ashburner M. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 2005;6:R44.1–R44.12. doi: 10.1186/gb-2005-6-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masson P., Hulo C., de Castro E., Foulger R., Poux S., Bridge A., Lomax J., Bougueleret L., Xenarios I., Le Mercier P. An Integrated Ontology Resource to Explore and Study Host-Virus Relationships. PLoS ONE. 2014;9:e108075. doi: 10.1371/journal.pone.0108075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dietze H., Berardini T.Z., Foulger R.E., Hill D.P., Lomax J., Osumi-Sutherland D., Roncaglia P., Mungall C.J. TermGenie – a web-application for pattern-based ontology class generation. J. Biomed. Sem. 2014 doi: 10.1186/2041-1480-5-48. in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mungall C.J., Bada M., Berardini T.Z., Deegan J., Ireland A., Harris M.A., Hill D.P., Lomax J. Cross-product extensions of the Gene Ontology. J. Biomed. Inform. 2011;44:80–88. doi: 10.1016/j.jbi.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bard J., Rhee S., Ashburner M. An ontology for cell types. Genome Biol. 2005;6:R21.1–R21.5. doi: 10.1186/gb-2005-6-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dönitz J., Wingender E. The ontology-based answers (OBA) service: A connector for embedded usage of ontologies in applications. Front. Genet. 2012;3 doi: 10.3389/fgene.2012.00197. doi:10.3389/fgene.2012.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Natale D.A., Arighi C.N., Barker W.C., Blake J.A., Bult C.J., Caudy M., Drabkin H.J., D'Eustachio P., Evsikov A.V., Huang H., et al. The Protein Ontology: a structured representation of protein forms and complexes. Nucleic Acids Res. 2011;39:D539–D545. doi: 10.1093/nar/gkq907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaudet P., Livstone M.S., Lewis S.E., Thomas P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011;12:449–462. doi: 10.1093/bib/bbr042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huntley R., Harris M., Alam-Faruque Y., Blake J., Carbon S., Dietze H., Dimmer E., Foulger R., Hill D., Khodiyar V., et al. A method for increasing expressivity of Gene Ontology annotations using a compositional approach. BMC Bioinformatics. 2014;15:155. doi: 10.1186/1471-2105-15-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimmer E.C., Huntley R.P., Alam-Faruque Y., Sawford T., O'Donovan C., Martin M.J., Bely B., Browne P., Mun Chan W., Eberhardt R., et al. The UniProt-GO annotation database in 2011. Nucleic Acids Res. 2012;40:D565–D570. doi: 10.1093/nar/gkr1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tripathi S., Christie K.R., Balakrishnan R., Huntley R., Hill D.P., Thommesen L., Blake J.A., Kuiper M., Lægreid A. Gene Ontology annotation of sequence-specific DNA binding transcription factors: setting the stage for a large-scale curation effort. Database. 2013;2013:bat062. doi: 10.1093/database/bat062. doi:10.1093/database/bat062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wick H., Drabkin H., Ngu H., Sackman M., Fournier C., Haggett J., Blake J., Bianchi D., Slonim D. DFLAT: functional annotation for human development. BMC Bioinformatics. 2014;15:45. doi: 10.1186/1471-2105-15-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monaco M.K., Stein J., Naithani S., Wei S., Dharmawardhana P., Kumari S., Amarasinghe V., Youens-Clark K., Thomason J., Preece J., et al. Gramene 2013: comparative plant genomics resources. Nucleic Acids Res. 2014;42:D1193–D1199. doi: 10.1093/nar/gkt1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mi H., Muruganujan A., Thomas P.D. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]